 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Peramaiyan Rajendran | -- | 2175 | 2023-03-16 11:04:24 | | | |
| 2 | Peter Tang | Meta information modification | 2175 | 2023-03-17 02:37:17 | | |
Video Upload Options
Stroke is one of the main causes of mortality and disability, and it is due to be included in monetary implications on wellbeing frameworks around the world. Ischemic stroke is caused by interference in cerebral blood flow, leading to a deficit in the supply of oxygen to the affected region. It accounts for nearly 80–85% of all cases of stroke. Oxidative stress has a significant impact on the pathophysiologic cascade in brain damage leading to stroke. In the acute phase, oxidative stress mediates severe toxicity, and it initiates and contributes to late-stage apoptosis and inflammation. Oxidative stress conditions occur when the antioxidant defense in the body is unable to counteract the production and aggregation of reactive oxygen species (ROS).
1. Introduction
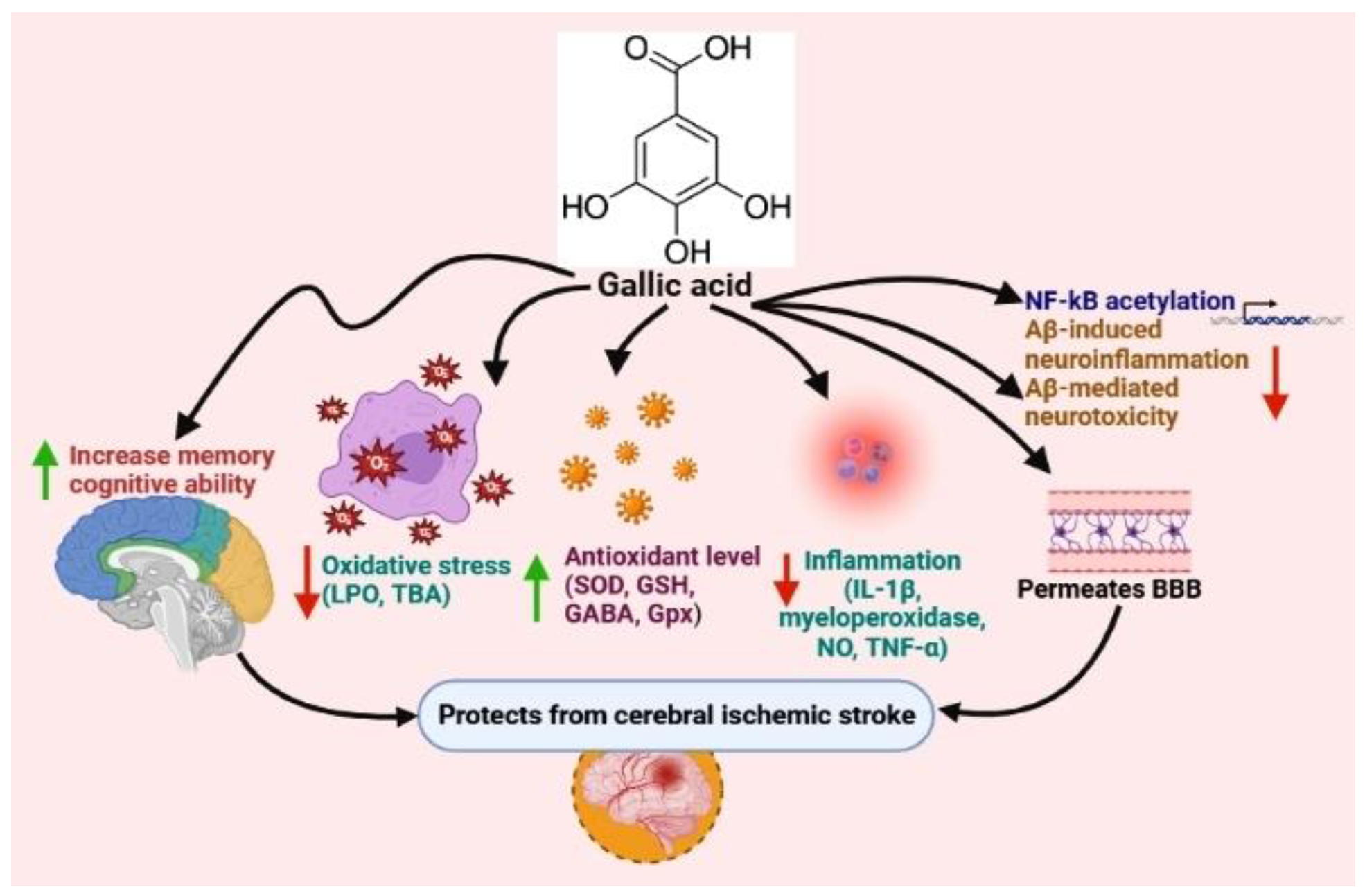
2. Decreased Blood Flow Contributes to the Pathogenesis of Ischemic Stroke
3. Oxidative Stress and Stroke
|
Name of the Polyphenol |
Mode of Study |
Major Findings |
Refs. |
|
Resveratrol |
Mouse and Rat |
Oxidative stress inhibition, anti-inflammation, brain damage and apoptosis inhibition |
[37][38][39][40][41][42][43][44][45][46][47][48][49][50][51] |
|
Gallic acid |
Mouse and Rat |
Oxidative stress reduction, anti-inflammation |
|
|
Quercetin |
Mouse and Rat |
Oxidative stress inhibition, MMP9 reduction |
|
|
Kaempferol |
Mouse and Rat |
Oxidative stress inhibition, anti-inflammation, mitochondrial dysfunction suppression |
|
|
Mangiferin |
Mouse and Rat |
Oxidative stress inhibition, anti-inflammation |
|
|
Epigallocatechin |
Mouse and Rat |
Oxidative stress inhibition, anti-inflammation |
|
|
Pinocembrin |
Mouse and Rat |
Oxidative stress inhibition, anti-inflammation |
4. Effects of Polyphenols on Stroke
References
- Campbell, B.C.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Prim. 2019, 5, 70.
- Xian, Y.; Holloway, R.G.; Chan, P.S.; Noyes, K.; Shah, M.N.; Ting, H.H.; Chappel, A.R.; Peterson, E.D.; Friedman, B. Association between stroke center hospitalization for acute ischemic stroke and mortality. Jama 2011, 305, 373–380.
- Johnson, W.; Onuma, O.; Owolabi, M.; Sachdev, S. Stroke: A global response is needed. Bull. World Health Organ. 2016, 94, 634.
- Afshari, L.; Amani, R.; Soltani, F.; Haghighizadeh, M.H.; Afsharmanesh, M.R. The relation between serum Vitamin D levels and body antioxidant status in ischemic stroke patients: A case–control study. Adv. Biomed. Res. 2015, 4, 13.
- Rodrigo, R.; Fernández-Gajardo, R.; Gutiérrez, R.; Manuel Matamala, J.; Carrasco, R.; Miranda-Merchak, A.; Feuerhake, W. Oxidative stress and pathophysiology of ischemic stroke: Novel therapeutic opportunities. CNS Neurol. Disord.-Drug Targets (Former. Curr. Drug Targets-CNS Neurol. Disord.) 2013, 12, 698–714.
- Choe, H.; Hwang, J.-Y.; Yun, J.A.; Kim, J.-M.; Song, T.-J.; Chang, N.; Kim, Y.-J.; Kim, Y. Intake of antioxidants and B vitamins is inversely associated with ischemic stroke and cerebral atherosclerosis. Nutr. Res. Pract. 2016, 10, 516–523.
- Rajendran, P.; Abdelsalam, S.A.; Renu, K.; Veeraraghavan, V.; Ben Ammar, R.; Ahmed, E.A. Polyphenols as Potent Epigenetics Agents for Cancer. Int. J. Mol. Sci. 2022, 23, 11712.
- Mollica, A.; Scioli, G.; Della Valle, A.; Cichelli, A.; Novellino, E.; Bauer, M.; Kamysz, W.; Llorent-Martínez, E.J.; Fernández-de Córdova, M.L.; Castillo-López, R. Phenolic analysis and in vitro biological activity of red wine, pomace and grape seeds oil derived from Vitis vinifera L. cv. Montepulciano d’Abruzzo. Antioxidants 2021, 10, 1704.
- Stefanucci, A.; Mollica, A. Phenolic analysis and in vitro biological activity of red wine, pomace and grape seeds oil derived from Vitis vinifera L. Cv. montepulciano d’Abruzzo. Nutr. Sci. Diet. 2012, 8, 45.
- Mahomoodally, M.F.; Mollica, A.; Stefanucci, A.; Aumeeruddy, M.Z.; Poorneeka, R.; Zengin, G. Volatile components, pharmacological profile, and computational studies of essential oil from Aegle marmelos (Bael) leaves: A functional approach. Ind. Crops Prod. 2018, 126, 13–21.
- Abbas, M.; Saeed, F.; Anjum, F.M.; Afzaal, M.; Tufail, T.; Bashir, M.S.; Ishtiaq, A.; Hussain, S.; Suleria, H.A.R. Natural polyphenols: An overview. Int. J. Food Prop. 2017, 20, 1689–1699.
- Cananzi, S.G.; Mayhan, W.G. In utero exposure to alcohol alters reactivity of cerebral arterioles. J. Cereb. Blood Flow Metab. 2019, 39, 332–341.
- Faraci, F.M.; Sobey, C.G. Role of potassium channels in regulation of cerebral vascular tone. J. Cereb. Blood Flow Metab. 1998, 18, 1047–1063.
- Mayhan, W.G.; Mayhan, J.F.; Sun, H.; Patel, K.P. In vivo properties of potassium channels in cerebral blood vessels during diabetes mellitus. Microcirculation 2004, 11, 605–613.
- Saha, P.S.; Knecht, T.M.; Arrick, D.M.; Watt, M.J.; Scholl, J.L.; Mayhan, W.G. Prenatal exposure to alcohol impairs responses of cerebral arterioles to activation of potassium channels: Role of oxidative stress. Alcohol. Clin. Exp. Res. 2022.
- Dayal, S.; Baumbach, G.L.; Arning, E.; Bottiglieri, T.; Faraci, F.M.; Lentz, S.R. Deficiency of superoxide dismutase promotes cerebral vascular hypertrophy and vascular dysfunction in hyperhomocysteinemia. PLoS ONE 2017, 12, e0175732.
- Mayhan, W.G.; Arrick, D.M.; Sharpe, G.M.; Sun, H. Age-related alterations in reactivity of cerebral arterioles: Role of oxidative stress. Microcirculation 2008, 15, 225–236.
- Kitagawa, K.; Matsumoto, M.; Mabuchi, T.; Yagita, Y.; Ohtsuki, T.; Hori, M.; Yanagihara, T. Deficiency of intercellular adhesion molecule 1 attenuates microcirculatory disturbance and infarction size in focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1998, 18, 1336–1345.
- Huang, J.; Choudhri, T.F.; Winfree, C.J.; McTaggart, R.A.; Kiss, S.; Mocco, J.; Kim, L.J.; Protopsaltis, T.S.; Zhang, Y.; Pinsky, D.J. Postischemic cerebrovascular E-selectin expression mediates tissue injury in murine stroke. Stroke 2000, 31, 3047–3053.
- Herz, J.; Sabellek, P.; Lane, T.E.; Gunzer, M.; Hermann, D.M.; Doeppner, T.R. Role of neutrophils in exacerbation of brain injury after focal cerebral ischemia in hyperlipidemic mice. Stroke 2015, 46, 2916–2925.
- Ziebell, J.M.; Adelson, P.D.; Lifshitz, J. Microglia: Dismantling and rebuilding circuits after acute neurological injury. Metab. Brain Dis. 2015, 30, 393–400.
- Doll, D.N.; Barr, T.L.; Simpkins, J.W. Cytokines: Their role in stroke and potential use as biomarkers and therapeutic targets. Aging Dis. 2014, 5, 294.
- Kim, J.Y.; Kawabori, M.; Yenari, M.A. Innate inflammatory responses in stroke: Mechanisms and potential therapeutic targets. Curr. Med. Chem. 2014, 21, 2076–2097.
- Sheng, W.S.; Hu, S.; Feng, A.; Rock, R.B. Reactive oxygen species from human astrocytes induced functional impairment and oxidative damage. Neurochem. Res. 2013, 38, 2148–2159.
- Roberts, R.A.; Smith, R.A.; Safe, S.; Szabo, C.; Tjalkens, R.B.; Robertson, F.M. Toxicological and pathophysiological roles of reactive oxygen and nitrogen species. Toxicology 2010, 276, 85–94.
- Allen, C.L.; Bayraktutan, U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int. J. Stroke 2009, 4, 461–470.
- Scapagnini, G.; Sonya, V.; Nader, A.G.; Calogero, C.; Zella, D.; Fabio, G. Modulation of Nrf2/ARE pathway by food polyphenols: A nutritional neuroprotective strategy for cognitive and neurodegenerative disorders. Mol. Neurobiol. 2011, 44, 192–201.
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102.
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344.
- Morris-Blanco, K. Mechanisms of Mitochondrial Regulation and Ischemic Neuroprotection by the PKC? Pathway; University of Miami: Miami, FL, USA, 2014.
- Tullio, F.; Perrelli, M.-G.; Femminò, S.; Penna, C.; Pagliaro, P. Mitochondrial sources of ROS in cardio protection and ischemia/reperfusion injury. Ann. Cardiovasc. Dis. 2016, 1, 1–15.
- Webster, K.A. Mitochondrial membrane permeabilization and cell death during myocardial infarction: Roles of calcium and reactive oxygen species. Future Cardiol. 2012, 8, 863–884.
- Ferreira, I.L.; Duarte, C.B.; Carvalho, A.P. Ca2+ influx through glutamate receptor-associated channels in retina cells correlates with neuronal cell death. Eur. J. Pharmacol. 1996, 302, 153–162.
- Parpura, V.; Verkhratsky, A. Homeostatic function of astrocytes: Ca2+ and Na+ signalling. Transl. Neurosci. 2012, 3, 334–344.
- Brustovetsky, T.; Pellman, J.J.; Yang, X.-F.; Khanna, R.; Brustovetsky, N. Collapsin response mediator protein 2 (CRMP2) interacts with N-methyl-D-aspartate (NMDA) receptor and Na+/Ca2+ exchanger and regulates their functional activity. J. Biol. Chem. 2014, 289, 7470–7482.
- Radak, D.; Resanovic, I.; Isenovic, E.R. Link between oxidative stress and acute brain ischemia. Angiology 2014, 65, 667–676.
- Agrawal, M.; Kumar, V.; Kashyap, M.P.; Khanna, V.K.; Randhawa, G.S.; Pant, A.B. Ischemic insult induced apoptotic changes in PC12 cells: Protection by trans resveratrol. Eur. J. Pharmacol. 2011, 666, 5–11.
- Alquisiras-Burgos, I.; Ortiz-Plata, A.; Franco-Pérez, J.; Millán, A.; Aguilera, P. Resveratrol reduces cerebral edema through inhibition of de novo SUR1 expression induced after focal ischemia. Exp. Neurol. 2020, 330, 113353.
- Arteaga, O.; Revuelta, M.; Urigüen, L.; Alvarez, A.; Montalvo, H.; Hilario, E. Pretreatment with resveratrol prevents neuronal injury and cognitive deficits induced by perinatal hypoxia-ischemia in rats. PLoS ONE 2015, 10, e0142424.
- Bonsack, F.; Alleyne, C.H., Jr.; Sukumari-Ramesh, S. Resveratrol attenuates neurodegeneration and improves neurological outcomes after intracerebral hemorrhage in mice. Front. Cell. Neurosci. 2017, 11, 228.
- Faggi, L.; Pignataro, G.; Parrella, E.; Porrini, V.; Vinciguerra, A.; Cepparulo, P.; Cuomo, O.; Lanzillotta, A.; Mota, M.; Benarese, M. Synergistic association of valproate and resveratrol reduces brain injury in ischemic stroke. Int. J. Mol. Sci. 2018, 19, 172.
- Hou, Y.; Wang, K.; Wan, W.; Cheng, Y.; Pu, X.; Ye, X. Resveratrol provides neuroprotection by regulating the JAK2/STAT3/PI3K/AKT/mTOR pathway after stroke in rats. Genes Dis. 2018, 5, 245–255.
- Lanzillotta, A.; Pignataro, G.; Branca, C.; Cuomo, O.; Sarnico, I.; Benarese, M.; Annunziato, L.; Spano, P.; Pizzi, M. Targeted acetylation of NF-kappaB/RelA and histones by epigenetic drugs reduces post-ischemic brain injury in mice with an extended therapeutic window. Neurobiol. Dis. 2013, 49, 177–189.
- Lu, X.; Dong, J.; Zheng, D.; Li, X.; Ding, D.; Xu, H. Reperfusion combined with intraarterial administration of resveratrol-loaded nanoparticles improved cerebral ischemia–reperfusion injury in rats. Nanomed. Nanotechnol. Biol. Med. 2020, 28, 102208.
- Pan, S.; Li, S.; Hu, Y.; Zhang, H.; Liu, Y.; Jiang, H.; Fang, M.; Li, Z.; Xu, K.; Zhang, H. Resveratrol post-treatment protects against neonatal brain injury after hypoxia-ischemia. Oncotarget 2016, 7, 79247.
- Qian, C.; Jin, J.; Chen, J.; Li, J.; Yu, X.; Mo, H.; Chen, G. SIRT1 activation by resveratrol reduces brain edema and neuronal apoptosis in an experimental rat subarachnoid hemorrhage model. Mol. Med. Rep. 2017, 16, 9627–9635.
- Shao, A.W.; Wu, H.J.; Chen, S.; Ammar, A.b.; Zhang, J.M.; Hong, Y. Resveratrol attenuates early brain injury after subarachnoid hemorrhage through inhibition of NF-κB-dependent inflammatory/MMP-9 pathway. CNS Neurosci. Ther. 2014, 20, 182.
- Teertam, S.K.; Jha, S. Up-regulation of Sirt1/miR-149-5p signaling may play a role in resveratrol induced protection against ischemia via p53 in rat brain. J. Clin. Neurosci. 2020, 72, 402–411.
- Wan, D.; Zhou, Y.; Wang, K.; Hou, Y.; Hou, R.; Ye, X. Resveratrol provides neuroprotection by inhibiting phosphodiesterases and regulating the cAMP/AMPK/SIRT1 pathway after stroke in rats. Brain Res. Bull. 2016, 121, 255–262.
- West, T.; Atzeva, M.; Holtzman, D.M. Pomegranate polyphenols and resveratrol protect the neonatal brain against hypoxic-ischemic injury. Dev. Neurosci. 2007, 29, 363–372.
- Zhou, X.-M.; Zhou, M.-L.; Zhang, X.-S.; Zhuang, Z.; Li, T.; Shi, J.-X.; Zhang, X. Resveratrol prevents neuronal apoptosis in an early brain injury model. J. Surg. Res. 2014, 189, 159–165.
- Mirshekari Jahangiri, H.; Sarkaki, A.; Farbood, Y.; Dianat, M.; Goudarzi, G. Gallic acid affects blood-brain barrier permeability, behaviors, hippocampus local EEG, and brain oxidative stress in ischemic rats exposed to dusty particulate matter. Environ. Sci. Pollut. Res. 2020, 27, 5281–5292.
- Sun, J.; Li, Y.-z.; Ding, Y.-h.; Wang, J.; Geng, J.; Yang, H.; Ren, J.; Tang, J.-y.; Gao, J. Neuroprotective effects of gallic acid against hypoxia/reoxygenation-induced mitochondrial dysfunctions in vitro and cerebral ischemia/reperfusion injury in vivo. Brain Res. 2014, 1589, 126–139.
- Zhao, Y.; Li, D.; Zhu, Z.; Sun, Y. Improved neuroprotective effects of gallic acid-loaded chitosan nanoparticles against ischemic stroke. Rejuvenation Res. 2020, 23, 284–292.
- Knekt, P.; Isotupa, S.; Rissanen, H.; Heliövaara, M.; Järvinen, R.; Häkkinen, S.; Aromaa, A.; Reunanen, A. Quercetin intake and the incidence of cerebrovascular disease. Eur. J. Clin. Nutr. 2000, 54, 415–417.
- Lee, J.-K.; Kwak, H.-J.; Piao, M.-S.; Jang, J.-W.; Kim, S.-H.; Kim, H.-S. Quercetin reduces the elevated matrix metalloproteinases-9 level and improves functional outcome after cerebral focal ischemia in rats. Acta Neurochir. 2011, 153, 1321–1329.
- Lei, X.; Chao, H.; Zhang, Z.; Lv, J.; Li, S.; Wei, H.; Xue, R.; Li, F.; Li, Z. Neuroprotective effects of quercetin in a mouse model of brain ischemic/reperfusion injury via anti-apoptotic mechanisms based on the Akt pathway. Mol. Med. Rep. 2015, 12, 3688–3696.
- Lin, X.; Lin, C.-H.; Zhao, T.; Zuo, D.; Ye, Z.; Liu, L.; Lin, M.-T. Quercetin protects against heat stroke-induced myocardial injury in male rats: Antioxidative and antiinflammatory mechanisms. Chem.-Biol. Interact. 2017, 265, 47–54.
- Tota, S.; Awasthi, H.; Kamat, P.K.; Nath, C.; Hanif, K. Protective effect of quercetin against intracerebral streptozotocin induced reduction in cerebral blood flow and impairment of memory in mice. Behav. Brain Res. 2010, 209, 73–79.
- Li, W.-H.; Cheng, X.; Yang, Y.-L.; Liu, M.; Zhang, S.-S.; Wang, Y.-H.; Du, G.-H. Kaempferol attenuates neuroinflammation and blood brain barrier dysfunction to improve neurological deficits in cerebral ischemia/reperfusion rats. Brain Res. 2019, 1722, 146361.
- López-Sánchez, C.; Martín-Romero, F.J.; Sun, F.; Luis, L.; Samhan-Arias, A.K.; García-Martínez, V.; Gutiérrez-Merino, C. Blood micromolar concentrations of kaempferol afford protection against ischemia/reperfusion-induced damage in rat brain. Brain Res. 2007, 1182, 123–137.
- Wu, B.; Luo, H.; Zhou, X.; Cheng, C.-y.; Lin, L.; Liu, B.-l.; Liu, K.; Li, P.; Yang, H. Succinate-induced neuronal mitochondrial fission and hexokinase II malfunction in ischemic stroke: Therapeutical effects of kaempferol. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 2307–2318.
- Yang, Y.-L.; Cheng, X.; Li, W.-H.; Liu, M.; Wang, Y.-H.; Du, G.-H. Kaempferol attenuates LPS-induced striatum injury in mice involving anti-neuroinflammation, maintaining BBB integrity, and down-regulating the HMGB1/TLR4 pathway. Int. J. Mol. Sci. 2019, 20, 491.
- Yu, L.; Chen, C.; Wang, L.-F.; Kuang, X.; Liu, K.; Zhang, H.; Du, J.-R. Neuroprotective effect of kaempferol glycosides against brain injury and neuroinflammation by inhibiting the activation of NF-κB and STAT3 in transient focal stroke. PLoS ONE 2013, 8, e55839.
- Du, S.; Liu, H.; Lei, T.; Xie, X.; Wang, H.; He, X.; Tong, R.; Wang, Y. Mangiferin: An effective therapeutic agent against several disorders. Mol. Med. Rep. 2018, 18, 4775–4786.
- Feng, X.; Xue, J.H.; Xie, K.X.; Liu, S.P.; Zhong, H.P.; Wang, C.C.; Feng, X.Q. Beneficial effect of Mangiferin against sleep deprivation-induced neurodegeneration and memory impairment in mice. Biomed. Res. (0970-938X) 2017, 28, 769–777.
- Kim, S.-J.; Sung, M.-S.; Heo, H.; Lee, J.-H.; Park, S.-W. Mangiferin protects retinal ganglion cells in ischemic mouse retina via SIRT1. Curr. Eye Res. 2016, 41, 844–855.
- Márquez, L.; García-Bueno, B.; Madrigal, J.L.; Leza, J.C. Mangiferin decreases inflammation and oxidative damage in rat brain after stress. Eur. J. Nutr. 2012, 51, 729–739.
- Prabhu, S.; Jainu, M.; Sabitha, K.; Devi, C.S. Role of mangiferin on biochemical alterations and antioxidant status in isoproterenol-induced myocardial infarction in rats. J. Ethnopharmacol. 2006, 107, 126–133.
- Xi, J.-S.; Wang, Y.-F.; Long, X.-X.; Ma, Y. Mangiferin potentiates neuroprotection by isoflurane in neonatal hypoxic brain injury by reducing oxidative stress and activation of phosphatidylinositol-3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) signaling. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 7459.
- Yang, Z.; Weian, C.; Susu, H.; Hanmin, W. Protective effects of mangiferin on cerebral ischemia–reperfusion injury and its mechanisms. Eur. J. Pharmacol. 2016, 771, 145–151.
- Aneja, R.; Hake, P.W.; Burroughs, T.J.; Denenberg, A.G.; Wong, H.R.; Zingarelli, B. Epigallocatechin, a green tea polyphenol, attenuates myocardial ischemia reperfusion injury in rats. Mol. Med. 2004, 10, 55–62.
- Bai, Q.; Lyu, Z.; Yang, X.; Pan, Z.; Lou, J.; Dong, T. Epigallocatechin-3-gallate promotes angiogenesis via up-regulation of Nfr2 signaling pathway in a mouse model of ischemic stroke. Behav. Brain Res. 2017, 321, 79–86.
- Koh, S.-H.; Lee, S.M.; Kim, H.Y.; Lee, K.-Y.; Lee, Y.J.; Kim, H.-T.; Kim, J.; Kim, M.-H.; Hwang, M.S.; Song, C. The effect of epigallocatechin gallate on suppressing disease progression of ALS model mice. Neurosci. Lett. 2006, 395, 103–107.
- Lim, S.H.; Kim, H.S.; Kim, Y.K.; Kim, T.-M.; Im, S.; Chung, M.E.; Hong, B.Y.; Ko, Y.J.; Kim, H.W.; Lee, J.I. The functional effect of epigallocatechin gallate on ischemic stroke in rats. Acta Neurobiol. Exp. (Wars) 2010, 70, 40–46.
- Park, D.-J.; Kang, J.-B.; Koh, P.-O. Epigallocatechin gallate alleviates neuronal cell damage against focal cerebral ischemia in rats. J. Vet. Med. Sci. 2020, 82, 639–645.
- Park, J.-W.; Hong, J.-S.; Lee, K.-S.; Kim, H.-Y.; Lee, J.-J.; Lee, S.-R. Green tea polyphenol (−)-epigallocatechin gallate reduces matrix metalloproteinase-9 activity following transient focal cerebral ischemia. J. Nutr. Biochem. 2010, 21, 1038–1044.
- Yao, C.; Zhang, J.; Liu, G.; Chen, F.; Lin, Y. Neuroprotection by (-)-epigallocatechin-3-gallate in a rat model of stroke is mediated through inhibition of endoplasmic reticulum stress. Mol. Med. Rep. 2014, 9, 69–72.
- Zhang, F.; Li, N.; Jiang, L.; Chen, L.; Huang, M. Neuroprotective effects of (−)-epigallocatechin-3-gallate against focal cerebral ischemia/reperfusion injury in rats through attenuation of inflammation. Neurochem. Res. 2015, 40, 1691–1698.
- Zhang, J.-C.; Xu, H.; Yuan, Y.; Chen, J.-Y.; Zhang, Y.-J.; Lin, Y.; Yuan, S.-Y. Delayed treatment with green tea polyphenol EGCG promotes neurogenesis after ischemic stroke in adult mice. Mol. Neurobiol. 2017, 54, 3652–3664.
- Habtemariam, S. The Nrf2/HO-1 axis as targets for flavanones: Neuroprotection by pinocembrin, naringenin, and eriodictyol. Oxidative Med. Cell. Longev. 2019, 2019.
- Lan, X.; Han, X.; Li, Q.; Li, Q.; Gao, Y.; Cheng, T.; Wan, J.; Zhu, W.; Wang, J. Pinocembrin protects hemorrhagic brain primarily by inhibiting toll-like receptor 4 and reducing M1 phenotype microglia. Brain Behav. Immun. 2017, 61, 326–339.
- Liu, R.; Gao, M.; Yang, Z.-H.; Du, G.-H. Pinocembrin protects rat brain against oxidation and apoptosis induced by ischemia–reperfusion both in vivo and in vitro. Brain Res. 2008, 1216, 104–115.
- Meng, F.; Liu, R.; Gao, M.; Wang, Y.; Yu, X.; Xuan, Z.; Sun, J.; Yang, F.; Wu, C.; Du, G. Pinocembrin attenuates blood–brain barrier injury induced by global cerebral ischemia–reperfusion in rats. Brain Res. 2011, 1391, 93–101.
- Pei, B.; Sun, J. Pinocembrin alleviates cognition deficits by inhibiting inflammation in diabetic mice. J. Neuroimmunol. 2018, 314, 42–49.
- Shen, X.; Liu, Y.; Luo, X.; Yang, Z. Advances in biosynthesis, pharmacology, and pharmacokinetics of pinocembrin, a promising natural small-molecule drug. Molecules 2019, 24, 2323.
- Su, Q.; Sun, Y.; Ye, Z.; Yang, H.; Kong, B.; Li, L. Pinocembrin protects endothelial cells from oxidized LDL-induced injury. Cytokine 2018, 111, 475–480.
- Wu, C.-X.; Liu, R.; Gao, M.; Zhao, G.; Wu, S.; Wu, C.-F.; Du, G.-H. Pinocembrin protects brain against ischemia/reperfusion injury by attenuating endoplasmic reticulum stress induced apoptosis. Neurosci. Lett. 2013, 546, 57–62.
- Tresserra-Rimbau, A.; Arranz, S.; Vallverdu-Queralt, A. New insights into the benefits of polyphenols in chronic diseases. Oxidative Med. Cell. Longev. 2017, 2017.
- Simonyi, A.; Wang, Q.; Miller, R.L.; Yusof, M.; Shelat, P.B.; Sun, A.Y.; Sun, G.Y. Polyphenols in cerebral ischemia. Mol. Neurobiol. 2005, 31, 135–147.
- Rajendran, P.; Rengarajan, T.; Nandakumar, N.; Divya, H.; Nishigaki, I. Mangiferin in cancer chemoprevention and treatment: Pharmacokinetics and molecular targets. J. Recept. Signal Transduct. 2015, 35, 76–84.
- Russo, G.L.; Tedesco, I.; Spagnuolo, C.; Russo, M. Antioxidant polyphenols in cancer treatment: Friend, foe or foil? Semin. Cancer Biol. 2017, 46, 1–13.
- Tzachristas, A.; Pasvanka, K.; Calokerinos, A.; Proestos, C. Polyphenols: Natural antioxidants to be used as a quality tool in wine authenticity. Appl. Sci. 2020, 10, 5908.
- Basli, A.; Soulet, S.; Chaher, N.; Mérillon, J.-M.; Chibane, M.; Monti, J.-P.; Richard, T. Wine polyphenols: Potential agents in neuroprotection. Oxidative Med. Cell. Longev. 2012, 2012.
- Sun, A.Y.; Chen, Y.-M. Oxidative stress and neurodegenerative disorders. J. Biomed. Sci. 1998, 5, 401–414.
- Fazel Nabavi, S.; M Dean, O.; Turner, A.; Sureda, A.; Daglia, M.; Mohammad Nabavi, S. Oxidative stress and post-stroke depression: Possible therapeutic role of polyphenols? Curr. Med. Chem. 2015, 22, 343–351.
- Pacifici, F.; Rovella, V.; Pastore, D.; Bellia, A.; Abete, P.; Donadel, G.; Santini, S.; Beck, H.; Ricordi, C.; Daniele, N.D. Polyphenols and ischemic stroke: Insight into one of the best strategies for prevention and treatment. Nutrients 2021, 13, 1967.
- Cheng, Y.-C.; Sheen, J.-M.; Hu, W.L.; Hung, Y.-C. Polyphenols and oxidative stress in atherosclerosis-related ischemic heart disease and stroke. Oxidative Med. Cell. Longev. 2017, 2017.
- Parrella, E.; Gussago, C.; Porrini, V.; Benarese, M.; Pizzi, M. From Preclinical Stroke Models to Humans: Polyphenols in the Prevention and Treatment of Stroke. Nutrients 2020, 13, 85.
- Yamagata, K. Polyphenols regulate endothelial functions and reduce the risk of cardiovascular disease. Curr. Pharm. Des. 2019, 25, 2443–2458.
- Zhou, Y.; Jiang, Z.; Lu, H.; Xu, Z.; Tong, R.; Shi, J.; Jia, G. Recent Advances of Natural Polyphenols Activators for Keap1-Nrf2 Signaling Pathway. Chem. Biodivers. 2019, 16, e1900400.
- Gao, K.; Liu, M.; Ding, Y.; Yao, M.; Zhu, Y.; Zhao, J.; Cheng, L.; Bai, J.; Wang, F.; Cao, J. A phenolic amide (LyA) isolated from the fruits of Lycium barbarum protects against cerebral ischemia–reperfusion injury via PKCε/Nrf2/HO-1 pathway. Aging (Albany NY) 2019, 11, 12361.
- Martínez-Huélamo, M.; Rodríguez-Morató, J.; Boronat, A.; De la Torre, R. Modulation of Nrf2 by olive oil and wine polyphenols and neuroprotection. Antioxidants 2017, 6, 73.
- S Panickar, K.; Jang, S. Dietary and plant polyphenols exert neuroprotective effects and improve cognitive function in cerebral ischemia. Recent Pat. Food Nutr. Agric. 2013, 5, 128–143.
- Xue, R.; Wu, G.; Wei, X.; Lv, J.; Fu, R.; Lei, X.; Zhang, Z.; Li, W.; He, J.; Zhao, H. Tea polyphenols may attenuate the neurocognitive impairment caused by global cerebral ischemia/reperfusion injury via anti-apoptosis. Nutr. Neurosci. 2016, 19, 63–69.
- Wang, T.; Wang, F.; Yu, L.; Li, Z. Nobiletin alleviates cerebral ischemic-reperfusion injury via MAPK signaling pathway. Am. J. Transl. Res. 2019, 11, 5967.
- Lu, H.; Wang, B.; Cui, N.; Zhang, Y. Artesunate suppresses oxidative and inflammatory processes by activating Nrf2 and ROS-dependent p38 MAPK and protects against cerebral ischemia-reperfusion injury. Mol. Med. Rep. 2018, 17, 6639–6646.
- Khan, H.; Sureda, A.; Belwal, T.; Çetinkaya, S.; Süntar, İ.; Tejada, S.; Devkota, H.P.; Ullah, H.; Aschner, M. Polyphenols in the treatment of autoimmune diseases. Autoimmun. Rev. 2019, 18, 647–657.
- Ding, S.; Jiang, H.; Fang, J. Regulation of immune function by polyphenols. J. Immunol. Res. 2018, 2018.

