Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | J Timothy Timothy Wright | -- | 2823 | 2023-03-17 03:39:35 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Wright, J.T. Enamel Phenotypes. Encyclopedia. Available online: https://encyclopedia.pub/entry/42287 (accessed on 16 June 2026).
Wright JT. Enamel Phenotypes. Encyclopedia. Available at: https://encyclopedia.pub/entry/42287. Accessed June 16, 2026.
Wright, John Timothy. "Enamel Phenotypes" Encyclopedia, https://encyclopedia.pub/entry/42287 (accessed June 16, 2026).
Wright, J.T. (2023, March 17). Enamel Phenotypes. In Encyclopedia. https://encyclopedia.pub/entry/42287
Wright, John Timothy. "Enamel Phenotypes." Encyclopedia. Web. 17 March, 2023.
Copy Citation
Dental enamel is a specialized tissue that has adapted over millions of years of evolution to enhance the survival of a variety of species. In humans, enamel evolved to form the exterior protective layer for the crown of the exposed tooth crown. Its unique composition, structure, physical properties and attachment to the underlying dentin tissue allow it to be a resilient, although not self-repairing, tissue. The process of enamel formation, known as amelogenesis, involves epithelial-derived cells called ameloblasts that secrete a unique extracellular matrix that influences the structure of the mineralizing enamel crystallites.
enamel
development
phenotype
gene
epigenetic
environment
pathology
1. Introduction
Enamel, the hardest tissue in the body, covers the crown of human teeth and serves in a variety of capacities that are critical for normal tooth function and longevity [1]. Enamel has tremendous wear and fracture resistance due to its unique structure and composition. It can withstand the tremendous forces and pressures that are required for masticating the diverse foods consumed by humans [2]. Not only is this outer protective enamel layer extraordinarily resistant to wear and fracture, it also serves as an outstanding insulator against thermal and chemical challenges that occur during nutrient consumption [3]. This resilient outer covering of the dental crown serves to protect the tooth and allows it to function and be resistant to failure even when challenged by the rigors of mastication, chemical insults, thermal fluctuations, biofilm colonization and decades of use.
Enamel is derived from the oral epithelium that invaginates during tooth development [4]. Enamel has no capacity for repair or regeneration as the cells that form this tissue are lost during the developmental process and tooth eruption [1]. The process of enamel formation, amelogenesis, can be perturbed by a variety of genetic and environmental influences [1].
2. Normal Enamel Development
Tooth formation in humans begins around the 8th week of gestation with the invagination of the oral epithelium that interacts with the underlying ectomesenchyme cells to initiate tooth bud development [4]. These two tissues interact through molecular signals including growth factors that direct the epithelial component to transform into the future cells that will give rise to the enamel-forming cells, the ameloblasts [5]. The forming tooth germs progress through a series of developmental stages that have been named for key events. These have been termed: (1) initiation—stage of initial tissue invagination and proliferation, (2) morphogenesis—development of three-dimensional crown form and differentiated cells, (3) matrix secretion—formation of extracellular matrix, (4) eruption—the tooth root elongates, and the crown emerges into the oral cavity [6]. Tooth formation can be modified or terminated at any of these developmental stages by genetic alterations and/or environmental stressors. There are numerous excellent reviews that describe the normal processes of tooth development and their molecular signaling and regulation [7][8].
The enamel organ, which is responsible for the formation of enamel, contains several epithelium-derived cell types that contribute to the development of the dental crown and enamel [9]. The inner and outer enamel epithelium are generated at the cervical loop, which serves as a dental epithelial stem cell niche for the developing tooth. The inner enamel epithelium gives rise to the ameloblasts [9]. The ameloblasts are initially adjacent to the mesenchymal tissue and odontoblasts that form the dentin. Epithelial-derived cells known as the stratum intermedium form a single cell layer on the non-secretory side of the ameloblasts. The stratum intermedium cells appear to be critical for normal enamel development, including stabilization of the ameloblast layer, although their role appears to be more complex than is currently [10] understood. The preameloblasts change their morphology as they differentiate into tall columnar cells that will secrete a unique extracellular matrix and then degrade and remove most of this matrix while creating a microenvironment necessary to allow for mineralization of the enamel [1][11]. Once the full thickness of the enamel has been laid down, the ameloblasts continue to degrade and remove the extracellular matrix in an orderly fashion by secreting specific proteinases (MMP20, KLK4) [11]. Removal of the extracellular matrix provides room for crystallite growth, allowing the enamel to achieve its highly mineralized state of about 96% mineral by weight [12][13]. The mineral component of enamel is carbonate-substituted hydroxyapatite that contains numerous trace elements, some of which, such as fluoride, can alter the enamel solubility and structure of the crystallites [14]. Enamel has a unique structure that varies between species and reflects the evolutionary development and modification in response to functional needs placed on the dentition [15].
Given the complexity of human enamel and the temporal and spatial requirements to develop such a refined tissue, it is not surprising that there are numerous known etiologies leading to abnormal enamel development. These alterations can occur due to genetic and epigenetic influences and can result from environmental exposures and stressors [16][17]. Conditions altering enamel development can result in changes in the amount, composition and or structure of the tissue (Figure 1) [16][18]. The enamel phenotypes (Figure 1) are diverse, with a decreased amount (enamel hypoplasia) and reduced mineral content (enamel hypomineralization) being the predominate malformations observed. Non-pathological variants, such as SNPS in genes coding for proteins that contribute to enamel formation (e.g., AMELX), have been associated with dental caries, presumably due to subtle changes in the enamel structure and composition [19][20][21]. Developmental defects of enamel (DDE) are common in the general population (up to 80%) and vary markedly in phenotype with a variety of hypoplastic and hypomineralized defects [22][23][24]. DDE are associated with pathologies and altered disease risk, including dental caries and enamel fracturing [25][26]. Morbidities including dental hypersensitivity, altered esthetics, dental caries, and enamel or even tooth loss are associated with DDE [27][28]. DDE also can be associated with co-morbidities such as abnormal tooth eruption, dental cysts, and dental malocclusions as well as with systemic manifestations and syndromes [28][29].
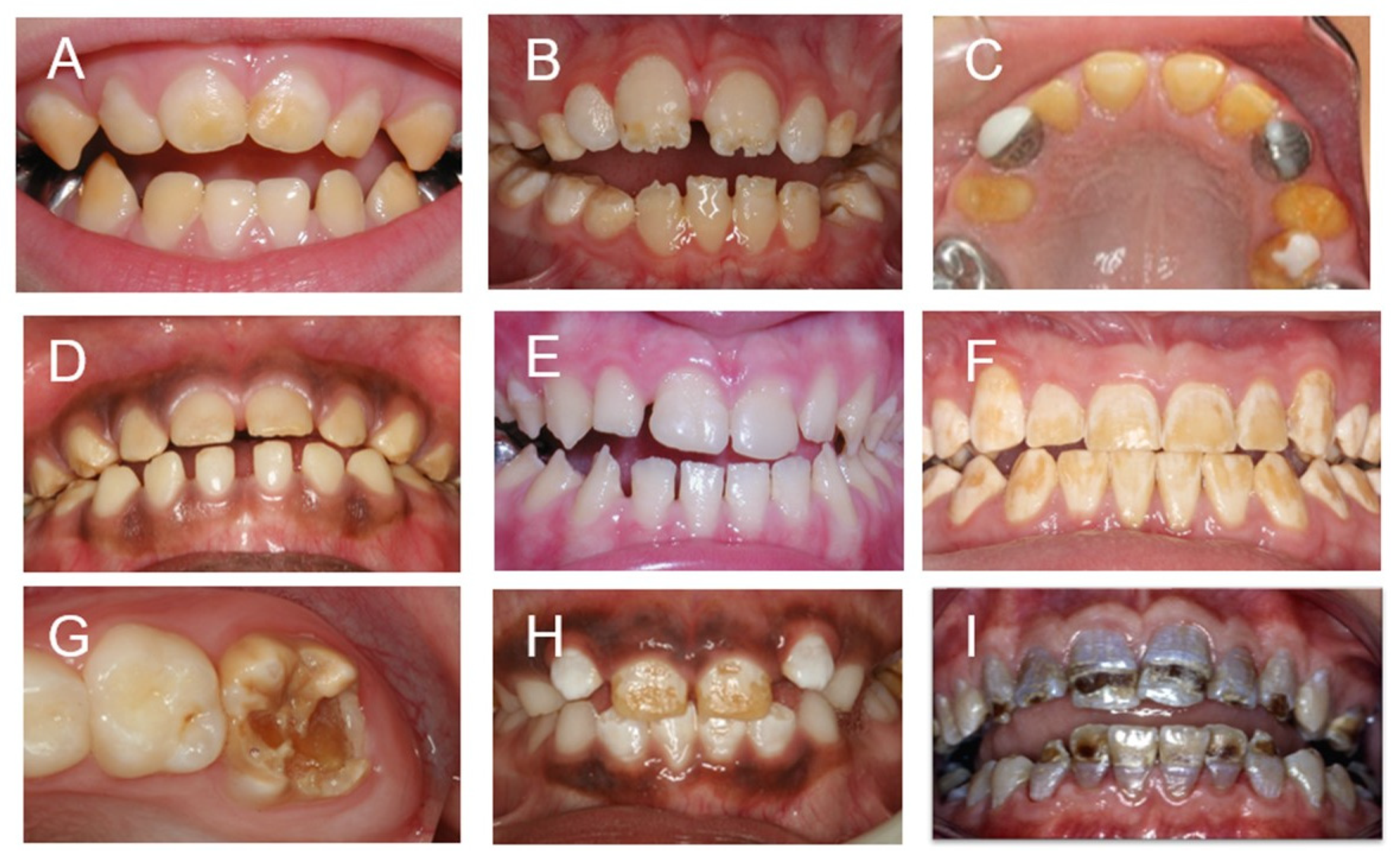
Figure 1. This panel illustrates the variable hypoplastic and hypomineralized phenotypes that can result from genetic and environmental etiologies: (A–C) syndrome-associated DDE; (A) hypomineralized enamel—#226750. KOHLSCHUTTER–TONZ SYNDROME; (B) hypoplastic enamel—#277440. VITAMIN D-DEPENDENT RICKETS; (C) hypoplastic enamel—#601216 DENTAL ANOMALIES AND SHORT STATURE; (D–F) are amelogenesis imperfectas; (D) hypomineralized enamel—#204700. AMELOGENESIS IMPERFECTA, HYPOMATURATION TYPE; (E) hypoplastic enamel—#104500. AMELOGENESIS IMPERFECTA, TYPE IB; (F) hypomineralized enamel—# 301200. AMELOGENESIS IMPERFECTA, TYPE IE; (G–I) environmental associated DDE; (G) hypomineralized enamel—MOLAR HYPOMINERALIZTION; (H) hypomineralized enamel—DENTAL FLUOROSIS; (I) combined hypoplastic hypomineralized enamel—KIDNEY DYSFUNTION and TETRACYCLINE.
Animal studies have added greatly to the knowledge of both normal and pathological enamel formation. The advent of transgenic animals has allowed for detailed studies of gene expression, protein function, pathogenic mechanisms, and detailed phenotyping that would not be possible in humans [30][31][32]. For example, many studies assessed tooth enamel genes (e.g., AmelX, Enam, Mmp20) and different mutations, the resulting proteins, and how and why these changes were associated with different enamel phenotypes [33][34][35]. Studies of the mouse and human transcriptomes of ameloblasts have demonstrated the multitude and diversity of genes involved in amelogenesis [36][37]. While animal studies have added greatly to the knowledge and understanding of enamel defects and their etiologies, it is beyond the scope of this manuscript to evaluate the many different animal and molecular models described in the literature. The following sections provide an overview of developmental defects of human enamel and the current knowledge of their etiologies and phenotypes.
3. Genetically Determined Enamel Defects
Enamel formation is exquisitely controlled and highly regulated at the molecular level with over 10,000 genes being expressed by amelobasts during amelogenesis [37]. Additionally, there are hundreds if not more microRNAs that appear to help regulate gene expression and that appear to be important for normal enamel formation [38][39]. Some of the gene products are unique to enamel, but most genes and regulator elements involved in enamel formation are also functional in cells other than ameloblasts [37]. The AMELX gene that codes for the most prevalent enamel matrix protein, amelogenin, is thought to function only in enamel, although some investigators suggest there could be a function beyond enamel formation [40]. There are many examples of genes associated with enamel defects that are also causative of pathology in other tissues such as skin and hair [41]. Historically, hereditary enamel defects have been separated into syndromic and non-syndromic conditions [42][43]. Classification of the non-syndromic hereditary enamel defects were named using the nosology of the amelogenesis imperfectas (AI) [43]. Syndrome-associated enamel defects were not referred to as AI using the Carl Witkop Jr. nosology [42]. The AI conditions were subdivided based on mode of inheritance, phenotype, and perceived development mechanism (i.e., hypoplastic, hypomaturation, hypocalcified). This nosology for enamel defects developed by Carl Witkop Jr. in the 1950s and 1960s and refined in the late 1980s was remarkably insightful given that it was developed prior to knowledge of any of the AI-associated genes and remains in use to this day [42][43]. Revision of the AI nosology using the current knowledge of genes and molecular pathways has been proposed; however, a system has yet to be fully developed and adopted for general use [44][45].
Enamel phenotypes can be subtle and variable (as can systemic phenotypes), making the classification of hereditary enamel defects into syndromic and non-syndromic challenging, and terminology is used inconsistently in the literature [27][46]. In this overview, conditions are categorized using Online Mendelian Inheritance in Man designations along with genotype and phenotype information that has been augmented by searches in the literature to help clarify descriptions of phenotypes and molecular etiology.
Genes known to be associated with hereditary enamel defects code for proteins with diverse functions such as transcription factors, growth factors, extracellular matrix proteins, ion channels, cell structure and cell motility proteins [16]. The physiology of the ameloblasts and surrounding cells is regulated by genes that are critical for normal cellular function as well as those required for the unique processes of amelogenesis. For example, ameloblasts express genes coding for cellular connections and channels for shuttling ions and water, which help create the microenvironment necessary for enamel mineralization [36]. Given the diversity of proteins and functions necessary for the creation of enamel, it is not surprising that there are myriad genetic causes of enamel defects.
4. Environmental Etiologies of Developmental Defects of Enamel
Developmental enamel defects that result from environmental causes are numerous in etiology and are diverse in phenotype. Given the timing, orchestration, and regulation of enamel formation, it is not surprising that the amount, composition, and structure of enamel can be perturbed by environmental stressors [47]. The teeth involved and the extent and characteristics of DDE are influenced by the timing of the event in relationship to the developmental stage of enamel, the duration, magnitude and type of stressor. Opacities and hypomineralization defects of the enamel are seen most often with enamel hypoplasia being less common based on epidemiological studies [22][47]. Excellent reviews on the topic of environmental etiologies of enamel defects have been published [17][48][49]. A recent scoping review identified 114 factors associated with DDE [17]. Factors associated with DDE can be clustered or grouped according to timing of the environmental exposure (i.e., prenatal, neonatal, postnatal) and by the type of stressor or mechanism of cellular insult (i.e., metabolism, blood supply, hypoxia, immune response, xenobiotic) [17][50]. One of the most studied and most common DDEs is dental fluorosis that results from ingesting excessive fluoride during the time of enamel formation. The prevalence of dental fluorosis varies in populations around the globe with very mild and above fluorosis reportedly affecting about 60% of the population in the United States [51]. Fluoride exposure can affect cellular physiology and the enamel mineral composition and structure in a dose-dependent manner [52][53][54][55]. The enamel phenotype varies from mild opacities to severe hypomineralization that results in enamel discoloration, fracturing, and loss [56][57]. While the cause of dental fluorosis is due to consumption and systemic exposure to fluoride, the exact mechanisms leading to the enamel defect and phenotype continue to be investigated. The contribution of genetics to the risk and resistance for developing dental fluorosis was observed in animals and has subsequently been increasingly studied in humans [58]. This will be explored further in the following section on gene interactions with environmental exposures.
Molar hypomineralization (MH), commonly referred to as molar–incisor hypomineralization, is a DDE associated with a variety of environmental etiologies including childhood illnesses that occurs between birth and 3 years of age [59][60][61]. Maternal illness is associated with MH, indicating that prenatal stressors can play a role as well as perinatal and postnatal factors such as prematurity, caesarean section birth, kidney disease, urinary tract infection, and gastric disorders, to name a few [62]. The prevalence varies between populations with a worldwide average prevalence of 13% [63][64][65]. The enamel phenotype varies from mild (enamel opacity and discoloration) to severe (marked hypomineralization with enamel loss upon tooth eruption) [66][67]. Hypomineralization of the primary second molars is associated with hypomineralization of the first permanent molars [68]. It has been suggested in a number of studies that the etiology and variability of phenotype associated with MH may be influenced by a variety of genetic factors [69][70][71]. Given the broad influence of genetics and genetic variation in the human genome and gene environment interactions, it is becoming generally accepted that MH is a complex condition resulting from a combination of environmental and genetic factors [72]. Gene–environment interactions such as those involved in conditions such as fluorosis and MH will be further reviewed in the following section of enamel development and epigenetics.
5. Gene and Environment Interactions Influencing Enamel Development
It is now believed that most human phenotypes have both a genetic and environmental contribution, although the understanding of those contributions and the genetics-to-phenotypes relationship is far from complete [73]. The traits that the researchers observe clinically are the result of a variety of interactions and mechanisms resulting from gene–gene and gene–environment interactions (G x E). For some developmental defects in humans, the relationships of G x E are much clearer than they are for tooth development and enamel formation. The mechanisms for gene–gene interactions and G X E that can modify development are diverse and include environmental exposure of a substance with specific proteins, disruption of signaling pathways, epigenetics and microRNA modification [74]. How much contribution is genetic versus environment varies from one phenotype to another and has been investigated using a variety of methodological approaches. For example, there is 100% concordance for fetal alcohol syndrome in monozygotic twins compared with 64% in dizygotic twins, indicating genetic modulation of this clinical outcome [75]. Twin studies assessing enamel defects show varying results as to the contribution of heritability to the enamel phenotype [76][77][78]. Two twin studies assessing enamel defects in the primary dentition show conflicting results as to twin concordance [76][77]. A study of MH involving the first permanent molars suggested greater concordance of the trait in twins but also greater risk associated with environmental determinants, including family income level and gestational hemorrhage [78]. The influence of genetic variation has been investigated for several DDE including fluorosis and MH.
Studies of skeletal fluorosis indicate that fluoride increases osteoblast activity and increases expression of Wnt3A in a rat model, suggesting that fluoride affects the Wnt-βcatenin signaling pathway [79]. Other molecular mechanisms influenced by fluoride and potentially contributing to skeletal fluorosis include Hedgehog, Notch, parathyroid hormone, endoplasmic reticulum stress and epigenetics [79]. DNA methylation of the P16 gene promoter in a fluorosis model resulted in decreased P16 protein expression that is important in cell cycle regulation [79]. Fluoride can cause hypermethylation of BMP1, MMP11 and other genes based on studies in a human osteosarcoma cell model [80]. Ameloblast cell models show that fluoride exposure influences calcium signaling pathways, apoptosis and causes endoplasmic reticulum stress [81][82]. While there are multiple studies examining SNPs in genes associated with fluorosis, studies examining the epigenetic contributions are limited [83]. One study of humans in a region with endemic coal-burning associate fluorosis-found hypermethylation rates of the O6–methylguanine–DNA–methyltransferase gene, a DNA repair gene affecting the severity of fluorosis [84].
There are few studies evaluating epigenetics and DNA methylation in relation to dental caries and hypomineralization of teeth. One twin study, suggesting multiple genes known to be associated with tooth formation, showed differential methylation in children with dental caries and hypomineralized second primary molars [85]. A study using cheek cells found no difference in global methylation of DNA in children with MH [86]. While these epigenetic studies are quite preliminary, collectively, they suggest the potential contribution of epigenetics to DDE either as an association, or possibly mechanistically, by altering the expression of genes important to normal enamel development. Examination of the role of bisphenol A exposure and the formation of enamel defects suggest that it may affect multiple biological processes, including having an epigenetic effect [87].
MicroRNAs are known to play an important role in regulating gene expression during enamel formation and DDE [88]. Evaluation of the role of microRNAs in gene regulation related to AI indicates that miR-16-5p and miR-27b-3p are likely involved through dysregulation of mouse AI genes [89]. Are the MicroRNAs involved in tooth formation influenced by environmental exposures as another contributor to DDE variance? This is an interesting question. One study assessing microRNA expression by rat fetuses having prenatal fluoride exposure shows differential expression of multiple microRNAs known to be involved in tooth formation and fluorosis pathogenesis [89]. Differential microRNA expression in the fluoride-exposed enamel organ was associated with functional annotation of target genes identified with pathways including the calcium signaling pathway and MAPK signaling pathway [89].
References
- Lacruz, R.S.; Habelitz, S.; Wright, J.T.; Paine, M.L. Dental Enamel Formation and Implications for Oral Health and Disease. Physiol. Rev. 2017, 97, 939–993.
- Ungar, P. Materials science: Strong teeth, strong seeds. Nature 2008, 452, 703–705.
- Wilmers, J.; Bargmann, S. Nature’s design solutions in dental enamel: Uniting high strength and extreme damage resistance. Acta Biomater 2020, 107, 1–24.
- Rathee, M.; Jain, P. Embryology, Teeth. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022.
- Thesleff, I. Differentiation of ameloblasts and its regulation by epithelial-mesenchymal interactions. In Dental Enamel: Formation to Destruction; Robinson, C., Kirkham, J., Shore, R., Eds.; CRC Press: Boca Raton, FL, USA, 1995; pp. 1–19.
- Balic, A.; Thesleff, I. Tissue Interactions Regulating Tooth Development and Renewal. Curr. Top. Dev. Biol. 2015, 115, 157–186.
- Thesleff, I.; Nieminen, P. Tooth morphogenesis and cell differentiation. Curr. Opin. Cell Biol. 1996, 8, 844–850.
- Yu, T.; Klein, O.D. Molecular and cellular mechanisms of tooth development, homeostasis and repair. Development 2020, 147, dev184754.
- Thesleff, I.; Vaahtokari, A.; Kettunen, P.; Åberg, T. Epithelial-mesenchymal signaling during tooth development. Connect. Tissue Res. 1995, 32, 9–15.
- Liu, H.; Yan, X.; Pandya, M.; Luan, X.; Diekwisch, T.G. Daughters of the Enamel Organ: Development, Fate, and Function of the Stratum Intermedium, Stellate Reticulum, and Outer Enamel Epithelium. Stem Cells Dev. 2016, 25, 1580–1590.
- Hu, J.C.; Yamakoshi, Y.; Yamakoshi, F.; Krebsbach, P.H.; Simmer, J.P. Proteomics and genetics of dental enamel. Cells Tissues Organs 2005, 181, 219–231.
- Robinson, C.; Briggs, H.; Atkinson, P.; Weatherell, J. Chemical changes during formation and maturation of human deciduous enamel. Arch. Oral Biol. 1981, 26, 1027–1033.
- Wright, J.T.; Hall, K.; Yamauchi, M. The protein composition of normal and developmentally defective enamel. In Dental Enamel; John Wiley & Sons: Chichester, UK, 1997; pp. 85–99.
- Weatherell, J.A.; Robinson, C.; Hiller, C.R. Distribution of carbonate in thin sections of dental enamel. Caries Res. 1968, 2, 1–9.
- Brown, K.S. Evolution and development of the dentition. Birth Defects Orig. Artic. Ser. 1983, 19, 29–66.
- Wright, J.T.; Carrion, I.A.; Morris, C. The molecular basis of hereditary enamel defects in humans. J. Dent. Res. 2015, 94, 52–61.
- Collignon, A.M.; Vergnes, J.N.; Germa, A.; Azogui, S.; Breinig, S.; Hollande, C.; Bonnet, A.-L.; Nabet, C. Factors and Mechanisms Involved in Acquired Developmental Defects of Enamel: A Scoping Review. Front. Pediatr. 2022, 10, 836708.
- Mucci, L.A.; Bjorkman, L.; Douglass, C.W.; Pedersen, N.L. Environmental and heritable factors in the etiology of oral diseases—A population-based study of Swedish twins. J. Dent. Res. 2005, 84, 800–805.
- Shungin, D.; Haworth, S.; Divaris, K.; Agler, C.S.; Kamatani, Y.; Lee, M.K.; Grinde, K.; Hindy, G.; Alaraudanjoki, V.; Pesonen, P.; et al. Genome-wide analysis of dental caries and periodontitis combining clinical and self-reported data. Nat. Commun. 2019, 10, 2773.
- Lips, A.; Antunes, L.S.; Antunes, L.A.; Pintor, A.V.; Santos, D.A.; Bachinski, R.; Küchler, E.C.; Alves, G.G. Salivary protein polymorphisms and risk of dental caries: A systematic review. Braz. Oral Res. 2017, 31, e41.
- Chisini, L.A.; Cademartori, M.G.; Conde, M.C.M.; Costa, F.D.S.; Salvi, L.C.; Tovo-Rodrigues, L.; Correa, M.B. Single nucleotide polymorphisms of taste genes and caries: A systematic review and meta-analysis. Acta Odontol. Scand. 2021, 79, 147–155.
- Brook, A.H.; Smith, J.M. The aetiology of developmental defects of enamel: A prevalence and family study in east London, U.K. Conn. Tis. Res. 1998, 39, 455–460.
- Ruschel, H.C.; Vargas-Ferreira, F.; Tovo, M.F.; Kramer, P.F.; Feldens, C.A. Developmental defects of enamel in primary teeth: Highly prevalent, unevenly distributed in the oral cavity and not associated with birth weight. Eur. Arch. Paediatr. Dent. 2019, 20, 241–248.
- Vargas-Ferreira, F.; Peres, M.A.; Dumith, S.C.; Thomson, W.M.; Demarco, F.F. Association of Pre- Peri- and Postnatal Factors with Developmental Defects of Enamel in Schoolchildren. J. Clin. Pediatr. Dent. 2018, 42, 125–134.
- Li, Y.; Navia, J.M.; Bian, J.Y. Caries experience in deciduous dentition of rural Chinese children 3–5 years old in relation to the presence or absence of enamel hypoplasia. Caries Res. 1996, 30, 8–15.
- Hong, L.; Levy, S.M.; Warren, J.J.; Broffitt, B. Association between enamel hypoplasia and dental caries in primary second molars: A cohort study. Caries Res. 2009, 43, 345–353.
- Wright, J.T.; Torain, M.; Long, K.; Seow, K.; Crawford, P.; Aldred, M.J.; Hart, P.S.; Hart, T.C. Amelogenesis imperfecta: Genotype-phenotype studies in 71 families. Cells Tissues Organs 2011, 194, 279–283.
- Poulsen, S.; Gjorup, H.; Haubek, D.; Haukali, G.; Hintze, H.; Løvschall, H.; Errboe, M. Amelogenesis imperfecta—A systematic literature review of associated dental and oro-facial abnormalities and their impact on patients. Acta Odontol. Scand. 2008, 66, 193–199.
- de la Dure-Molla, M.; Quentric, M.; Yamaguti, P.M.; Acevedo, A.-C.; Mighell, A.J.; Vikkula, M.; Huckert, M.; Berdal, A.; Bloch-Zupan, A. Pathognomonic oral profile of Enamel Renal Syndrome (ERS) caused by recessive FAM20A mutations. Orphanet. J. Rare Dis. 2014, 9, 84.
- Nunez, S.M.; Chun, Y.P.; Ganss, B.; Hu, Y.; Richardson, A.S.; Schmitz, J.E.; Fajardo, R.; Yang, J.; Hu, J.C.-C.; Simmer, J.P. Maturation stage enamel malformations in Amtn and Klk4 null mice. Matrix Biol. 2016, 52–54, 219–233.
- Ji, Y.; Li, C.; Tian, Y.; Gao, Y.; Dong, Z.; Xiang, L.; Xu, Z.; Gao, Y.; Zhang, L. Maturation stage enamel defects in Odontogenesis-associated phosphoprotein (Odaph) deficient mice. Dev. Dyn. 2021, 250, 1505–1517.
- Winkler, R.; Quaas, M.; Glasmacher, S.; Wolfrum, U.; Thalheim, T.; Galle, J.; Krohn, K.; Magin, T.M.; Aust, G. The Adhesion G-Protein-Coupled Receptor GPR115/ADGRF4 Regulates Epidermal Differentiation and Associates with Cytoskeletal KRT1. Cells 2022, 11, 3151.
- Gibson, C.W.; Yuan, Z.A.; Hall, B.; Longenecker, G.; Chen, E.; Thyagarajan, T.; Sreenath, T.; Wright, J.T.; Decker, S.; Piddington, R.; et al. Amelogenin-deficient mice display an amelogenesis imperfecta phenotype. J. Biol. Chem. 2001, 276, 31871–31875.
- Chen, E.; Yuan, Z.A.; Wright, J.T.; Hong, S.P.; Li, Y.; Collier, P.M.; Hall, B.; D’Angelo, M.; Decker, S.; Piddington, R.; et al. The Small Bovine Amelogenin LRAP Fails to Rescue the Amelogenin Null Phenotype. Calcif. Tissue Int. 2003, 73, 487–495.
- Wright, J.T.; Hart, T.C.; Hart, P.S.; Simmons, D.; Suggs, C.; Daley, B.; Simmer, J.; Hu, J.; Bartlett, J.D.; Li, Y.; et al. Human and mouse enamel phenotypes resulting from mutation or altered expression of AMEL, ENAM, MMP20 and KLK4. Cells Tissues Organs 2009, 189, 224–229.
- Lacruz, R.S.; Smith, C.E.; Chen, Y.B.; Hubbard, M.J.; Hacia, J.G.; Paine, M.L. Gene-expression analysis of early- and late-maturation-stage rat enamel organ. Eur. J. Oral Sci. 2011, 119 (Suppl. S1), 149–157.
- Hu, S.; Parker, J.; Wright, J.T. Towards unraveling the human tooth transcriptome: The dentome. PLoS ONE 2015, 10, e0124801.
- Cao, H.; Wang, J.; Li, X.; Florez, S.; Huang, Z.; Venugopalan, S.; Elangovan, S.; Skobe, Z.; Margolis, H.; Martin, J.; et al. MicroRNAs play a critical role in tooth development. J. Dent. Res. 2010, 89, 779–784.
- Yin, K.; Lin, W.; Guo, J.; Sugiyama, T.; Snead, M.L.; Hacia, J.G.; Paine, M.L. MiR-153 Regulates Amelogenesis by Targeting Endocytotic and Endosomal/lysosomal Pathways-Novel Insight into the Origins of Enamel Pathologies. Sci. Rep. 2017, 7, 44118.
- Gruenbaum-Cohen, Y.; Tucker, A.S.; Haze, A.; Shilo, D.; Taylor, A.L.; Shay, B.; Sharpe, P.T.; Mitsiadis, T.A.; Ornoy, A.; Blumenfeld, A.; et al. Amelogenin in cranio-facial development: The tooth as a model to study the role of amelogenin during embryogenesis. J. Exp. Zool. Part B Mol. Dev. Evol. 2009, 312B, 445–457.
- Peschel, N.; Wright, J.T.; Koster, M.I.; Clarke, A.J.; Tadini, G.; Fete, M.; Hadj-Rabia, S.; Sybert, V.P.; Norderyd, J.; Maier-Wohlfart, S.; et al. Molecular Pathway-Based Classification of Ectodermal Dysplasias: First Five-Yearly Update. Genes 2022, 13, 2327.
- Witkop, C.J. Hereditary defects in enamel and dentin. Acta Genet. 1957, 7, 236–239.
- Witkop, C.J. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: Problems in classification. J. Oral Pathol. Medi. 1988, 17, 547–553.
- Aldred, M.J.; Crawford, P.J.M. Amelogenesis imperfecta-towards a new classification. Oral Dis. 1995, 1, 2–5.
- Smith, C.E.L.; Poulter, J.A.; Antanaviciute, A.; Kirkham, J.; Brookes, S.J.; Inglehearn, C.F.; Mighell, A.J. Amelogenesis Imperfecta; Genes, Proteins, and Pathways. Front. Physiol. 2017, 8, 435.
- Nitayavardhana, I.; Theerapanon, T.; Srichomthong, C.; Piwluang, S.; Wichadakul, D.; Porntaveetus, T.; Shotelersuk, V. Four novel mutations of FAM20A in amelogenesis imperfecta type IG and review of literature for its genotype and phenotype spectra. Mol. Genet. Genom. 2020, 295, 923–931.
- Suckling, G.; Brown, R.; Herbison, G. The prevalence of developmental defects of enamel in 696 nine-year-old New Zealand children participating in a health development study. Community Dent. Health 1985, 2, 303–313.
- Suckling, G.; Pearce, E. Developmental defects of enamel in a group of New Zealand children: Their prevalence and some associated etiological factors. Community Dent. Oral Epidemiol. 1984, 12, 177–184.
- Seow, W.K. Developmental defects of enamel and dentine: Challenges for basic science research and clinical management. Aust. Dent. J. 2014, 59 (Suppl. 1), 143–154.
- Brook, A.H.; Fearne, J.M.; Smith, J.M. Environmental causes of enamel defects. Ciba Found. Symp. 1997, 205, 212–221, discussion 21-5.
- Wiener, R.C.; Shen, C.; Findley, P.; Tan, X.; Sambamoorthi, U. Dental Fluorosis over Time: A comparison of National Health and Nutrition Examination Survey data from 2001–2002 and 2011–2012. J. Dent. Hyg. 2018, 92, 23–29.
- Yanagisawa, T.; Takuma, S.; Fejerskov, O. Ultrastructure and composition of enamel in human dental fluorosis. Adv. Dent. Res. 1989, 3, 203–210.
- Richards, A.; Fejerskov, O.; Baelum, V. Enamel fluoride in relation to severity of human dental fluorosis. Adv. Dent. Res. 1989, 3, 147–153.
- Racz, R.; Foldes, A.; Bori, E.; Zsembery, Á.; Harada, H.; Steward, M.C.; DenBesten, P.; Bronckers, A.L.J.J.; Gerber, G.; Varga, G. No Change in Bicarbonate Transport but Tight-Junction Formation Is Delayed by Fluoride in a Novel Ameloblast Model. Front. Physiol. 2017, 8, 940.
- Zou, T.; Ma, L.; Gu, L.; Xi, S.; Zhang, K.; Guo, X. Role of Wnt/β-catenin signaling pathway in ameloblast differentiation in relevance to dental fluorosis. Chem. Biol. Interact. 2022, 367, 110145.
- Fejerskov, O.; Yanagisawa, T.; Tohda, H.; Larsen, M.J.; Josephsen, K.; Mosha, H.J. Posteruptive changes in human dental fluorosis--a histological and ultrastructural study. Proc. Finn. Dent. Soc. 1991, 87, 607–619.
- Fejerskov, O.; Thylstrup, A.; Larsen, M.J. Clinical and structural features and possible pathogenic mechanisms of dental fluorosis. Scand, J. Dent. Res. 1977, 85, 510–534.
- Everett, E.T.; Yin, Z.; Yan, D.; Zou, F. Fine mapping of dental fluorosis quantitative trait loci in mice. Eur. J. Oral Sci. 2011, 119 (Suppl. S1), 8–12.
- Weerheijm, K.L.; Duggal, M.; Mejare, I.; Papagiannoulis, L.; Koch, G.; Martens, L.C.; Hallonsten, A.-L. Judgement criteria for molar incisor hypomineralisation (MIH) in epidemiologic studies: A summary of the European meeting on MIH held in Athens, 2003. Eur. J. Paediatr. Dent. 2003, 4, 110–113.
- Ghanim, A.; Manton, D.; Bailey, D.; Marino, R.; Morgan, M. Risk factors in the occurrence of molar-incisor hypomineralization amongst a group of Iraqi children. Int. J. Paediatr. Dent. 2013, 23, 197–206.
- Ne, Y.G.S.; Frazao, D.R.; Lopes, G.O.; Fagundes, N.C.F.; Souza-Rodrigues, R.D.; Paula-Silva, F.W.G.; Maia, L.C.; Lima, R.R. Association between respiratory diseases and molar-incisor hypomineralization: A systematic review and meta-analysis. Front. Med. 2022, 9, 990421.
- Garot, E.; Rouas, P.; Somani, C.; Taylor, G.D.; Wong, F.; Lygidakis, N.A. An update of the aetiological factors involved in molar incisor hypomineralisation (MIH): A systematic review and meta-analysis. Eur. Arch. Paediatr. Dent. 2022, 23, 23–38.
- Abdelaziz, M.; Krejci, I.; Banon, J. Prevalence of Molar Incisor Hypomineralization in over 30,000 Schoolchildren in Switzerland. J. Clin. Pediatr. Dent. 2022, 46, 1–5.
- Biondi, A.M.; Cortese, S.G.; Martinez, K.; Ortolani, A.M.; Sebelli, P.M.F.; Ienco, M.; Paván, V.H.; Mendel, N.; Bertolino, M.; Hecht, P. Prevalence of molar incisor hypomineralization in the city of Buenos Aires. Acta Odontol. Latinoam. 2011, 24, 81–85.
- Schwendicke, F.; Elhennawy, K.; Reda, S.; Bekes, K.; Manton, D.J.; Krois, J. Global burden of molar incisor hypomineralization. J. Dent. 2018, 68, 10–18.
- Mathu-Muju, K.; Wright, J.T. Diagnosis and treatment of molar incisor hypomineralization. Compend. Contin. Educ. Dent. 2006, 27, 604–610, quiz 11.
- Da Costa-Silva, C.M.; Ambrosano, G.M.; Jeremias, F.; De Souza, J.F.; Mialhe, F.L. Increase in severity of molar-incisor hypomineralization and its relationship with the colour of enamel opacity: A prospective cohort study. Int. J. Paediatr. Dent. 2011, 21, 333–341.
- Negre-Barber, A.; Montiel-Company, J.M.; Boronat-Catala, M.; Catala-Pizarro, M.; Almerich-Silla, J.M. Hypomineralized Second Primary Molars as Predictor of Molar Incisor Hypomineralization. Sci. Rep. 2016, 6, 31929.
- Jeremias, F.; Pierri, R.A.; Souza, J.F.; Fragelli, C.M.B.; Restrepo, M.; Finoti, L.S.; Bussaneli, D.G.; Cordeiro, R.C.; Secolin, R.; Maurer-Morelli, C.V.; et al. Family-Based Genetic Association for Molar-Incisor Hypomineralization. Caries Res. 2016, 50, 310–318.
- Bezamat, M.; Souza, J.F.; Silva, F.M.F.; Corrêa, E.G.; Fatturi, A.L.; Brancher, J.A.; Carvalho, F.M.; Cavallari, T.; Bertolazo, L.; Machado-Souza, C.; et al. Gene-environment interaction in molar-incisor hypomineralization. PLoS ONE 2021, 16, e0241898.
- Bussaneli, D.G.; Restrepo, M.; Fragelli, C.M.B.; Santos-Pinto, L.; Jeremias, F.; Cordeiro, R.D.C.L.; Bezamat, M.; Vieira, A.R.; Scarel-Caminaga, R. Genes Regulating Immune Response and Amelogenesis Interact in Increasing the Susceptibility to Molar-Incisor Hypomineralization. Caries Res. 2019, 53, 217–227.
- Bussaneli, D.G.; Vieira, A.R.; Santos-Pinto, L.; Restrepo, M. Molar-incisor hypomineralisation: An updated view for aetiology 20 years later. Eur. Arch. Paediatr. Dent. 2022, 23, 193–198.
- Brandes, N.; Weissbrod, O.; Linial, M. Open problems in human trait genetics. Genome Biol. 2022, 23, 131.
- Lovely, C.; Rampersad, M.; Fernandes, Y.; Eberhart, J. Gene-environment interactions in development and disease. Wiley Interdiscip. Rev. Dev. Biol. 2017, 6, e247.
- Streissguth, A.P.; Dehaene, P. Fetal alcohol syndrome in twins of alcoholic mothers: Concordance of diagnosis and IQ. Am. J. Med. Genet. 1993, 47, 857–861.
- Taji, S.S.; Seow, W.K.; Townsend, G.C.; Holcombe, T. Enamel hypoplasia in the primary dentition of monozygotic and dizygotic twins compared with singleton controls. Int. J. Paediatr. Dent. 2011, 21, 175–184.
- Silva, M.J.; Kilpatrick, N.M.; Craig, J.M.; Manton, D.; Leong, P.; Burgner, D.; Scurrah, K. Etiology of Hypomineralized Second Primary Molars: A Prospective Twin Study. J. Dent. Res. 2019, 98, 77–83.
- Teixeira, R.; Andrade, N.S.; Queiroz, L.C.C.; Mendes, F.M.; Moura, M.S.; Moura, L.F.A.D.; Lima, M.D.M. Exploring the association between genetic and environmental factors and molar incisor hypomineralization: Evidence from a twin study. Int. J. Paediatr. Dent. 2018, 28, 198–206.
- Qiao, L.; Liu, X.; He, Y.; Zhang, J.; Huang, H.; Bian, W.; Chilufya, M.M.; Zhao, Y.; Han, J. Progress of Signaling Pathways, Stress Pathways and Epigenetics in the Pathogenesis of Skeletal Fluorosis. Int. J. Mol. Sci. 2021, 22, 11932.
- Daiwile, A.P.; Tarale, P.; Sivanesan, S.; Naoghare, P.K.; Bafana, A.; Parmar, D.; Kannan, K. Role of fluoride induced epigenetic alterations in the development of skeletal fluorosis. Ecotoxicol. Environ. Saf. 2019, 169, 410–417.
- Suzuki, M.; Bartlett, J.D. Sirtuin1 and autophagy protect cells from fluoride-induced cell stress. Biochim. Biophys. Acta 2014, 1842, 245–255.
- Kubota, K.; Lee, D.H.; Tsuchiya, M.; Young, C.S.; Everett, E.T.; Martinez-Mier, E.A.; Snead, M.L.; Nguyen, L.; Urano, F.; Bartlett, J.D. Fluoride induces endoplasmic reticulum stress in ameloblasts responsible for dental enamel formation. J. Biol. Chem. 2005, 280, 23194–23202.
- Balasubramanian, S.; Perumal, E. A systematic review on fluoride-induced epigenetic toxicity in mammals. Crit. Rev. Toxicol. 2022, 52, 449–468.
- Wu, C.X.; Wang, Y.H.; Li, Y.; Guan, Z.Z.; Qi, X.L. Changes of DNA repair gene methylation in blood of chronic fluorosis patients and rats. J. Trace. Elem. Med. Biol. 2018, 50, 223–228.
- Silva, M.J.; Mohandas, N.; Craig, J.M.; Manton, D.J.; Saffery, R.; Southey, M.C.; Burgner, D.P.; Lucas, J.; Kilpatrick, N.M.; Hopper, J.L.; et al. DNA methylation in childhood dental caries and hypomineralization. J. Dent. 2022, 117, 103913.
- Tynior, W.; Ilczuk-Rypula, D.; Hudy, D.; Strzelczyk, J.K. Is Aberrant DNA Methylation a Key Factor in Molar Incisor Hypomineralization? Curr. Issues Mol. Biol. 2022, 44, 2868–2878.
- Li, H.; Cui, D.; Zheng, L.; Zhou, Y.; Gan, L.; Liu, Y.; Pan, Y.; Zhou, X.; Wan, M. Bisphenol A Exposure Disrupts Enamel Formation via EZH2-Mediated H3K27me3. J. Dent. Res. 2021, 100, 847–857.
- Yin, K.; Hacia, J.G.; Zhong, Z.; Paine, M.L. Genome-wide analysis of miRNA and mRNA transcriptomes during amelogenesis. BMC Genom. 2014, 15, 998.
- Suzuki, A.; Yoshioka, H.; Liu, T.; Gull, A.; Singh, N.; Le, T.; Zhao, Z.; Iwata, J. Crucial Roles of microRNA-16-5p and microRNA-27b-3p in Ameloblast Differentiation Through Regulation of Genes Associated with Amelogenesis Imperfecta. Front. Genet. 2022, 13, 788259.
More
Information
Subjects:
Dentistry, Oral Surgery & Medicine
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Revision:
1 time
(View History)
Update Date:
17 Mar 2023
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No