Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hernando Vargas-uricoechea | -- | 14600 | 2023-03-24 01:25:45 | | | |
| 2 | Lindsay Dong | -9892 word(s) | 4708 | 2023-03-24 06:12:44 | | | | |
| 3 | Lindsay Dong | Meta information modification | 4708 | 2023-03-24 10:17:31 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Vargas-Uricoechea, H. Molecular Mechanisms in Autoimmune Thyroid Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/42495 (accessed on 28 July 2026).
Vargas-Uricoechea H. Molecular Mechanisms in Autoimmune Thyroid Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/42495. Accessed July 28, 2026.
Vargas-Uricoechea, Hernando. "Molecular Mechanisms in Autoimmune Thyroid Disease" Encyclopedia, https://encyclopedia.pub/entry/42495 (accessed July 28, 2026).
Vargas-Uricoechea, H. (2023, March 24). Molecular Mechanisms in Autoimmune Thyroid Disease. In Encyclopedia. https://encyclopedia.pub/entry/42495
Vargas-Uricoechea, Hernando. "Molecular Mechanisms in Autoimmune Thyroid Disease." Encyclopedia. Web. 24 March, 2023.
Copy Citation
The most common cause of acquired thyroid dysfunction is autoimmune thyroid disease, which is an organ-specific autoimmune disease with two presentation phenotypes: hyperthyroidism (Graves-Basedow disease) and hypothyroidism (Hashimoto’s thyroiditis). Hashimoto’s thyroiditis is distinguished by the presence of autoantibodies against thyroid peroxidase and thyroglobulin. Meanwhile, autoantibodies against the thyroid-stimulating hormone (TSH) receptor have been found in Graves-Basedow disease. Numerous susceptibility genes, as well as epigenetic and environmental factors, contribute to the pathogenesis of both diseases.
thyroid
autoimmunity
Graves-Basedow
Hashimoto
pathogenesis
1. Introduction
Autoimmune diseases (ADs) are a heterogeneous group of more than 100 pathological conditions that are characterized by an alteration in the regulation of inflammatory processes against one or multiple autoantigens [1][2].
ADs are usually classified as organ-specific (OS) or non-organ-specific (NOS), depending on whether they affect one organ or several; in NOS ADs, the autoimmune activity is systemic (as in systemic lupus erythematosus). In OS ADs, the immune response is directed toward single-organ antigens. The most common OS AD is autoimmune thyroid disease (AITD) [3][4][5][6].
The AITD spectrum includes Graves–Basedow disease (GBD) and Hashimoto’s thyroiditis (HT), with two extremes of clinical presentation: hyperthyroidism (in the case of GBD) and hypothyroidism (in the case of HT). However, cases can be identified in which, in the presence of thyroid autoimmunity, no clinical or biochemical manifestations of hypothyroidism or hyperthyroidism are identified.
Additionally, some subjects with HT may progress to GBD (or vice versa), and it is possible to identify individuals with simultaneous manifestations of both GBD and HT [7][8].
In fact, some individuals with biochemical findings of thyroid autoimmunity, typical of GBD (with the presence of thyroid ophthalmopathy), may present hypothyroidism (even requiring levothyroxine replacement). Similarly, some individuals with biochemical evidence of autoimmunity, typical of HT, may present with long-standing hyperthyroidism (with associated ophthalmopathy and dermopathy).
In GBD, there is a loss of immune tolerance, with the infiltration of T lymphocytes (TLs) in the thyroid and the activation of B lymphocytes (BLs), as well as an increase in the synthesis and secretion of autoantibodies directed against the TSH receptor (TSHR). Consequently, the interaction between the TSHR and its specific autoantibody (TRAb) causes an immune response that results in goiter, hyperthyroidism, ophthalmopathy, and dermopathy [9].
In HT, there is a cellular immune response with a high inflammatory load and apoptosis, which causes tissue destruction and thyroid dysfunction. HT also shares humoral mechanisms with GBD, with the presence of autoantibodies (Abs) against thyroid peroxidase (TPO) and thyroglobulin (Tg) [10].
It is generally accepted that AITD is the product of multiple environmental factors that act based on genetic susceptibility, together with some epigenetic mechanisms. However, the molecular mechanism by which immune dysfunction leads to the destruction of thyroid tissue remains largely unexplained [11][12][13][14][15].
2. Animal Models of AITD
The experimental models of ADs in animals are of two types: spontaneous, in which animals with or without genetic modifications develop the disease spontaneously, and induced, in which the outcome is developed artificially. In animal models of induced AITD, the strategy is based on the use of crude thyroid extracts, purified Tg or thyroid peroxidase (TPO), and selected ectodomains. Of these models, the best studied use nonobese diabetic (NOD) mice, which highlights the fact that NOD mice can develop different experimental ADs (including AITD) [16][17][18][19].
Animal models of AITD have greatly aided our understanding of its pathogenesis. The findings found with NOD mice concerning AITD are summarized Figure 1.
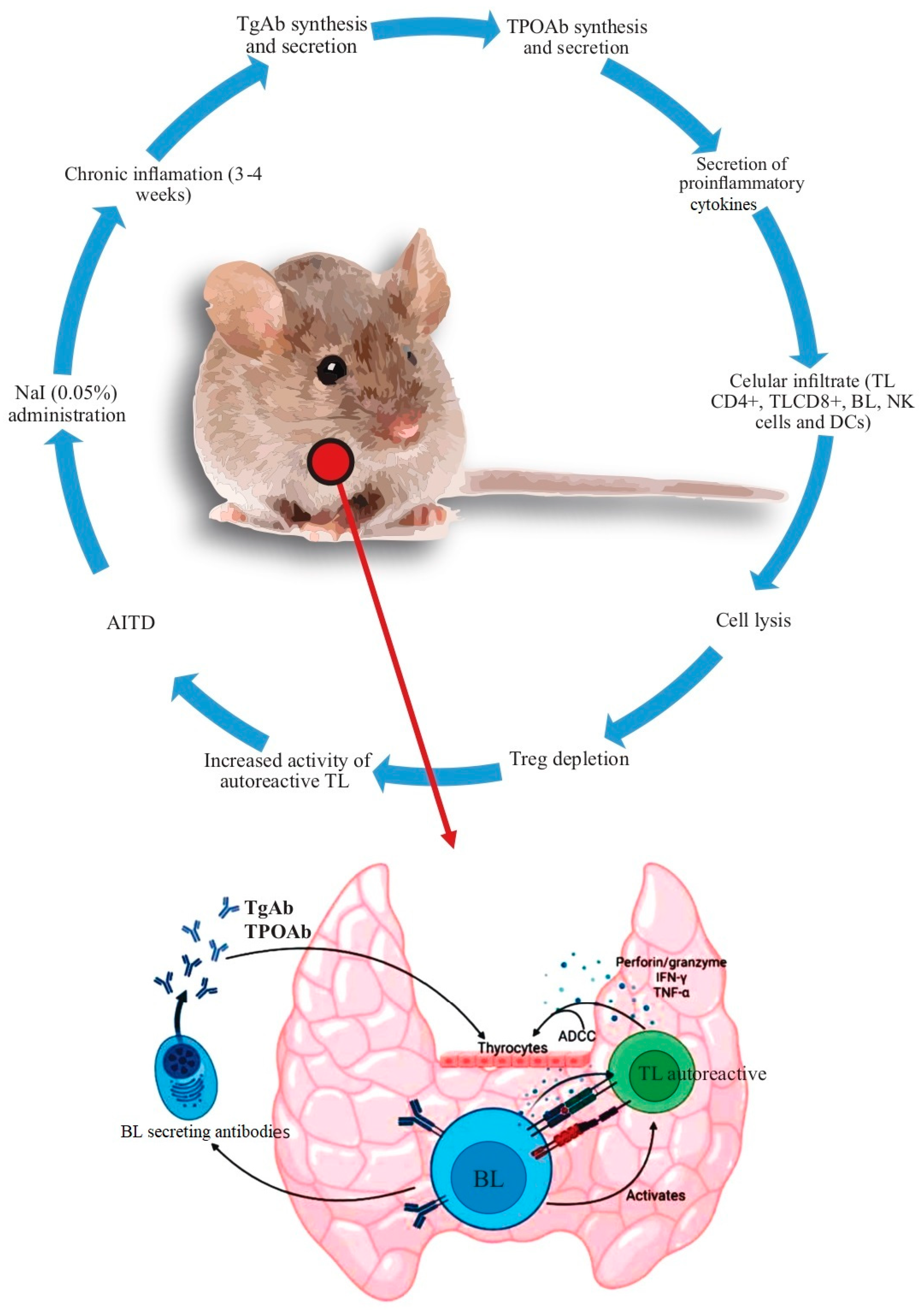
Figure 1. Summary of the mechanisms leading to AITD in the non–obese diabetic (NOD) mice. After the administration of 0.05% NaI in the drinking water, there were findings of chronic inflammation in the following 3–4 weeks. Subsequently, the synthesis of autoantibodies increases (initially against Tg and later against TPO). At the same time, there is an increase in the secretion of proinflammatory cytokines and cell infiltration [mediated initially by TL (CD4+) and then by TL (CD8+), macrophages, and, finally, by BL, although other cells, such as TL, NK, and DCs, among others, also participate in this process]. Additionally, there is an increased production of ADCC, perforin/granzyme, IFN–γ, and TNF–α. TReg depletion increases the severity of the immune response, with loss of immune tolerance and the development of AITD. Abbreviations: ADCC: antibody dependent cell cytotoxicity, BL: B lymphocyte, DCs: dendritic cells, NK: natural killer, Tg: thyroglobulin, TL: T lymphocyte, TPO: thyroid peroxidase, Treg: regulatory T lymphocyte.
3. Genetic Factors
AITD is considered a familial disease, as a family history is found among GBD patients in >60% of cases, and accumulations of GBD and HT have also been demonstrated (in relatives of the index case). Similarly, in monozygotic (MZ) twins, it has been found that if GBD is present in one twin, then HT can develop in the other (which suggests that the genetic factors that predispose to one of the two diseases can potentially increase the risk of the other) [20][21][22][23][24][25][26][27].
3.1. Susceptibility Genes for AITD Associated with Immune System
3.1.1. HLA-DR3
The human leukocyte antigen (HLA) system is a cluster of gene complexes encoding the major histocompatibility complex (MHC) proteins. The cell-mediated adaptive immune response is regulated by the HLA, and its primary function is to present endogenous and exogenous antigens to TLs for recognition and response. The HLA molecules that present antigens to TLs are divided into two main classes: HLA class I (HLA-I) and HLA class II (HLA-II). HLA-I molecules play an essential role in the immune defense against intracellular pathogens, whereas HLA-II molecules are predominantly involved in displaying peptides from extracellular pathogens [28].
HLA polymorphisms (SNPs) determine the HLA diversity and its association with various diseases, as they can determine the specificity of the binding to a specific antigen and the initiation of the immune response, as well as influence the differentiation of TLs in the thymus, modifying the regulation of the response [28][29].
The HLA also controls cytokine synthesis and secretion; therefore, certain HLA susceptibility alleles could lead to AITD (probably by preferentially regulating the Th2 (in GBD) and Th1 (in HT) pathways). HLA-A*68 and HLA-B*08 have also been found to confer GBD susceptibility, while HLA-A*33, HLA-DQB1*0201, and HLA-DQA1*0201 appear to have protective functions [30].
3.1.2. Molecular Mechanisms That May Explain Predisposition to AITD concerning HLA
By binding to specific antigenic peptides, the HLA can recognize thyroid autoantigens as “foreign”, which causes an immune response that is mediated by autoreactive TLs (CD4+/CD8+). In the presence of environmental or infectious factors, this response is magnified and produced by TL and BL activation, which promotes the synthesis and secretion of cytokines and autoantibodies and causes AITD [28][29].
Similarly, the distal extracellular domain of HLA-II is an antigen-binding groove (containing AITD-associated amino acids); thus, any amino acid changes at these sites may alter the interaction between the antigen/HLA and the receptor of TLs, magnifying the immune response to foreign and self-antigens. Finally, it has been postulated that the elevated familial risk of GBD presupposes that there must be one or more associated genetic components in the case of HLA that must be linked to the degenerate motif in the DRB1 gene product (that is, the amino acid at position 74 in the MHC II chain).
3.1.3. PTPN22 Gene
After HLA, the PTPN22 gene is the one that most predisposes to ADs. The PTPN22 gene is located on chromosome 1p13.3–p13.1, and it encodes lymphoid-specific tyrosine phosphatase (LYP). LYP is capable of suppressing kinases that mediate TL activation and regulation, plays an important role in BL signaling, and is involved at multiple levels in the TL receptor signaling and activation cascade [31][32][33][34].
Thus, the minor allele 1858T in the PTPN22 locus has a strong and consistent genetic association with AD. The cytosine changes to thymidine at nucleotide 1858, resulting in an amino acid change from arginine to tryptophan at codon 620 (R620W), which is located in the polyproline-binding motif P1. C1858T has been reported as a susceptibility locus associated with several ADs and AITD [31][32][33][34].
3.1.4. Molecular Mechanisms That Can Explain Susceptibility to AITD concerning PTPN22 SNPs
The PTPN22 gene can modulate the responses of TLs through the regulation of the antigen-presenting cell (APC) function, the downregulation of the Treg expansion at the peripheral level, or the transcriptional suppression of TLs (via transcription factors such as Foxp3). Additionally, PTPN22 can influence the differentiation and proliferation of BLs, and it participates in the escape of autoreactive BLs to the periphery and in the enhancement of autoantibody development [35][36][37].
Moreover, LYP variants are characterized by the expression of losses or gains in their functions; thus, the C1858T variant optimizes the activity of PTPN22, and, as a consequence, an increase in TL and BL receptor signaling can occur, which modifies their functions and the secretion of cytokines [38].
3.1.5. Cluster of Differentiation 40 (CD40) Gene
This gene is a member of the TNF-receptor superfamily. The encoded protein is a receptor on APCs, and it is essential for mediating a broad variety of immune and inflammatory responses. CD40 can be detected on APCs, and it is expressed in granulocytes, endothelial cells, smooth muscle cells, fibroblasts, and epithelial cells [39].
The interaction of CD40L with its respective receptor on BLs (CD40) is of critical importance for immunoglobulin isotype switching during the immune response. CD40L-induced signaling in these cells leads to the upregulation of adhesion and co-stimulatory molecules, as well as the production of proinflammatory cytokines, chemokines, growth factors, and matrix metalloproteinases [40][41].
3.1.6. Molecular Mechanisms That May Explain Susceptibility to AITD concerning CD40 SNPs
CD40 signaling may potentially contribute to AITD in several ways: at the level of the TL selection phenomenon in the thymus, which allows autoreactive TL clones to escape deletion; in secondary lymphoid organs, where TLs are primed by BLs and other APCs; and in the thyroid, where CD40 signaling leads to the production of proinflammatory cytokines and chemokines that contribute to tissue destruction and inflammatory cell entry, with a potential increase in the risk of AITD [42][43].
3.1.7. The Cytotoxic T Lymphocyte-Associated Factor 4 (CTLA4) Gene
CTLA4 is a negative regulator of the TL-mediated immune response, while CD152 is the product of the expression of the gene that codes for the synthesis of CTLA4. CTLA4 competes with the CD28 molecule (to bind to CD80 and CD86, which are co-stimulatory molecules on the surfaces of APCs). CD28 is constitutively expressed on TLs, and upon binding to CD80 and CD86, it emits a positive signal that results in TL activation [44].
When CTLA4 is expressed on the surfaces of TLs, it binds to CD80 and CD86 (with a higher affinity than CD28) and inhibits the positive signals of the CD28-CD80/CD86 interaction, causing independent negative signals and thereby limiting interleukin-2 production (IL-2) and TL proliferation and survival [44][45].
3.1.8. Molecular Mechanisms That May Explain Susceptibility to AITD concerning CTLA4 SNPs
CTLA4 has an inhibitory effect on immune responses that competes with the co-stimulatory molecules on APCs. The balance between the binding of CTLA4/CD28 to its common ligand (B7) plays an important role in determining the immune response. Therefore, factors that regulate the expression or activation of CTLA4 can affect this balance, with the consequent loss of control of the immune responses, which leads to autoimmunity. CTLA4 downregulates TL activation because CD152 expression on TLs increases in the late stage of immune activation (and competes with CD28 for binding to B7).
In the early stage of CD152 activation, CTLA4 binds to the intracellular domain and regulates the negative signaling that inhibits TL activation; consequently, the reduced CTLA4 expression can lead to a hyperactive and self-destructive immune response. Additionally, the variants described above can reduce the expression of the CTLA4 gene and the amount of CTLA4 expressed in immune cells, although they can also change its structure, affecting its inhibitory effect on TL activation. This can translate into an uncontrolled immune response with the increased production and secretion of thyroid autoantibodies, and eventually, AITD [46][47].
3.1.9. The FOXP3 Gene
The FOXP3 gene provides instructions for producing the forkhead box P3 (Foxp3) protein. The Foxp3 protein attaches (binds) to specific regions of DNA and helps control the activities of the genes that are involved in regulating the immune system. FOXP3 is a key gene in the development of Tregs [48][49].
Several SNPs have been studied to evaluate their possible associations with AITD. For example, a meta-analysis revealed a possible association between AITD and the FOXP3-3279 SNP. For its part, the FOXP3 (GT)n microsatellite has also been evaluated; however, no association with AITD was found [50].
3.1.10. Molecular Mechanisms That Can Explain Susceptibility to AITD concerning FOXP3 SNPs
Genetic variations in the FOXP3 gene may promote AITD by weakening the inhibitory function of Tregs and promoting an autoimmune response. Upon its expression, a self-regulating transcriptional circuit stabilizes the expression of FOXP3 to consolidate the differentiation of Tregs and activate the suppressive function. Therefore, in AITD, autoreactive TLs are more resistant to suppression, not so much because of the low number of Tregs but because of their regulatory inability. Likewise, in the loss of immune tolerance, alterations in FOXP3 acetylation could disrupt the transcriptional and epigenetic regulation of FOXP3, which results in less Treg generation and a weaker suppressor function. In this sense, a deficiency of Tregs (rather than an absolute numerical deficiency) could be considered, where a functional deficit would better explain its association with AITD [51][52].
3.1.11. α Chain of IL-2R (IL-2Rα) Gene
The IL-2Rα gene is located on chromosome 10, and it was the first to be defined at the molecular level due to its unique ability to independently bind IL-2. IL-2Rα (also known as CD25) is a glycoprotein that plays an essential role in the TL response to IL-2, which is the main growth factor for these cells (CD25 expression is important for proliferation, longer life expectancy, and TL function) [53].
CD25 occurs on the surfaces of maturing TLs and BLs, undergoes transient expression on activated TLs and BLs, and occurs constitutively on Tregs, which inhibit the activation of autoreactive TLs [54].
3.1.12. Molecular Mechanisms That May Explain the Susceptibility to AITD concerning IL-2Rα SNPs
Given the “quantal theory” of TL activation, the cells of the immune system recognize and react to different antigens (self and foreign), proliferating and differentiating into effector cells in an all-or-nothing (quantum) manner. In addition, the theory establishes that these cells make this decision only after “counting” the number of receptors for the antigen that has been activated, which ultimately determines the number of activated IL-2R molecules, and this number is what determines the quantum decision to progress through the cell cycle and undergo DNA replication and the related cytokinesis, which is the basis for clonal expansion. According to the above, there are several IL-2R molecules expressed on the TL surface that are considered “critical” for the cellular response to stimuli; thus, any potential epigenetic modifications in the promoter region of this gene could affect the expression of the gene and therefore constitute a possible regulatory mechanism.
3.2. Thyroid-Specific AITD Susceptibility Genes
Although AITD has been associated with immunomodulatory genes, this does not fully explain the specific autoimmune component directed toward the thyroid. Therefore, antigen-specific genes (TSHR, Tg, TPO, among others) are a group of candidate genes that are potentially associated with AITD.
3.2.1. The TSHR Gene
TSHR is encoded by a gene located at 14q31 and is part of the glycoprotein hormone receptors, which are a subgroup of G protein-coupled receptors (class A) [55][56][57][58].
TSHR has a large extracellular domain, seven transmembrane domains, and a small intracellular domain. The endogenous TSHR ligand is TSH; therefore, such binding activates several coupled signaling pathways, which promote the expression of “effector” genes that control the growth and differentiation of thyrocytes and the synthesis and secretion of thyroid hormones [57][58][59].
TSHR is considered one of the major thyroid autoantigens, specifically in GBD. In the presence of its autoantibody (TRAb), the activation of signaling cascades is stimulated, simulating the effect of constant TSH stimulation on the thyroid, which clinically results in hyperthyroidism, although biologically, there are other types of stimuli for TSHR activation (for example, through the autonomous activation of the TSHR (induced by somatic or germline mutations in the TSHR gene) and thyrostimulin) [60][61].
Although thyrostimulin is a potent stimulator of the thyrocyte function, its influence on thyroid physiology is not fully understood, although it likely plays a paracrine role in the anterior pituitary, and in other tissues that express the TSHR [61][62][63].
3.2.2. Molecular Mechanisms That May Explain Predisposition to AITD concerning TSHR SNPs
Several mechanisms may explain the associations between THSR SNPs and AITD, specifically in GBD. For instance, two mechanisms modify the peripheral tolerance and central tolerance. One of the mechanisms proposes that intron 1 SNPs associated with GBD participate in the regulation of the alternative splicing of the TSHR mRNA in the thyroid. Because the carriers of the risk alleles for GBD have higher relative expressions of some variants, such as ST4 and ST5 (soluble isoforms), these isoforms may have greater immunogenic potentials, as they are associated with the loss of peripheral tolerance [64].
The other mechanism is due to the modulation of the expression of the TSHR in the thymus, which highlights the role of central tolerance, and it is influenced by the expressions of autoantigens (in this case, the TSHR) inside the thymus (in the process of the negative selection of clones of autoreactive TLs); therefore, the TSHR SNPs could influence the level of the expression itself in the thymus, which is a potential trigger of thyroid autoimmunity.
3.2.3. The Tg Gene
The Tg gene is located on chromosome 8q24. This gene contains more than 16,000 SNPs, and around 10 of them are considered pathogenic, as they are found in germ cells. This characteristic suggests that this gene could be associated with AITD [65].
Tg is a hyperglycosylated protein that is expressed in thyrocytes and secreted into the follicular lumen, where it accumulates. Dimeric Tg undergoes an iodination process to form different tyrosine residues. This process is regulated by iodine intake from the diet. Once iodinated, Tg is transported to the cytosol to be subsequently metabolized, releasing T3 and T4. Therefore, Tg is a precursor for the synthesis of thyroid hormones. TSH and the transporter protein responsible for storing iodine in the colloid intervene in this process [66][67][68].
Some Tg SNPs and allelic variations have been associated with AITD. For example, a study carried out on 56 families revealed seven major loci and other minor loci for AITD. One of the minor loci was on chromosome 8q24. Additionally, a microsatellite was found within intron 27 of the Tg gene. This microsatellite showed a strong association with AITD [69].
3.2.4. Molecular Mechanisms That May Explain Predisposition to AITD concerning Tg SNPs
Tg variants may predispose to AITD by altering Tg degradation in endosomes, which may give rise to a repertoire of pathogenic Tg peptides. Genetic interactions between HLA-DRβ-Arg74 and some Tg variants have been shown to increase the risk of presenting GBD. Additionally, it has also been shown that some Tg peptides can bind to the HLA-DRβ-Arg74 pockets, with one of these peptides being a major epitope of TLs.
3.2.5. The TPO Gene
The TPO gene is located on chromosome 2p25 and encodes a glycosylated hemoprotein of 933 amino acids. The full-length TPO (TPO 1) protein, which consists of 933 amino acids, contains a large extracellular domain, a short transmembrane domain, and an intracellular C-terminal region [70][71].
Even though TPO is considered one of the major thyroid antigens, there are not many studies that evaluate TPO SNPs and their possible associations with AITD. For instance, the association between the T1936C, T2229C, and A2257C TPO SNPs and TPOAb levels was evaluated, and it was found that, in the presence of the C allele of T1936C, the TPOAb levels were significantly increased [72].
3.2.6. Molecular Mechanisms That May Explain Predisposition to AITD concerning TPO SNPs
Genetic studies suggest that TPO gene mutations with autosomal recessive inheritance are one of the most common causes of AITD, with several different inactivating mutations identified in patients with total iodide organification defects. AITD may result from several mechanisms, including the total absence of TPO activity, the inability of TPO to bind to the heme cofactor, the inability to interact with the Tg substrate, and abnormal subcellular localization. Although TPOAbs are valid clinical biomarkers of AITD, they are generally considered to be secondary to the thyroid damage inflicted by TLs [73].
4. Epigenetic Mechanisms in AITD
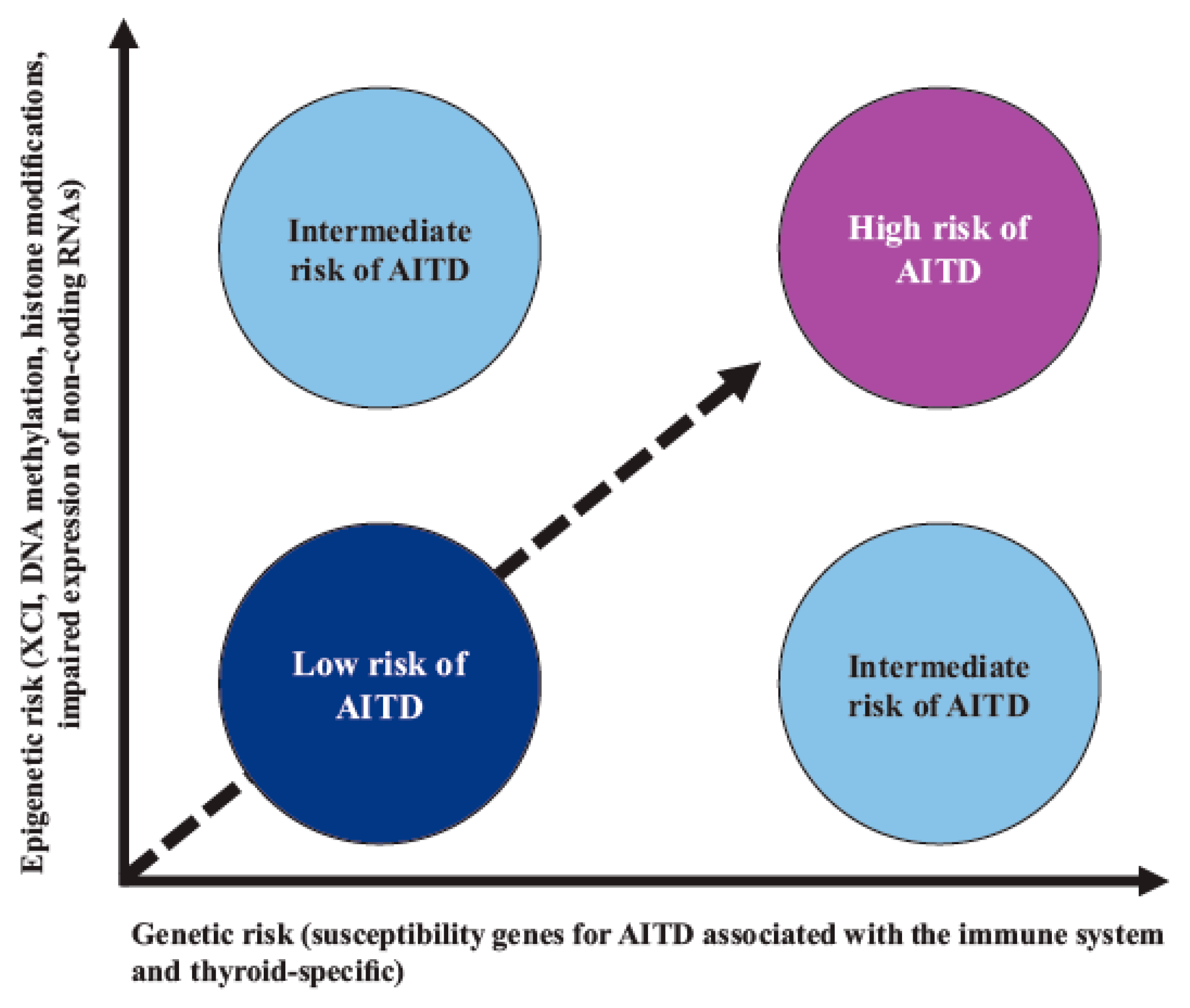
In AITD, studies have shown that the vast majority of the “candidate” gene SNPs are associated with a given risk for the disease; however, this risk is low, indicating that, for a given genetic risk factor, other nongenetic (epigenetic) modifiers are probably necessary to trigger AITD (Figure 2) [74][75][76].
Figure 2. Interaction between genetic susceptibility and epigenetic factors in AITD. Genetic risk by itself confers a low-moderate risk for AITD; however, risk is increased when there is synergy with epigenetic modifications of regulatory regions that are capable of controlling gene expression. Abbreviations: AITD: autoimmune thyroid disease, XCI: X-chromosome inactivation.
Among the epigenetic factors associated with AITD, the skewed X-chromosome inactivation (XCI) SNPs of the genes involved in DNA methylation, the DNA methyltransferases (DNMTs) genes, or the methylenetetrahydrofolate reductase (MTHFR) and methionine synthase reductase (MTRR) genes stand out. Other SNPs have also been implicated, such as the immunoregulatory factors ADRB2 (hypermethylated), ICAM1 (hypomethylated), and B3GNT2 (hypermethylated), as well as histone modifications and the impaired expression of noncoding RNAs, among other factors [77][78].
5. Nongenetic Factors in AITD
5.1. Role of Nongenetic (and Infectious) Factors in AITD
A significant number of nongenetic factors have been associated with AITD, which can be divided into infectious and noninfectious. The infectious factors have almost always been evaluated retrospectively (based on the measurement of antibodies against microorganisms), especially for Y. enterocolitica, H. pylori, B. burgdorferi, Hepatitis C virus (HCV), Hantavirus, Saccharomyces, T. gondii, human immunodeficiency virus (HIV), and the gut microbiota [37].
Molecular Mechanisms That May Explain Predisposition to AITD concerning Nongenetic (Infectious) Factors
The pathophysiological mechanisms that explain possible associations between infectious factors and AITD are still unclear [79][80][81][82].
-
Molecular mimicry (the activation of autoreactive TLs by microorganism peptides that are structurally similar to self-peptides);
-
Viral and bacterial superantigens (the activation of autoreactive TLs that express particular Vβ segments) and the enhanced processing and presentation of autoantigens (by APCs recruited to an inflammatory site and followed by autoreactive lymphocyte priming);
-
The bystander effect (enhanced cytokine production that induces the expansion of autoreactive TLs);
-
The activation of lymphocytes by lymphotropic viruses (an infection of BLs resulting in BL proliferation and excess antibody production);
-
The formation of circulating immune complexes and cytokine storms.
5.2. Role of Nongenetic (and Noninfectious) Factors in AITD
These factors can be classified as nutritional or nonnutritional. The nutritional factors associated with AITD include iron deficiency, iodine excess, selenium deficiency, vitamin D deficiency, and gluten consumption [83].
5.3. Other Nongenetic and Nonnutritional Factors Related to AITD
Fetal cell microchimerism (FCM) and maternal cell microchimerism (MCM) are phenomena that occur during pregnancy, in which the export of cells, including some from the immune systems, can occur either from the fetus to the mother (FCM) or vice versa (MCM) [84].
This implies that an individual can have a specific proportion of cells from a genetically different organism. This bidirectional traffic of transplacental cells begins in the second week of gestation and increases as the pregnancy progresses. These allogeneic cells can persist in the host for years or decades [85][86].
One way to determine the presence of FCM is by demonstrating the presence of male cells in a woman with a previous male pregnancy (employing polymerase chain reaction or fluorescence in situ hybridization analysis) [87][88].
6. Thyroid Autoantibodies
6.1. TRAbs
TRAbs are IgG-type antibodies, and they can be classified into two classes: those that stimulate the TSH receptor (TSAbs) and those that block it (TBAbs). TSAbs stimulate the TSHR and are highly prevalent in GBD patients. TBAbs increase the risk of hypothyroidism and are detectable in between 10 and 90% of individuals with HT, although they can also be found in a minority of GBD patients. TRAbs have also been detected that can bind to the TSHR but do not alter thyroid function; thus, these TRAbs are neutral (TNAbs) [89][90][91].
6.2. TPOAbs
There is no convincing evidence that indicates that TPOAbs initiate the phenomenon of thyroid autoimmunity. This has a simple explanation, which is that these antibodies cannot penetrate the tight junctions between thyroid cells, and therefore they cannot bind to their autoantigen (TPO), which is located apically. However, they could play a role in the autoimmune response in cases in which the tight junctions are disrupted as a consequence of TL-mediated damage [92].
6.3. TgAbs
The role of TgAbs in the pathogenesis of AITD is less conclusive than those found with TRAbs and TPOAbs; however, more than 90% of HT patients are TgAbs-positive. One explanatory hypothesis is antibody-dependent cell-mediated cytotoxicity, in which cells containing the antigen are killed by TLs (NK) and macrophages. Furthermore, in animal models of AITD, TgAbs are detected earlier (leading to the subsequent discovery of the presence of TPOAbs), which suggests that the tolerances of BLs and TLs are likely initially altered towards Tg and later towards TPO [93][94].
6.4. Pendrin and NIS Antibodies
The role of pendrin and NIS antibodies in the pathogenesis of AITD is conflicting, which suggests that these antibodies are present in some patients with AITD, notwithstanding that their clinical importance in the pathogenesis of AITD and on thyroid function is yet to be determined. So far, their measurement does not seem to offer any diagnostic, treatment, or prognostic benefits [95][96].
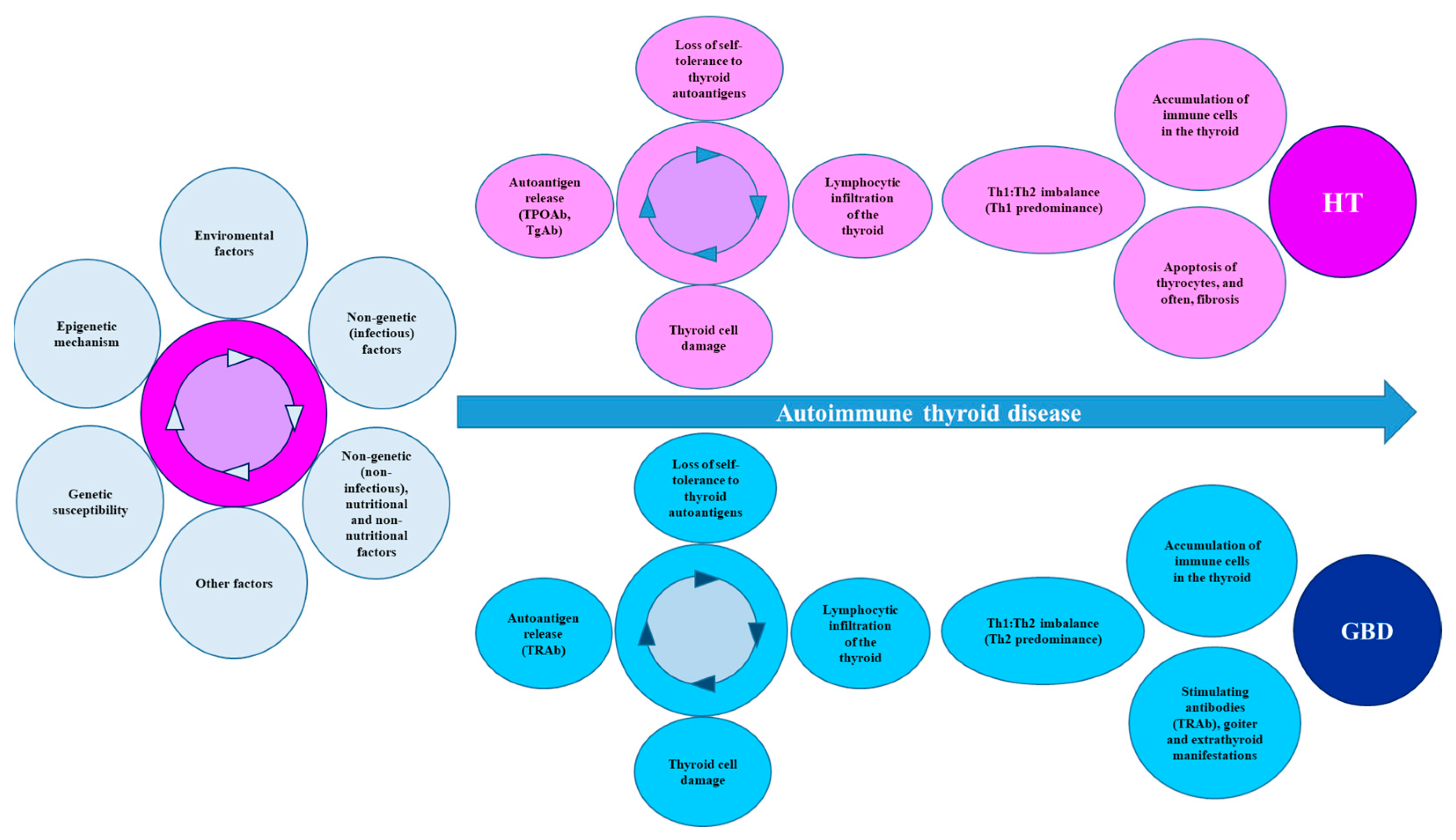
7. Summary of Molecular Mechanisms Leading to AITD
AITD is triggered by a variety of factors (genetic, nongenetic, epigenetic, and environmental). Among the susceptibility genes associated with the immune system, the following stand out: HLA-DR3; PTPN22; CD40; FOXP3; CTLA-4; and IL-2Rα, although thyroid-specific susceptibility genes have been described (TSHR, Tg, and TPO). Some SNPs in these genes play key roles that help explain (at least in part) the increased risk for AITD. For example, the SNPs in FOXP3 and IL-2Rα are involved in peripheral tolerance mechanisms, while the SNPs in the CD40, CTLA-4, and HLA genes compromise the activation of TLs and the antigenic presentation; therefore, the SNPs in immunoregulatory genes can potentially alter the functioning or normal development of the central and peripheral tolerance mechanisms and the interaction of TLs with APCs. Some SNPs in other genes involved in the synthesis of cytokines with potential inflammatory effects have also been described and associated with an increased risk of AITD.
Although genetic susceptibility can potentially explain the pathogenesis of AITD, the genetic risk by itself is low; however, this risk is increased when there is synergism with some components or epigenetic modifications in the region’s regulators that are capable of controlling the gene expression. These epigenetic modifications include the XCI SNPs of the genes involved in DNA methylation, DNMT genes, or MTHFR and MTRR genes. Histone modifications and the impaired expressions of noncoding RNAs have also been implicated.
Environmental factors can be infectious and noninfectious and, in turn, nutritional and nonnutritional. These factors can, by mechanisms not yet elucidated, increase the susceptibility to AITD. However, the key point for the development of AITD is the infiltration of the thyroid by APCs, which may be induced by environmental factors.
Considering that the thyroid follicular cells from individuals with AITD can also abnormally express HLA-II (induced by IFN-γ), the phenomenon of thyroid autoantigen presentation, which facilitates the activation of TLs, is feasible. Likewise, there is also the thyroid infiltration of BLs, cytotoxic TLs, and TLs (CD4+). The interaction with APCs leads to the activation of TLs (CD4+) and the differentiation towards Tregs and Th (Th1, Th2, and Th17), with an imbalance in the Th1:Th2 ratio. For HT, the predominance is towards a Th1 response, whereas in GBD, the predominance is towards a Th2 response.
In addition, an attenuated Treg response has also been found, which may increase the proinflammatory activity of Th17. These mechanisms involve cytokines/chemokines and/or cytotoxins. For HT, apoptosis and subsequent fibrosis lead to the presence of hypothyroidism, while in GBD, the persistent stimulation of the TSHR by its autoantibody (TRAb) induces hyperthyroidism, goiter, and extrathyroidal manifestations (Figure 3).
Figure 3. Summary of the mechanisms that lead to AITD. AITD is the product of multiple environmental factors that act on the basis of genetic susceptibility, together with some epigenetic mechanisms, leading to a loss of immune tolerance, with destruction of thyroid tissue and increased synthesis and secretion of autoantibodies. Finally, the Th1:Th2 imbalance directs the clinical and biochemical manifestations towards HT or GBD. Abbreviations: AITD: autoimmune thyroid disease, GBD: Graves-Basedow disease, HT: Hashimoto’s disease, Tg: thyroglobulin, TgAb: Tg autoantibodies, TPO: thyroid peroxidase, TPOAbs: TPO autoantibodies, TRAbs: thyroid stimulating hormone receptor autoantibodies.
References
- Bieber, K.; Hundt, J.E.; Yu, X.; Ehlers, M.; Petersen, F.; Karsten, C.M.; Köhl, J.; Kridin, K.; Kalies, K.; Kasprick, A.; et al. Autoimmune pre-disease. Autoimmun. Rev. 2023, 22, 103236.
- Wang, L.; Wang, F.S.; Gershwin, M.E. Human autoimmune diseases: A comprehensive update. J. Intern. Med. 2015, 278, 369–395.
- Moroncini, G.; Calogera, G.; Benfaremo, D.; Gabrielli, A. Biologics in Inflammatory Immune-mediated Systemic Diseases. Curr. Pharm. Biotechnol. 2017, 18, 1008–1016.
- Ceccarelli, F.; Govoni, M.; Piga, M.; Cassone, G.; Cantatore, F.P.; Olivieri, G.; Cauli, A.; Favalli, E.G.; Atzeni, F.; Gremese, E.; et al. Arthritis in Systemic Lupus Erythematosus: From 2022 International GISEA/OEG Symposium. J. Clin. Med. 2022, 11, 6016.
- Bach, J.F. The hygiene hypothesis in autoimmunity: The role of pathogens and commensals. Nat. Rev. Immunol. 2018, 18, 105–120.
- McLeod, D.S.; Cooper, D.S. The incidence and prevalence of thyroid autoimmunity. Endocrine 2012, 42, 252–265.
- Ralli, M.; Angeletti, D.; Fiore, M.; D’Aguanno, V.; Lambiase, A.; Artico, M.; de Vincentiis, M.; Greco, A. Hashimoto’s thyroiditis: An update on pathogenic mechanisms, diagnostic protocols, therapeutic strategies, and potential malignant transformation. Autoimmun. Rev. 2020, 19, 102649.
- Hoang, T.D.; Stocker, D.J.; Chou, E.L.; Burch, H.B. 2022 Update on Clinical Management of Graves Disease and Thyroid Eye Disease. Endocrinol. Metab. Clin. North Am. 2022, 51, 287–304.
- Bartalena, L.; Piantanida, E.; Gallo, D.; Ippolito, S.; Tanda, M.L. Management of Graves’ hyperthyroidism: Present and future. Expert Rev. Endocrinol. Metab. 2022, 17, 153–166.
- Ragusa, F.; Fallahi, P.; Elia, G.; Gonnella, D.; Paparo, S.R.; Giusti, C.; Churilov, L.P.; Ferrari, S.M.; Antonelli, A. Hashimotos’ thyroiditis: Epidemiology, pathogenesis, clinic and therapy. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101367.
- Qiu, K.; Li, K.; Zeng, T.; Liao, Y.; Min, J.; Zhang, N.; Peng, M.; Kong, W.; Chen, L.L. Integrative Analyses of Genes Associated with Hashimoto’s Thyroiditis. J. Immunol. Res. 2021, 2021, 8263829.
- Kuś, A.; Chaker, L.; Teumer, A.; Peeters, R.P.; Medici, M. The Genetic Basis of Thyroid Function: Novel Findings and New Approaches. J. Clin. Endocrinol. Metab. 2020, 105, dgz225.
- Lee, H.J.; Li, C.W.; Hammerstad, S.S.; Stefan, M.; Tomer, Y. Immunogenetics of autoimmune thyroid diseases: A comprehensive review. J. Autoimmun. 2015, 64, 82–90.
- Guarneri, F.; Benvenga, S. Environmental factors and genetic background that interact to cause autoimmune thyroid disease. Curr. Opin. Endocrinol. Diabetes Obes. 2007, 14, 398–409.
- Koch, C.A.; Antonelli, A. Immunoendocrinology: When (neuro)endocrinology and immunology meet. Rev. Endocr. Metab. Disord. 2018, 19, 277–282.
- Yu, X.; Petersen, F. A methodological review of induced animal models of autoimmune diseases. Autoimmun. Rev. 2018, 17, 473–479.
- Topping, L.M.; Romero-Castillo, L.; Urbonaviciute, V.; Bolinsson, H.; Clanchy, F.I.; Holmdahl, R.; Bäckström, B.T.; Williams, R.O. Standardization of Antigen-Emulsion Preparations for the Induction of Autoimmune Disease Models. Front. Immunol. 2022, 13, 892251.
- Lam-Tse, W.K.; Lernmark, A.; Drexhage, H.A. Animal models of endocrine/organ-specific autoimmune diseases: Do they really help us to understand human autoimmunity? Springer Semin. Immunopathol. 2002, 24, 297–321.
- Kong, Y.M. Experimental models for autoimmune thyroid disease: Recent developments. In Autoimmune Endocrinopathies (Contemporary Endocrinology); Volpe´, R., Ed.; Humana Press: Totowa, NJ, USA, 2004; pp. 91–111.
- Hall, R.; Stanbury, J.B. Familial studies of autoimmune thyroiditis. Clin. Exp. Immunol. 1967, 2, 719–725.
- Carey, C.; Skosey, C.; Pinnamaneni, K.M.; Barsano, C.P.; DeGroot, L.J. Thyroid abnormalities in children of parents who have Graves’ disease: Possible pre-Graves’ disease. Metabolism 1980, 29, 369–376.
- Burek, C.L.; Hoffman, W.H.; Rose, N.R. The presence of thyroid autoantibodies in children and adolescents with, A.I.TD and in their siblings and parents. Clin. Immunol. Immunopathol. 1982, 25, 395–404.
- Brix, T.H.; Kyvik, K.O.; Christensen, K.; Hegedüs, L. Evidence for a major role of heredity in Graves’ disease: A population-based study of two Danish twin cohorts. J. Clin. Endocrinol. Metab. 2001, 86, 930–934.
- Hansen, P.S.; Brix, T.H.; Iachine, I.; Kyvik, K.O.; Hegedüs, L. The relative importance of genetic and environmental effects for the early stages of thyroid autoimmunity: A study of healthy Danish twins. Eur. J. Endocrinol. 2006, 154, 29–38.
- Skov, J.; Eriksson, D.; Kuja-Halkola, R.; Höijer, J.; Gudbjörnsdottir, S.; Svensson, A.M.; Magnusson, P.K.E.; Ludvigsson, J.F.; Kämpe, O.; Bensing, S. Co-aggregation and heritability of organ-specific autoimmunity: A population-based twin study. Eur. J. Endocrinol. 2020, 182, 473–480.
- Tomer, Y.; Ban, Y.; Concepcion, E.; Barbesino, G.; Villanueva, R.; Greenberg, D.A.; Davies, T.F. Common. and unique susceptibility loci in Graves and Hashimoto diseases: Results of whole-genome screening in a data set of 102 multiplex families. Am. J. Hum. Genet. 2003, 73, 736–747.
- Aust, G.; Krohn, K.; Morgenthaler, N.G.; Schröder, S.; Schütz, A.; Edelmann, J.; Brylla, E. Graves’ disease and Hashimoto’s thyroiditis in monozygotic twins: Case study as well as transcriptomic and immunohistological analysis of thyroid tissues. Eur. J. Endocrinol. 2006, 154, 13–20.
- Sollid, L.M.; Pos, W.; Wucherpfennig, K.W. Molecular Mechanisms for contribution of MHC molecules to autoimmune diseases. Curr. Opin. Immunol. 2014, 31, 24–30.
- Wamala, D.; Buteme, H.K.; Kirimunda, S.; Kallenius, G.; Joloba, M. Association between human leukocyte antigen class II and pulmonary tuberculosis due to mycobacterium tuberculosis in Uganda. BMC Infect. Dis. 2016, 16, 23.
- Mehraji, Z.; Farazmand, A.; Esteghamati, A.; Noshad, S.; Sadr, M.; Amirzargar, S.; Yekaninejad, M.S.; Amirzargar, A. Association of Human Leukocyte Antigens Class I and II with Graves’ Disease in Iranian Population. Iran. J. Immunol. 2017, 14, 223–230.
- Cohen, S.; Dadi, H.; Shaoul, E.; Sharfe, N.; Roifman, C.M. Cloning and characterization of a lymphoid-specific, inducible human protein tyrosine phosphatase, Lyp. Blood 1999, 93, 2013–2024.
- Siminovitch, K.A. PTPN22 and autoimmune disease. Nat. Genet. 2004, 36, 1248–1249.
- Tizaoui, K.; Kim, S.H.; Jeong, G.H.; Kronbichler, A.; Lee, K.S.; Lee, K.H.; Shin, J.I. Association of PTPN22 1858C/T Polymorphism with Autoimmune Diseases: A Systematic Review and Bayesian Approach. J. Clin. Med. 2019, 8, 347.
- Jassim, B.A.; Lin, J.; Zhang, Z.Y. PTPN22, structure, function, and developments in inhibitor discovery with applications for immunotherapy. Expert Opin. Drug. Discov. 2022, 17, 825–837.
- Burn, G.L.; Svensson, L.; Sanchez-Blanco, C.; Saini, M.; Cope, A.P. Why is PTPN22 a good candidate susceptibility gene for autoimmune disease? FEBS Lett. 2011, 585, 3689–3698.
- Tizaoui, K.; Shin, J.I.; Jeong, G.H.; Yang, J.W.; Park, S.; Kim, J.H.; Hwang, S.Y.; Park, S.J.; Koyanagi, A.; Smith, L. Genetic Polymorphism of PTPN22 in Autoimmune Diseases: A Comprehensive Review. Medicina 2022, 58, 1034.
- Bogusławska, J.; Godlewska, M.; Gajda, E.; Piekiełko-Witkowska, A. Cellular and molecular basis of thyroid autoimmunity. Eur. Thyroid J. 2022, 11, e210024.
- Pyzik, A.; Grywalska, E.; Matyjaszek-Matuszek, B.; Rolinski, J. Immune disorders in Hashimoto’s thyroiditis: What do we know so far? J. Immunol. Res. 2015, 2015, 979167.
- Salomon, R.; Dahan, R. Next Generation CD40 Agonistic Antibodies for Cancer Immunotherapy. Front. Immunol. 2022, 13, 940674.
- Karnell, J.L.; Rieder, S.A.; Ettinger, R.; Kolbeck, R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv. Drug. Deliv. Rev. 2019, 141, 92–103.
- Laman, J.D.; Claassen, E.; Noelle, R.J. Functions of CD40 and Its Ligand, gp39 (CD40L). Crit. Rev. Immunol. 2017, 37, 371–420.
- Li, M.; Sun, H.; Liu, S.; Yu, J.; Li, Q.; Liu, P.; Shen, H.; Sun, D. CD40 C/T-1 polymorphism plays different roles in Graves’ disease and Hashimoto’s thyroiditis: A meta-analysis. Endocr. J. 2012, 59, 1041–1050.
- Chand Dakal, T.; Dhabhai, B.; Agarwal, D.; Gupta, R.; Nagda, G.; Meena, A.R.; Dhakar, R.; Menon, A.; Mathur, R.; Mona; et al. Mechanistic basis of co-stimulatory CD40-CD40L ligation mediated regulation of immune responses in cancer and autoimmune disorders. Immunobiology 2020, 225, 151899.
- Chennamadhavuni, A.; Abushahin, L.; Jin, N.; Presley, C.J.; Manne, A. Risk Factors and Biomarkers for Immune-Related Adverse Events: A Practical Guide to Identifying High-Risk Patients and Rechallenging Immune Checkpoint Inhibitors. Front. Immunol. 2022, 13, 779691.
- Van Coillie, S.; Wiernicki, B.; Xu, J. Molecular and Cellular Functions of CTLA-4. Adv. Exp. Med. Biol. 2020, 1248, 7–32.
- Narooie-Nejad, M.; Taji, O.; Kordi Tamandani, D.M.; Kaykhaei, M.A. Association of CTLA-4 gene polymorphisms -318C/T and +49A/G and Hashimoto’s thyroidits in Zahedan, Iran. Biomed. Rep. 2017, 6, 108–112.
- Xiaoheng, C.; Yizhou, M.; Bei, H.; Huilong, L.; Xin, W.; Rui, H.; Lu, L.; Zhiguo, D. General and Specific Genetic Polymorphism of Cytokines-Related Gene in AITD. Mediat. Inflamm. 2017, 2017, 3916395.
- Mazzieri, A.; Montanucci, P.; Basta, G.; Calafiore, R. The role behind the scenes of Tregs and Th17s in Hashimoto’s thyroiditis: Toward a pivotal role of FOXP3 and BACH2. Front. Immunol. 2022, 13, 1098243.
- Ramirez, R.N.; Chowdhary, K.; Leon, J.; Mathis, D.; Benoist, C. FoxP3 associates with enhancer-promoter loops to regulate Treg-specific gene expression. Sci. Immunol. 2022, 7, eabj9836.
- Lee, M.G.; Bae, S.C.; Lee, Y.H. Association between FOXP3 polymorphisms and susceptibility to autoimmune diseases: A meta-analysis. Autoimmunity 2015, 48, 445–452.
- Effraimidis, G.; Wiersinga, W.M. Mechanisms in endocrinology: Autoimmune thyroid disease: Old and new players. Eur. J. Endocrinol. 2014, 170, R241–R252.
- Frommer, L.; Kahaly, G.J. Type 1 Diabetes and Autoimmune Thyroid Disease-The Genetic Link. Front. Endocrinol. 2021, 12, 618213.
- Li, Y.; Li, X.; Geng, X.; Zhao, H. The IL-2A receptor pathway and its role in lymphocyte differentiation and function. Cytokine Growth Factor Rev. 2022, 67, 66–79.
- Damoiseaux, J. The IL-2-IL-2 receptor pathway in health and disease: The role of the soluble IL-2 receptor. Clin. Immunol. 2020, 218, 108515.
- Schuppert, F.; Deiters, S.; Rambusch, E.; Sierralta, W.; Dralle, H.; Mühlen, A.V.Z. TSH-receptor expression and human thyroid disease: Relation to clinical, endocrine, and molecular thyroid parameters. Thyroid 1996, 6, 575–587.
- Szkudlinski, M.W.; Fremont, V.; Ronin, C.; Weintraub, B.D. Thyroid-stimulating hormone and thyroid-stimulating hormone receptor structure-function relationships. Physiol. Rev. 2002, 82, 473–502.
- Chu, Y.D.; Yeh, C.T. The Molecular Function and Clinical Role of Thyroid Stimulating Hormone Receptor in Cancer Cells. Cells 2020, 9, 1730.
- Akamizu, T. Antithyrotropin Receptor Antibody: An Update. Thyroid 2001, 11, 1123–1134.
- Kleinau, G.; Worth, C.L.; Kreuchwig, A.; Biebermann, H.; Marcinkowski, P.; Scheerer, P.; Krause, G. Structural-Functional Features of the Thyrotropin Receptor: A Class A G-Protein-Coupled Receptor at Work. Front. Endocrinol. 2017, 8, 86.
- Davies, T.F.; Ando, T.; Lin, R.Y.; Tomer, Y.; Latif, R. Thyrotropin receptor-associated diseases: From adenomata to Graves disease. J. Clin. Investig. 2005, 115, 1972–1983.
- Karponis, D.; Ananth, S. The role of thyrostimulin and its potential clinical significance. Endocr. Regul. 2017, 51, 117–128.
- van Zeijl, C.J.; Surovtseva, O.V.; Kwakkel, J.; van Beeren, H.C.; Bassett, J.H.; Williams, G.R.; Wiersinga, W.M.; Fliers, E.; Boelen, A. Thyrostimulin deficiency does not alter peripheral responses to acute inflammation-induced nonthyroidal illness. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E527–E537.
- Sun, S.C.; Hsu, P.J.; Wu, F.J.; Li, S.H.; Lu, C.H.; Luo, C.W. Thyrostimulin, but not thyroid-stimulating hormone (TSH), acts as a paracrine regulator to activate the TSH receptor in mammalian ovary. J. Biol. Chem. 2010, 285, 3758–3765.
- Pujol-Borrell, R.; Giménez-Barcons, M.; Marín-Sánchez, A.; Colobran, R. Genetics of Graves’ Disease: Special Focus on the Role of TSHR Gene. Horm. Metab. Res. 2015, 47, 753–766.
- Coscia, F.; Taler-Verčič, A.; Chang, V.T.; Sinn, L.; O’Reilly, F.J.; Izoré, T.; Renko, M.; Berger, I.; Rappsilber, J.; Turk, D.; et al. The structure of human thyroglobulin. Nature 2020, 578, 627–630.
- Di Jeso, B.; Arvan, P. Thyroglobulin From Molecular and Cellular Biology to Clinical Endocrinology. Endocr. Rev. 2016, 37, 2–36.
- Vono-Toniolo, J.; Rivolta, C.M.; Targovnik, H.M.; Medeiros-Neto, G.; Kopp, P. Naturally occurring mutations in the thyroglobulin gene. Thyroid 2005, 15, 1021–1033.
- Zhang, X.; Young, C.; Morishita, Y.; Kim, K.; Kabil, O.O.; Clarke, O.B.; Di Jeso, B.; Arvan, P. Defective Thyroglobulin: Cell Biology of Disease. Int. J. Mol. Sci. 2022, 23, 13605.
- Tomer, Y.; Greenberg, D.A.; Concepcion, E.; Ban, Y.; Davies, T.F. Thyroglobulin is a thyroid specific gene for the familial autoimmune thyroid diseases. J. Clin. Endocrinol. Metab. 2002, 87, 404–407.
- Williams, D.E.; Le, S.N.; Godlewska, M.; Hoke, D.E.; Buckle, A.M. Thyroid Peroxidase as an Autoantigen in Hashimoto’s Disease: Structure, Function, and Antigenicity. Horm. Metab. Res. 2018, 50, 908–921.
- Le, S.N.; Porebski, B.T.; McCoey, J.; Fodor, J.; Riley, B.; Godlewska, M.; Góra, M.; Czarnocka, B.; Banga, J.P.; Hoke, D.E.; et al. Modelling of Thyroid Peroxidase Reveals Insights into Its Enzyme Function and Autoantigenicity. PLoS ONE 2015, 10, e0142615.
- Faam, B.; Daneshpour, M.S.; Azizi, F.; Salehi, M.; Hedayati, M. Association between TPO gene polymorphisms and Anti-TPO level in Tehranian population: TLGS. Gene 2012, 498, 116–119.
- Medici, M.; Porcu, E.; Pistis, G.; Teumer, A.; Brown, S.J.; Jensen, R.A.; Rawal, R.; Roef, G.L.; Plantinga, T.S.; Vermeulen, S.H.; et al. Identification of novel genetic Loci associated with thyroid peroxidase antibodies and clinical thyroid disease. PLoS Genet. 2014, 10, e1004123.
- Jungel, A.; Ospelt, C.; Gay, S. What can we learn from epigenetics in the year 2009? Curr. Opin. Rheumatol. 2010, 22, 284–292.
- Yan, N.; Mu, K.; An, X.F.; Li, L.; Qin, Q.; Song, R.H.; Yao, Q.M.; Shao, X.Q.; Zhang, J.A. Aberrant Histone Methylation in Patients with Graves’ Disease. Int. J. Endocrinol. 2019, 2019, 1454617.
- Coppedè, F. Epigenetics and Autoimmune Thyroid Diseases. Front. Endocrinol. 2017, 8, 149.
- Zhou, F.; Wang, X.; Wang, L.; Sun, X.; Tan, G.; Wei, W.; Zheng, G.; Ma, X.; Tian, D.; Yu, H. Genetics, Epigenetics, Cellular Immunology, and Gut Microbiota: Emerging Links With Graves’ Disease. Front. Cell. Dev. Biol. 2022, 9, 794912.
- Karagianni, P.; Tzioufas, A.G. Epigenetic Perspectives on Systemic Autoimmune Disease. J. Autoimmun. 2019, 104, 102315.
- Luty, J.; Ruckemann-Dziurdzińska, K.; Witkowski, J.M.; Bryl, E. Immunological aspects of autoimmune thyroid disease-Complex interplay between cells and cytokines. Cytokine 2019, 116, 128–133.
- Li, Q.; Wang, B.; Mu, K.; Zhang, J.A. The pathogenesis of thyroid autoimmune diseases: New T lymphocytes-Cytokines circuits beyond the Th1-Th2 paradigm. J. Cell. Physiol. 2019, 234, 2204–2216.
- Bargiel, P.; Szczuko, M.; Stachowska, L.; Prowans, P.; Czapla, N.; Markowska, M.; Petriczko, J.; Kledzik, J.; Jędrzejczyk-Kledzik, A.; Palma, J.; et al. Microbiome Metabolites and Thyroid Dysfunction. J. Clin. Med. 2021, 10, 3609.
- Benvenga, S.; Guarneri, F. Molecular mimicry and autoimmune thyroid disease. Rev. Endocr. Metab. Disord. 2016, 17, 485–498.
- Rayman, M. Multiple nutritional factors and thyroid disease, with particular reference to autoimmune thyroid disease. Proc. Nutr. Soc. 2019, 78, 34–44.
- Khosrotehrani, K.; Johnson, K.L.; Cha, D.H.; Salomon, R.N.; Bianchi, D.W. Transfer of fetal cells with multilineage potential to maternal tissue. J. Am. Med. Assoc. 2004, 292, 75–80.
- O’Donoghue, K.; Chan, J.; de la Fuente, J.; Kennea, N.; Sandison, A.; Anderson, J.R.; Roberts, I.A.; Fisk, N.M. Microchimerism in female bone marrow and bone decades after fetal mesenchymal stem-cell trafficking in pregnancy. Lancet 2004, 364, 179–182.
- Lambert, N.; Nelson, J.L. Microchimerism in autoimmune disease: More questions than answers? Autoimmun. Rev. 2003, 2, 133–139.
- Klintschar, M.; Immel, U.D.; Kehlen, A.; Schwaiger, P.; Mustafa, T.; Mannweiler, S.; Regauer, S.; Kleiber, M.; Hoang-Wu, C. Fetal microchimerism in Hashimoto’s thyroiditis: A quantitative approach. Eur. J. Endocrinol. Eur. Fed. Endocr. Soc. 2006, 154, 237–241.
- Ando, T.; Imaizumi, M.; Graves, P.N.; Unger, P.; Davies, T.F. Intrathyroidal fetal microchimerism in Graves’ disease. J. Clin. Endocrinol. Metab. 2002, 87, 3315–3320.
- Dwivedi, S.N.; Kalaria, T.; Buch, H. Thyroid autoantibodies. J. Clin. Pathol. 2023, 76, 19–28.
- Gupta, A.K.; Kumar, S. Utility of Antibodies in the Diagnoses of Thyroid Diseases: A Review Article. Cureus 2022, 14, e31233.
- Napolitano, G.; Bucci, I.; Di Dalmazi, G.; Giuliani, C. Non-Conventional Clinical Uses of TSH Receptor Antibodies: The Case of Chronic Autoimmune Thyroiditis. Front. Endocrinol. 2021, 12, 769084.
- McLachlan, S.M.; Rapoport, B. Discoveries in Thyroid Autoimmunity in the Past Century. Thyroid 2022.
- Chen, C.R.; Hamidi, S.; Braley-Mullen, H.; Nagayama, Y.; Bresee, C.; Aliesky, H.A.; Rapoport, B.; McLachlan, S.M. Antibodies to thyroid peroxidase arise spontaneously with age in NOD.H-2h4 mice and appear after thyroglobulin antibodies. Endocrinology 2010, 151, 4583–4593.
- Suzuki, S.; Mitsunaga, M.; Miyoshi, M.; Hirakawa, S.; Nakagawa, O.; Miura, H.; Ofuji, T. Cytophilic antithyroglobulin antibody and antibody-dependent monocyte-mediated cytotoxicity in Hashimoto’s thyroiditis. J. Clin. Endocrinol. Metab. 1980, 51, 446–453.
- Czarnocka, B. Thyroperoxidase, thyroglobulin, Na(+)/I(-) symporter, pendrin in thyroid autoimmunity. Front. Biosci. Landmark Ed. 2011, 16, 783–802.
- Eleftheriadou, A.M.; Mehl, S.; Renko, K.; Kasim, R.H.; Schaefer, J.A.; Minich, W.B.; Schomburg, L. Re-visiting autoimmunity to sodium-iodide symporter and pendrin in thyroid disease. Eur. J. Endocrinol. 2020, 183, 571–580.
More
Information
Subjects:
Medicine, Research & Experimental
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
3 times
(View History)
Update Date:
24 Mar 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No