The most common cause of acquired thyroid dysfunction is autoimmune thyroid disease, which is an organ-specific autoimmune disease with two presentation phenotypes: hyperthyroidism (Graves-Basedow disease) and hypothyroidism (Hashimoto’s thyroiditis). Hashimoto’s thyroiditis is distinguished by the presence of autoantibodies against thyroid peroxidase and thyroglobulin. Meanwhile, autoantibodies against the thyroid-stimulating hormone (TSH) receptor have been found in Graves-Basedow disease. Numerous susceptibility genes, as well as epigenetic and environmental factors, contribute to the pathogenesis of both diseases.
- thyroid
- autoimmunity
- Graves-Basedow
- Hashimoto
- pathogenesis
1. Introduction
2. Animal Models of AITD
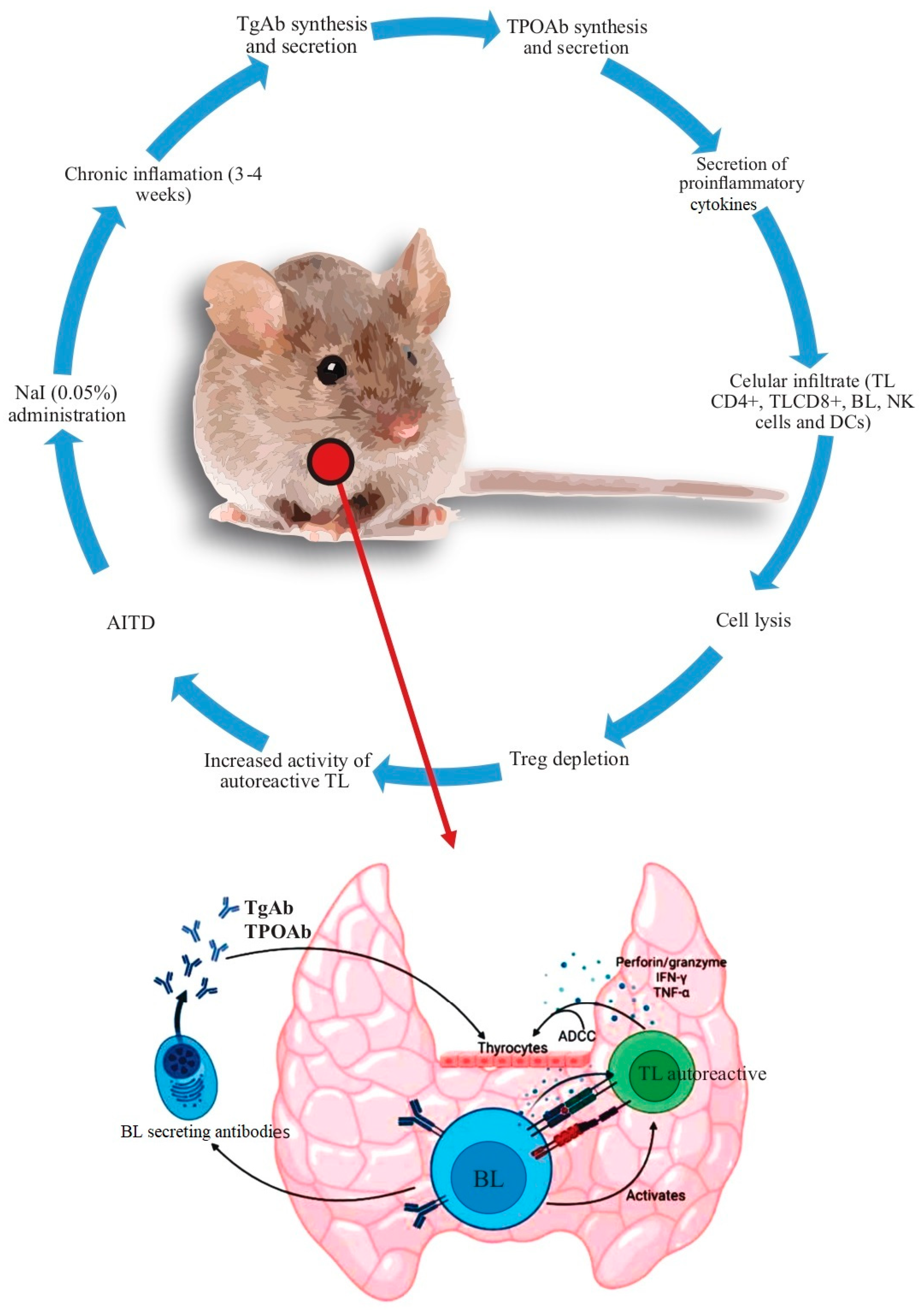
3. Genetic Factors
3.1. Susceptibility Genes for AITD Associated with Immune System
3.1.1. HLA-DR3
3.1.2. Molecular Mechanisms That May Explain Predisposition to AITD concerning HLA
3.1.3. PTPN22 Gene
3.1.4. Molecular Mechanisms That Can Explain Susceptibility to AITD concerning PTPN22 SNPs
3.1.5. Cluster of Differentiation 40 (CD40) Gene
3.1.6. Molecular Mechanisms That May Explain Susceptibility to AITD concerning CD40 SNPs
3.1.7. The Cytotoxic T Lymphocyte-Associated Factor 4 (CTLA4) Gene
3.1.8. Molecular Mechanisms That May Explain Susceptibility to AITD concerning CTLA4 SNPs
3.1.9. The FOXP3 Gene
3.1.10. Molecular Mechanisms That Can Explain Susceptibility to AITD concerning FOXP3 SNPs
3.1.11. α Chain of IL-2R (IL-2Rα) Gene
3.1.12. Molecular Mechanisms That May Explain the Susceptibility to AITD concerning IL-2Rα SNPs
3.2. Thyroid-Specific AITD Susceptibility Genes
3.2.1. The TSHR Gene
3.2.2. Molecular Mechanisms That May Explain Predisposition to AITD concerning TSHR SNPs
3.2.3. The Tg Gene
3.2.4. Molecular Mechanisms That May Explain Predisposition to AITD concerning Tg SNPs
3.2.5. The TPO Gene
3.2.6. Molecular Mechanisms That May Explain Predisposition to AITD concerning TPO SNPs
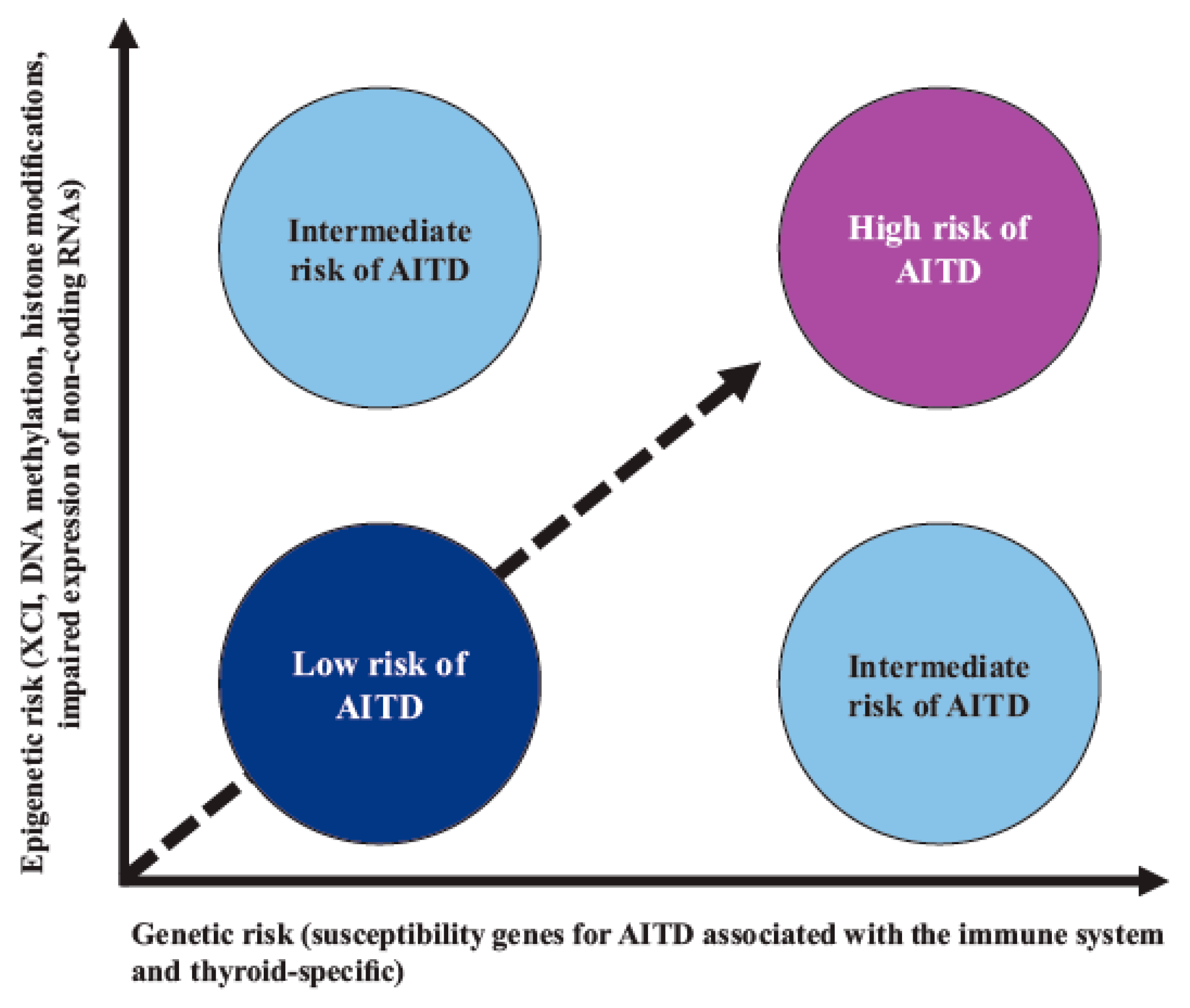
4. Epigenetic Mechanisms in AITD
5. Nongenetic Factors in AITD
5.1. Role of Nongenetic (and Infectious) Factors in AITD
Molecular Mechanisms That May Explain Predisposition to AITD concerning Nongenetic (Infectious) Factors
-
Molecular mimicry (the activation of autoreactive TLs by microorganism peptides that are structurally similar to self-peptides);
-
Viral and bacterial superantigens (the activation of autoreactive TLs that express particular Vβ segments) and the enhanced processing and presentation of autoantigens (by APCs recruited to an inflammatory site and followed by autoreactive lymphocyte priming);
-
The bystander effect (enhanced cytokine production that induces the expansion of autoreactive TLs);
-
The activation of lymphocytes by lymphotropic viruses (an infection of BLs resulting in BL proliferation and excess antibody production);
-
The formation of circulating immune complexes and cytokine storms.
5.2. Role of Nongenetic (and Noninfectious) Factors in AITD
5.3. Other Nongenetic and Nonnutritional Factors Related to AITD
6. Thyroid Autoantibodies
6.1. TRAbs
6.2. TPOAbs
6.3. TgAbs
6.4. Pendrin and NIS Antibodies
7. Summary of Molecular Mechanisms Leading to AITD
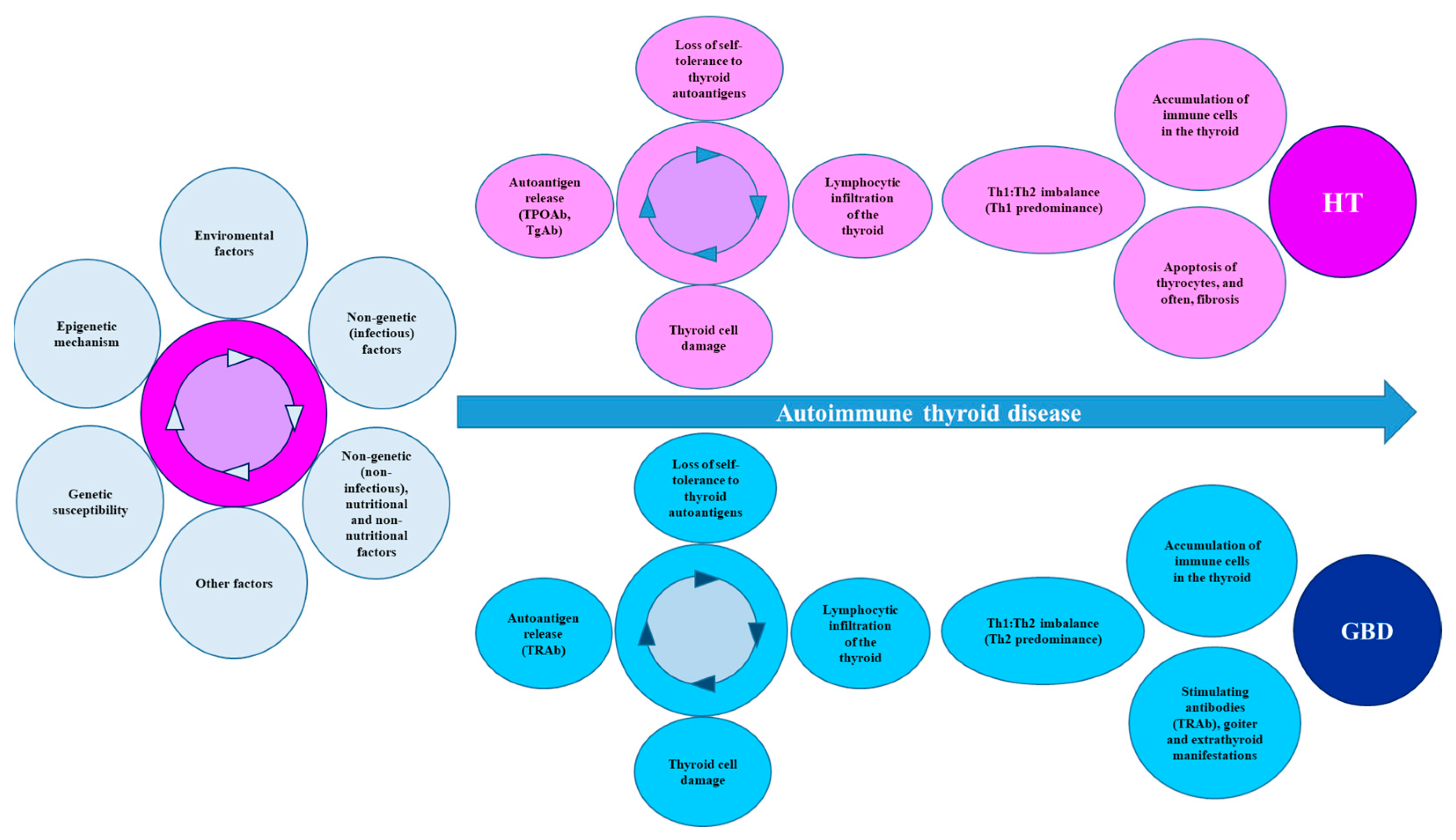
This entry is adapted from the peer-reviewed paper 10.3390/cells12060918
References
- Bieber, K.; Hundt, J.E.; Yu, X.; Ehlers, M.; Petersen, F.; Karsten, C.M.; Köhl, J.; Kridin, K.; Kalies, K.; Kasprick, A.; et al. Autoimmune pre-disease. Autoimmun. Rev. 2023, 22, 103236.
- Wang, L.; Wang, F.S.; Gershwin, M.E. Human autoimmune diseases: A comprehensive update. J. Intern. Med. 2015, 278, 369–395.
- Moroncini, G.; Calogera, G.; Benfaremo, D.; Gabrielli, A. Biologics in Inflammatory Immune-mediated Systemic Diseases. Curr. Pharm. Biotechnol. 2017, 18, 1008–1016.
- Ceccarelli, F.; Govoni, M.; Piga, M.; Cassone, G.; Cantatore, F.P.; Olivieri, G.; Cauli, A.; Favalli, E.G.; Atzeni, F.; Gremese, E.; et al. Arthritis in Systemic Lupus Erythematosus: From 2022 International GISEA/OEG Symposium. J. Clin. Med. 2022, 11, 6016.
- Bach, J.F. The hygiene hypothesis in autoimmunity: The role of pathogens and commensals. Nat. Rev. Immunol. 2018, 18, 105–120.
- McLeod, D.S.; Cooper, D.S. The incidence and prevalence of thyroid autoimmunity. Endocrine 2012, 42, 252–265.
- Ralli, M.; Angeletti, D.; Fiore, M.; D’Aguanno, V.; Lambiase, A.; Artico, M.; de Vincentiis, M.; Greco, A. Hashimoto’s thyroiditis: An update on pathogenic mechanisms, diagnostic protocols, therapeutic strategies, and potential malignant transformation. Autoimmun. Rev. 2020, 19, 102649.
- Hoang, T.D.; Stocker, D.J.; Chou, E.L.; Burch, H.B. 2022 Update on Clinical Management of Graves Disease and Thyroid Eye Disease. Endocrinol. Metab. Clin. North Am. 2022, 51, 287–304.
- Bartalena, L.; Piantanida, E.; Gallo, D.; Ippolito, S.; Tanda, M.L. Management of Graves’ hyperthyroidism: Present and future. Expert Rev. Endocrinol. Metab. 2022, 17, 153–166.
- Ragusa, F.; Fallahi, P.; Elia, G.; Gonnella, D.; Paparo, S.R.; Giusti, C.; Churilov, L.P.; Ferrari, S.M.; Antonelli, A. Hashimotos’ thyroiditis: Epidemiology, pathogenesis, clinic and therapy. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101367.
- Qiu, K.; Li, K.; Zeng, T.; Liao, Y.; Min, J.; Zhang, N.; Peng, M.; Kong, W.; Chen, L.L. Integrative Analyses of Genes Associated with Hashimoto’s Thyroiditis. J. Immunol. Res. 2021, 2021, 8263829.
- Kuś, A.; Chaker, L.; Teumer, A.; Peeters, R.P.; Medici, M. The Genetic Basis of Thyroid Function: Novel Findings and New Approaches. J. Clin. Endocrinol. Metab. 2020, 105, dgz225.
- Lee, H.J.; Li, C.W.; Hammerstad, S.S.; Stefan, M.; Tomer, Y. Immunogenetics of autoimmune thyroid diseases: A comprehensive review. J. Autoimmun. 2015, 64, 82–90.
- Guarneri, F.; Benvenga, S. Environmental factors and genetic background that interact to cause autoimmune thyroid disease. Curr. Opin. Endocrinol. Diabetes Obes. 2007, 14, 398–409.
- Koch, C.A.; Antonelli, A. Immunoendocrinology: When (neuro)endocrinology and immunology meet. Rev. Endocr. Metab. Disord. 2018, 19, 277–282.
- Yu, X.; Petersen, F. A methodological review of induced animal models of autoimmune diseases. Autoimmun. Rev. 2018, 17, 473–479.
- Topping, L.M.; Romero-Castillo, L.; Urbonaviciute, V.; Bolinsson, H.; Clanchy, F.I.; Holmdahl, R.; Bäckström, B.T.; Williams, R.O. Standardization of Antigen-Emulsion Preparations for the Induction of Autoimmune Disease Models. Front. Immunol. 2022, 13, 892251.
- Lam-Tse, W.K.; Lernmark, A.; Drexhage, H.A. Animal models of endocrine/organ-specific autoimmune diseases: Do they really help us to understand human autoimmunity? Springer Semin. Immunopathol. 2002, 24, 297–321.
- Kong, Y.M. Experimental models for autoimmune thyroid disease: Recent developments. In Autoimmune Endocrinopathies (Contemporary Endocrinology); Volpe´, R., Ed.; Humana Press: Totowa, NJ, USA, 2004; pp. 91–111.
- Hall, R.; Stanbury, J.B. Familial studies of autoimmune thyroiditis. Clin. Exp. Immunol. 1967, 2, 719–725.
- Carey, C.; Skosey, C.; Pinnamaneni, K.M.; Barsano, C.P.; DeGroot, L.J. Thyroid abnormalities in children of parents who have Graves’ disease: Possible pre-Graves’ disease. Metabolism 1980, 29, 369–376.
- Burek, C.L.; Hoffman, W.H.; Rose, N.R. The presence of thyroid autoantibodies in children and adolescents with, A.I.TD and in their siblings and parents. Clin. Immunol. Immunopathol. 1982, 25, 395–404.
- Brix, T.H.; Kyvik, K.O.; Christensen, K.; Hegedüs, L. Evidence for a major role of heredity in Graves’ disease: A population-based study of two Danish twin cohorts. J. Clin. Endocrinol. Metab. 2001, 86, 930–934.
- Hansen, P.S.; Brix, T.H.; Iachine, I.; Kyvik, K.O.; Hegedüs, L. The relative importance of genetic and environmental effects for the early stages of thyroid autoimmunity: A study of healthy Danish twins. Eur. J. Endocrinol. 2006, 154, 29–38.
- Skov, J.; Eriksson, D.; Kuja-Halkola, R.; Höijer, J.; Gudbjörnsdottir, S.; Svensson, A.M.; Magnusson, P.K.E.; Ludvigsson, J.F.; Kämpe, O.; Bensing, S. Co-aggregation and heritability of organ-specific autoimmunity: A population-based twin study. Eur. J. Endocrinol. 2020, 182, 473–480.
- Tomer, Y.; Ban, Y.; Concepcion, E.; Barbesino, G.; Villanueva, R.; Greenberg, D.A.; Davies, T.F. Common. and unique susceptibility loci in Graves and Hashimoto diseases: Results of whole-genome screening in a data set of 102 multiplex families. Am. J. Hum. Genet. 2003, 73, 736–747.
- Aust, G.; Krohn, K.; Morgenthaler, N.G.; Schröder, S.; Schütz, A.; Edelmann, J.; Brylla, E. Graves’ disease and Hashimoto’s thyroiditis in monozygotic twins: Case study as well as transcriptomic and immunohistological analysis of thyroid tissues. Eur. J. Endocrinol. 2006, 154, 13–20.
- Sollid, L.M.; Pos, W.; Wucherpfennig, K.W. Molecular Mechanisms for contribution of MHC molecules to autoimmune diseases. Curr. Opin. Immunol. 2014, 31, 24–30.
- Wamala, D.; Buteme, H.K.; Kirimunda, S.; Kallenius, G.; Joloba, M. Association between human leukocyte antigen class II and pulmonary tuberculosis due to mycobacterium tuberculosis in Uganda. BMC Infect. Dis. 2016, 16, 23.
- Mehraji, Z.; Farazmand, A.; Esteghamati, A.; Noshad, S.; Sadr, M.; Amirzargar, S.; Yekaninejad, M.S.; Amirzargar, A. Association of Human Leukocyte Antigens Class I and II with Graves’ Disease in Iranian Population. Iran. J. Immunol. 2017, 14, 223–230.
- Cohen, S.; Dadi, H.; Shaoul, E.; Sharfe, N.; Roifman, C.M. Cloning and characterization of a lymphoid-specific, inducible human protein tyrosine phosphatase, Lyp. Blood 1999, 93, 2013–2024.
- Siminovitch, K.A. PTPN22 and autoimmune disease. Nat. Genet. 2004, 36, 1248–1249.
- Tizaoui, K.; Kim, S.H.; Jeong, G.H.; Kronbichler, A.; Lee, K.S.; Lee, K.H.; Shin, J.I. Association of PTPN22 1858C/T Polymorphism with Autoimmune Diseases: A Systematic Review and Bayesian Approach. J. Clin. Med. 2019, 8, 347.
- Jassim, B.A.; Lin, J.; Zhang, Z.Y. PTPN22, structure, function, and developments in inhibitor discovery with applications for immunotherapy. Expert Opin. Drug. Discov. 2022, 17, 825–837.
- Burn, G.L.; Svensson, L.; Sanchez-Blanco, C.; Saini, M.; Cope, A.P. Why is PTPN22 a good candidate susceptibility gene for autoimmune disease? FEBS Lett. 2011, 585, 3689–3698.
- Tizaoui, K.; Shin, J.I.; Jeong, G.H.; Yang, J.W.; Park, S.; Kim, J.H.; Hwang, S.Y.; Park, S.J.; Koyanagi, A.; Smith, L. Genetic Polymorphism of PTPN22 in Autoimmune Diseases: A Comprehensive Review. Medicina 2022, 58, 1034.
- Bogusławska, J.; Godlewska, M.; Gajda, E.; Piekiełko-Witkowska, A. Cellular and molecular basis of thyroid autoimmunity. Eur. Thyroid J. 2022, 11, e210024.
- Pyzik, A.; Grywalska, E.; Matyjaszek-Matuszek, B.; Rolinski, J. Immune disorders in Hashimoto’s thyroiditis: What do we know so far? J. Immunol. Res. 2015, 2015, 979167.
- Salomon, R.; Dahan, R. Next Generation CD40 Agonistic Antibodies for Cancer Immunotherapy. Front. Immunol. 2022, 13, 940674.
- Karnell, J.L.; Rieder, S.A.; Ettinger, R.; Kolbeck, R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv. Drug. Deliv. Rev. 2019, 141, 92–103.
- Laman, J.D.; Claassen, E.; Noelle, R.J. Functions of CD40 and Its Ligand, gp39 (CD40L). Crit. Rev. Immunol. 2017, 37, 371–420.
- Li, M.; Sun, H.; Liu, S.; Yu, J.; Li, Q.; Liu, P.; Shen, H.; Sun, D. CD40 C/T-1 polymorphism plays different roles in Graves’ disease and Hashimoto’s thyroiditis: A meta-analysis. Endocr. J. 2012, 59, 1041–1050.
- Chand Dakal, T.; Dhabhai, B.; Agarwal, D.; Gupta, R.; Nagda, G.; Meena, A.R.; Dhakar, R.; Menon, A.; Mathur, R.; Mona; et al. Mechanistic basis of co-stimulatory CD40-CD40L ligation mediated regulation of immune responses in cancer and autoimmune disorders. Immunobiology 2020, 225, 151899.
- Chennamadhavuni, A.; Abushahin, L.; Jin, N.; Presley, C.J.; Manne, A. Risk Factors and Biomarkers for Immune-Related Adverse Events: A Practical Guide to Identifying High-Risk Patients and Rechallenging Immune Checkpoint Inhibitors. Front. Immunol. 2022, 13, 779691.
- Van Coillie, S.; Wiernicki, B.; Xu, J. Molecular and Cellular Functions of CTLA-4. Adv. Exp. Med. Biol. 2020, 1248, 7–32.
- Narooie-Nejad, M.; Taji, O.; Kordi Tamandani, D.M.; Kaykhaei, M.A. Association of CTLA-4 gene polymorphisms -318C/T and +49A/G and Hashimoto’s thyroidits in Zahedan, Iran. Biomed. Rep. 2017, 6, 108–112.
- Xiaoheng, C.; Yizhou, M.; Bei, H.; Huilong, L.; Xin, W.; Rui, H.; Lu, L.; Zhiguo, D. General and Specific Genetic Polymorphism of Cytokines-Related Gene in AITD. Mediat. Inflamm. 2017, 2017, 3916395.
- Mazzieri, A.; Montanucci, P.; Basta, G.; Calafiore, R. The role behind the scenes of Tregs and Th17s in Hashimoto’s thyroiditis: Toward a pivotal role of FOXP3 and BACH2. Front. Immunol. 2022, 13, 1098243.
- Ramirez, R.N.; Chowdhary, K.; Leon, J.; Mathis, D.; Benoist, C. FoxP3 associates with enhancer-promoter loops to regulate Treg-specific gene expression. Sci. Immunol. 2022, 7, eabj9836.
- Lee, M.G.; Bae, S.C.; Lee, Y.H. Association between FOXP3 polymorphisms and susceptibility to autoimmune diseases: A meta-analysis. Autoimmunity 2015, 48, 445–452.
- Effraimidis, G.; Wiersinga, W.M. Mechanisms in endocrinology: Autoimmune thyroid disease: Old and new players. Eur. J. Endocrinol. 2014, 170, R241–R252.
- Frommer, L.; Kahaly, G.J. Type 1 Diabetes and Autoimmune Thyroid Disease-The Genetic Link. Front. Endocrinol. 2021, 12, 618213.
- Li, Y.; Li, X.; Geng, X.; Zhao, H. The IL-2A receptor pathway and its role in lymphocyte differentiation and function. Cytokine Growth Factor Rev. 2022, 67, 66–79.
- Damoiseaux, J. The IL-2-IL-2 receptor pathway in health and disease: The role of the soluble IL-2 receptor. Clin. Immunol. 2020, 218, 108515.
- Schuppert, F.; Deiters, S.; Rambusch, E.; Sierralta, W.; Dralle, H.; Mühlen, A.V.Z. TSH-receptor expression and human thyroid disease: Relation to clinical, endocrine, and molecular thyroid parameters. Thyroid 1996, 6, 575–587.
- Szkudlinski, M.W.; Fremont, V.; Ronin, C.; Weintraub, B.D. Thyroid-stimulating hormone and thyroid-stimulating hormone receptor structure-function relationships. Physiol. Rev. 2002, 82, 473–502.
- Chu, Y.D.; Yeh, C.T. The Molecular Function and Clinical Role of Thyroid Stimulating Hormone Receptor in Cancer Cells. Cells 2020, 9, 1730.
- Akamizu, T. Antithyrotropin Receptor Antibody: An Update. Thyroid 2001, 11, 1123–1134.
- Kleinau, G.; Worth, C.L.; Kreuchwig, A.; Biebermann, H.; Marcinkowski, P.; Scheerer, P.; Krause, G. Structural-Functional Features of the Thyrotropin Receptor: A Class A G-Protein-Coupled Receptor at Work. Front. Endocrinol. 2017, 8, 86.
- Davies, T.F.; Ando, T.; Lin, R.Y.; Tomer, Y.; Latif, R. Thyrotropin receptor-associated diseases: From adenomata to Graves disease. J. Clin. Investig. 2005, 115, 1972–1983.
- Karponis, D.; Ananth, S. The role of thyrostimulin and its potential clinical significance. Endocr. Regul. 2017, 51, 117–128.
- van Zeijl, C.J.; Surovtseva, O.V.; Kwakkel, J.; van Beeren, H.C.; Bassett, J.H.; Williams, G.R.; Wiersinga, W.M.; Fliers, E.; Boelen, A. Thyrostimulin deficiency does not alter peripheral responses to acute inflammation-induced nonthyroidal illness. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E527–E537.
- Sun, S.C.; Hsu, P.J.; Wu, F.J.; Li, S.H.; Lu, C.H.; Luo, C.W. Thyrostimulin, but not thyroid-stimulating hormone (TSH), acts as a paracrine regulator to activate the TSH receptor in mammalian ovary. J. Biol. Chem. 2010, 285, 3758–3765.
- Pujol-Borrell, R.; Giménez-Barcons, M.; Marín-Sánchez, A.; Colobran, R. Genetics of Graves’ Disease: Special Focus on the Role of TSHR Gene. Horm. Metab. Res. 2015, 47, 753–766.
- Coscia, F.; Taler-Verčič, A.; Chang, V.T.; Sinn, L.; O’Reilly, F.J.; Izoré, T.; Renko, M.; Berger, I.; Rappsilber, J.; Turk, D.; et al. The structure of human thyroglobulin. Nature 2020, 578, 627–630.
- Di Jeso, B.; Arvan, P. Thyroglobulin From Molecular and Cellular Biology to Clinical Endocrinology. Endocr. Rev. 2016, 37, 2–36.
- Vono-Toniolo, J.; Rivolta, C.M.; Targovnik, H.M.; Medeiros-Neto, G.; Kopp, P. Naturally occurring mutations in the thyroglobulin gene. Thyroid 2005, 15, 1021–1033.
- Zhang, X.; Young, C.; Morishita, Y.; Kim, K.; Kabil, O.O.; Clarke, O.B.; Di Jeso, B.; Arvan, P. Defective Thyroglobulin: Cell Biology of Disease. Int. J. Mol. Sci. 2022, 23, 13605.
- Tomer, Y.; Greenberg, D.A.; Concepcion, E.; Ban, Y.; Davies, T.F. Thyroglobulin is a thyroid specific gene for the familial autoimmune thyroid diseases. J. Clin. Endocrinol. Metab. 2002, 87, 404–407.
- Williams, D.E.; Le, S.N.; Godlewska, M.; Hoke, D.E.; Buckle, A.M. Thyroid Peroxidase as an Autoantigen in Hashimoto’s Disease: Structure, Function, and Antigenicity. Horm. Metab. Res. 2018, 50, 908–921.
- Le, S.N.; Porebski, B.T.; McCoey, J.; Fodor, J.; Riley, B.; Godlewska, M.; Góra, M.; Czarnocka, B.; Banga, J.P.; Hoke, D.E.; et al. Modelling of Thyroid Peroxidase Reveals Insights into Its Enzyme Function and Autoantigenicity. PLoS ONE 2015, 10, e0142615.
- Faam, B.; Daneshpour, M.S.; Azizi, F.; Salehi, M.; Hedayati, M. Association between TPO gene polymorphisms and Anti-TPO level in Tehranian population: TLGS. Gene 2012, 498, 116–119.
- Medici, M.; Porcu, E.; Pistis, G.; Teumer, A.; Brown, S.J.; Jensen, R.A.; Rawal, R.; Roef, G.L.; Plantinga, T.S.; Vermeulen, S.H.; et al. Identification of novel genetic Loci associated with thyroid peroxidase antibodies and clinical thyroid disease. PLoS Genet. 2014, 10, e1004123.
- Jungel, A.; Ospelt, C.; Gay, S. What can we learn from epigenetics in the year 2009? Curr. Opin. Rheumatol. 2010, 22, 284–292.
- Yan, N.; Mu, K.; An, X.F.; Li, L.; Qin, Q.; Song, R.H.; Yao, Q.M.; Shao, X.Q.; Zhang, J.A. Aberrant Histone Methylation in Patients with Graves’ Disease. Int. J. Endocrinol. 2019, 2019, 1454617.
- Coppedè, F. Epigenetics and Autoimmune Thyroid Diseases. Front. Endocrinol. 2017, 8, 149.
- Zhou, F.; Wang, X.; Wang, L.; Sun, X.; Tan, G.; Wei, W.; Zheng, G.; Ma, X.; Tian, D.; Yu, H. Genetics, Epigenetics, Cellular Immunology, and Gut Microbiota: Emerging Links With Graves’ Disease. Front. Cell. Dev. Biol. 2022, 9, 794912.
- Karagianni, P.; Tzioufas, A.G. Epigenetic Perspectives on Systemic Autoimmune Disease. J. Autoimmun. 2019, 104, 102315.
- Luty, J.; Ruckemann-Dziurdzińska, K.; Witkowski, J.M.; Bryl, E. Immunological aspects of autoimmune thyroid disease-Complex interplay between cells and cytokines. Cytokine 2019, 116, 128–133.
- Li, Q.; Wang, B.; Mu, K.; Zhang, J.A. The pathogenesis of thyroid autoimmune diseases: New T lymphocytes-Cytokines circuits beyond the Th1-Th2 paradigm. J. Cell. Physiol. 2019, 234, 2204–2216.
- Bargiel, P.; Szczuko, M.; Stachowska, L.; Prowans, P.; Czapla, N.; Markowska, M.; Petriczko, J.; Kledzik, J.; Jędrzejczyk-Kledzik, A.; Palma, J.; et al. Microbiome Metabolites and Thyroid Dysfunction. J. Clin. Med. 2021, 10, 3609.
- Benvenga, S.; Guarneri, F. Molecular mimicry and autoimmune thyroid disease. Rev. Endocr. Metab. Disord. 2016, 17, 485–498.
- Rayman, M. Multiple nutritional factors and thyroid disease, with particular reference to autoimmune thyroid disease. Proc. Nutr. Soc. 2019, 78, 34–44.
- Khosrotehrani, K.; Johnson, K.L.; Cha, D.H.; Salomon, R.N.; Bianchi, D.W. Transfer of fetal cells with multilineage potential to maternal tissue. J. Am. Med. Assoc. 2004, 292, 75–80.
- O’Donoghue, K.; Chan, J.; de la Fuente, J.; Kennea, N.; Sandison, A.; Anderson, J.R.; Roberts, I.A.; Fisk, N.M. Microchimerism in female bone marrow and bone decades after fetal mesenchymal stem-cell trafficking in pregnancy. Lancet 2004, 364, 179–182.
- Lambert, N.; Nelson, J.L. Microchimerism in autoimmune disease: More questions than answers? Autoimmun. Rev. 2003, 2, 133–139.
- Klintschar, M.; Immel, U.D.; Kehlen, A.; Schwaiger, P.; Mustafa, T.; Mannweiler, S.; Regauer, S.; Kleiber, M.; Hoang-Wu, C. Fetal microchimerism in Hashimoto’s thyroiditis: A quantitative approach. Eur. J. Endocrinol. Eur. Fed. Endocr. Soc. 2006, 154, 237–241.
- Ando, T.; Imaizumi, M.; Graves, P.N.; Unger, P.; Davies, T.F. Intrathyroidal fetal microchimerism in Graves’ disease. J. Clin. Endocrinol. Metab. 2002, 87, 3315–3320.
- Dwivedi, S.N.; Kalaria, T.; Buch, H. Thyroid autoantibodies. J. Clin. Pathol. 2023, 76, 19–28.
- Gupta, A.K.; Kumar, S. Utility of Antibodies in the Diagnoses of Thyroid Diseases: A Review Article. Cureus 2022, 14, e31233.
- Napolitano, G.; Bucci, I.; Di Dalmazi, G.; Giuliani, C. Non-Conventional Clinical Uses of TSH Receptor Antibodies: The Case of Chronic Autoimmune Thyroiditis. Front. Endocrinol. 2021, 12, 769084.
- McLachlan, S.M.; Rapoport, B. Discoveries in Thyroid Autoimmunity in the Past Century. Thyroid 2022.
- Chen, C.R.; Hamidi, S.; Braley-Mullen, H.; Nagayama, Y.; Bresee, C.; Aliesky, H.A.; Rapoport, B.; McLachlan, S.M. Antibodies to thyroid peroxidase arise spontaneously with age in NOD.H-2h4 mice and appear after thyroglobulin antibodies. Endocrinology 2010, 151, 4583–4593.
- Suzuki, S.; Mitsunaga, M.; Miyoshi, M.; Hirakawa, S.; Nakagawa, O.; Miura, H.; Ofuji, T. Cytophilic antithyroglobulin antibody and antibody-dependent monocyte-mediated cytotoxicity in Hashimoto’s thyroiditis. J. Clin. Endocrinol. Metab. 1980, 51, 446–453.
- Czarnocka, B. Thyroperoxidase, thyroglobulin, Na(+)/I(-) symporter, pendrin in thyroid autoimmunity. Front. Biosci. Landmark Ed. 2011, 16, 783–802.
- Eleftheriadou, A.M.; Mehl, S.; Renko, K.; Kasim, R.H.; Schaefer, J.A.; Minich, W.B.; Schomburg, L. Re-visiting autoimmunity to sodium-iodide symporter and pendrin in thyroid disease. Eur. J. Endocrinol. 2020, 183, 571–580.