 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Duangnapa Kovanich | -- | 2259 | 2023-03-21 12:54:17 | | | |
| 2 | Dean Liu | -31 word(s) | 2228 | 2023-03-27 12:17:18 | | | | |
| 3 | Dean Liu | -28 word(s) | 2200 | 2023-03-30 08:16:22 | | |
Video Upload Options
cAMP is a second messenger that regulates a myriad of cellular functions in response to multiple extracellular stimuli. New developments in the field have provided exciting insights into how cAMP utilizes compartmentalization to ensure specificity when the message conveyed to the cell by an extracellular stimulus is translated into the appropriate functional outcome. cAMP signaling uses different mechanisms, including protein–protein interactions (PPIs) and protein phosphorylation, to relay, process, and translate signals into cellular responses. cAMP signaling compartmentalization heavily relies on the formation of local signaling domains where cAMP signaling components involved in a specific cellular response cluster together. Within such domains, cAMP signals are translated into specific cellular responses via the phosphorylation of target proteins. As such, mapping the domain interaction landscape and defining the downstream phosphorylation events are the key aspects of compartmentalized signaling studies.
1.Introduction
cAMP signaling uses different mechanisms, including protein–protein interactions (PPIs) and protein phosphorylation, to relay, process, and translate signals into cellular responses. cAMP signaling compartmentalization heavily relies on the formation of local signaling domains where cAMP signaling components involved in a specific cellular response cluster together. Compartmentalization is critically dependent on the assembly of multiple signaling components that come together via protein–protein interactions to become functional signaling units. Within such domains, cAMP signals are translated into specific cellular responses via the phosphorylation of target proteins. As such, mapping the domain interaction landscape and defining the downstream phosphorylation events are the key aspects of compartmentalized signaling studies. Typically, the proteomics analysis of PPIs and protein phosphorylation is usually performed in a quantitative manner, so the changes in phosphorylation events across experimental conditions can be compared, and true signalosome components can be distinguished from the background. Below researchers describe the proteomics toolbox can be utilized to identify the molecular components of these domains and to define the dynamic cellular cAMP signaling landscape.
2. Protein Interaction and Proximity Profiling—Basic Principles
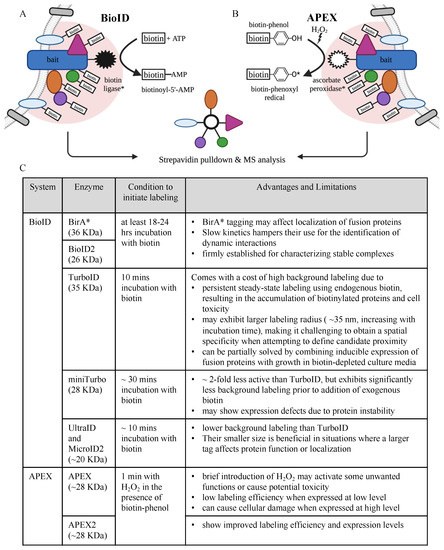
3. Phosphoproteomics
References
- Low, T.Y.; Syafruddin, S.E.; Mohtar, M.A.; Vellaichamy, A.; NS, A.R.; Pung, Y.F.; Tan, C.S.H. Recent progress in mass spectrometry-based strategies for elucidating protein-protein interactions. Cell. Mol. Life Sci. 2021, 78, 5325–5339.
- Lee, Y.-C.; Bååth, J.A.; Bastle, R.M.; Bhattacharjee, S.; Cantoria, M.J.; Dornan, M.; Gamero-Estevez, E.; Ford, L.; Halova, L.; Kernan, J.; et al. Impact of detergents on membrane protein complex isolation. J. Proteome. Res. 2018, 17, 348–358.
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810.
- Samavarchi-Tehrani, P.; Abdouni, H.; Samson, R.; Gingras, A.-C. A Versatile Lentiviral Delivery Toolkit for Proximity-dependent Biotinylation in Diverse Cell Types. Mol. Cell. Proteom. 2018, 17, 2256–2269.
- Vandemoortele, G.; De Sutter, D.; Moliere, A.; Pauwels, J.; Gevaert, K.; Eyckerman, S. A Well-Controlled BioID Design for Endogenous Bait Proteins. J. Proteome Res. 2019, 18, 95–106.
- Kim, D.I.; Kc, B.; Zhu, W.; Motamedchaboki, K.; Doye, V.; Roux, K.J. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. USA 2014, 111, E2453–E2461.
- Kim, D.I.; Jensen, S.C.; Noble, K.A.; Kc, B.; Roux, K.H.; Motamedchaboki, K.; Roux, K.J. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 2016, 27, 1188–1196.
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887.
- Kubitz, L.; Bitsch, S.; Zhao, X.; Schmitt, K.; Deweid, L.; Roehrig, A.; Barazzone, E.C.; Valerius, O.; Kolmar, H.; Béthune, J. Engineering of ultraID, a compact and hyperactive enzyme for proximity-dependent biotinylation in living cells. Commun. Biol. 2022, 5, 657.
- Johnson, B.S.; Chafin, L.; Farkas, D.; Adair, J.; Elhance, A.; Farkas, L.; Bednash, J.S.; Londino, J.D. MicroID2: A Novel Biotin Ligase Enables Rapid Proximity-Dependent Proteomics. Mol. Cell. Proteom. 2022, 21, 100256.
- May, D.G.; Scott, K.L.; Campos, A.R.; Roux, K.J. Comparative Application of BioID and TurboID for Protein-Proximity Biotinylation. Cells 2020, 9, 1070.
- Rhee, H.W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339, 1328–1331.
- Lam, S.S.; Martell, J.D.; Kamer, K.J.; Deerinck, T.J.; Ellisman, M.H.; Mootha, V.K.; Ting, A.Y. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 2015, 12, 51–54.
- Trinkle-Mulcahy, L. Recent advances in proximity-based labeling methods for interactome mapping. F1000Res 2019, 8, F1000.
- Bosch, J.A.; Chen, C.L.; Perrimon, N. Proximity-dependent labeling methods for proteomic profiling in living cells: An update. Wiley Interdiscip. Rev. Dev. Biol. 2021, 10, e392.
- Gingras, A.-C.; Abe, K.T.; Raught, B. Getting to know the neighborhood: Using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr. Opin. Chem. Biol. 2019, 48, 44–54.
- Chua, X.Y.; Aballo, T.; Elnemer, W.; Tran, M.; Salomon, A. Quantitative Interactomics of Lck-TurboID in Living Human T Cells Unveils T Cell Receptor Stimulation-Induced Proximal Lck Interactors. J. Proteome Res. 2021, 20, 715–726.
- de Graaf, E.L.; Giansanti, P.; Altelaar, A.F.; Heck, A.J. Single-step enrichment by Ti4+-IMAC and label-free quantitation enables in-depth monitoring of phosphorylation dynamics with high reproducibility and temporal resolution. Mol. Cell. Proteom. 2014, 13, 2426–2434.
- Humphrey, S.J.; Azimifar, S.B.; Mann, M. High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat. Biotechnol. 2015, 33, 990–995.
- Dephoure, N.; Gould, K.L.; Gygi, S.P.; Kellogg, D.R. Mapping and analysis of phosphorylation sites: A quick guide for cell biologists. Mol. Biol. Cell 2013, 24, 535–542.
- Low, T.Y.; Mohtar, M.A.; Lee, P.Y.; Omar, N.; Zhou, H.; Ye, M. Widening the bottleneck of phosphoproteomics: Evolving strategies for phosphopeptide enrichment. Mass Spectrom. Rev. 2021, 40, 309–333.
- Gauci, S.; Helbig, A.O.; Slijper, M.; Krijgsveld, J.; Heck, A.J.; Mohammed, S. Lys-N and trypsin cover complementary parts of the phosphoproteome in a refined SCX-based approach. Anal. Chem. 2009, 81, 4493–4501.
- Villén, J.; Gygi, S.P. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat. Protoc. 2008, 3, 1630–1638.
- Zhou, H.; Low, T.Y.; Hennrich, M.L.; van der Toorn, H.; Schwend, T.; Zou, H.; Mohammed, S.; Heck, A.J. Enhancing the identification of phosphopeptides from putative basophilic kinase substrates using Ti (IV) based IMAC enrichment. Mol. Cell. Proteom. 2011, 10, M110.006452.
- Humphrey, S.J.; Karayel, O.; James, D.E.; Mann, M. High-throughput and high-sensitivity phosphoproteomics with the EasyPhos platform. Nat. Protoc. 2018, 13, 1897–1916.
- Mertins, P.; Tang, L.C.; Krug, K.; Clark, D.J.; Gritsenko, M.A.; Chen, L.; Clauser, K.R.; Clauss, T.R.; Shah, P.; Gillette, M.A.; et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography–mass spectrometry. Nat. Protoc. 2018, 13, 1632–1661.
- Pinkse, M.W.; Uitto, P.M.; Hilhorst, M.J.; Ooms, B.; Heck, A.J. Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns. Anal. Chem. 2004, 76, 3935–3943.
- Matheron, L.; van den Toorn, H.; Heck, A.J.; Mohammed, S. Characterization of biases in phosphopeptide enrichment by Ti(4+)-immobilized metal affinity chromatography and TiO2 using a massive synthetic library and human cell digests. Anal. Chem. 2014, 86, 8312–8320.
- Yue, X.; Schunter, A.; Hummon, A.B. Comparing multistep immobilized metal affinity chromatography and multistep TiO2 methods for phosphopeptide enrichment. Anal. Chem. 2015, 87, 8837–8844.
- Giansanti, P.; Stokes, M.P.; Silva, J.C.; Scholten, A.; Heck, A.J. Interrogating cAMP-dependent kinase signaling in Jurkat T cells via a protein kinase A targeted immune-precipitation phosphoproteomics approach. Mol. Cell. Proteom. 2013, 12, 3350–3359.
- Potel, C.M.; Lemeer, S.; Heck, A.J.R. Phosphopeptide Fragmentation and Site Localization by Mass Spectrometry: An Update. Anal. Chem. 2019, 91, 126–141.
- Locard-Paulet, M.; Bouyssié, D.; Froment, C.; Burlet-Schiltz, O.; Jensen, L.J. Comparing 22 Popular Phosphoproteomics Pipelines for Peptide Identification and Site Localization. J. Proteome Res. 2020, 19, 1338–1345.
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29.
- Savage, S.R.; Zhang, B. Using phosphoproteomics data to understand cellular signaling: A comprehensive guide to bioinformatics resources. Clin. Proteom. 2020, 17, 27.
- Golkowski, M.; Shimizu-Albergine, M.; Suh, H.W.; Beavo, J.A.; Ong, S.E. Studying mechanisms of cAMP and cyclic nucleotide phosphodiesterase signaling in Leydig cell function with phosphoproteomics. Cell. Signal. 2016, 28, 764–778.
- Beltejar, M.-C.G.; Lau, H.-T.; Golkowski, M.G.; Ong, S.E.; Beavo, J.A. Analyses of PDE-regulated phosphoproteomes reveal unique and specific cAMP-signaling modules in T cells. Proc. Natl. Acad. Sci. USA 2017, 114, E6240–E6249.
- Subramaniam, G; Schleicher, K; Kovanich, D; Zerio, A; Folkmanaite, M; Chao, YC; Surdo, NC; Koschinski, A; Hu, J; Scholten, A; Heck, AJR; Ercu, M; Sholokh, A; Chan Park, K; Klussmann, E; Meraviglia, V; Bellin, M; Zanivan, S; Hester, S; Mohammed, S; Zaccolo M. Integrated Proteomics Unveils Nuclear PDE3A2 as a Regulator of Cardiac Myocyte Hypertrophy. Circ Res. 2023, doi: 10.1161/CIRCRESAHA.122.321448.

