Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mark Makarov | -- | 2681 | 2023-03-15 21:01:45 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Makarov, M.; Korkotian, E. Role of Mitochondria in the Central Nervous System. Encyclopedia. Available online: https://encyclopedia.pub/entry/42243 (accessed on 23 July 2026).
Makarov M, Korkotian E. Role of Mitochondria in the Central Nervous System. Encyclopedia. Available at: https://encyclopedia.pub/entry/42243. Accessed July 23, 2026.
Makarov, Mark, Eduard Korkotian. "Role of Mitochondria in the Central Nervous System" Encyclopedia, https://encyclopedia.pub/entry/42243 (accessed July 23, 2026).
Makarov, M., & Korkotian, E. (2023, March 15). Role of Mitochondria in the Central Nervous System. In Encyclopedia. https://encyclopedia.pub/entry/42243
Makarov, Mark and Eduard Korkotian. "Role of Mitochondria in the Central Nervous System." Encyclopedia. Web. 15 March, 2023.
Copy Citation
Disruption of the synapses leads to a deterioration in the communication of nerve cells and decreased plasticity, which is associated with a loss of cognitive functions and neurodegeneration. Maintaining proper synaptic activity depends on the qualitative composition of mitochondria, because synaptic processes require sufficient energy supply and fine calcium regulation.
mitophagy
autophagy
neurodegeneration
1. Introduction
For adequate interaction with the environment, adaptive behavior in society, and the realization of creative potential, every person needs a properly functioning nervous system (and the brain in particular). The elementary morphofunctional unit of the nervous system is the synapse; the formation of memory is associated with synaptic events. Morphological changes in the synapse correspond to functional requirements; this phenomenon is called neuroplasticity. This process requires careful control by the intracellular apparatus and enhanced energy supply. A normally functioning neuron and synapse are possible only in the presence of healthy mitochondria. In the synapse, mitochondria play the role not only of an energy station, but also as a calcium (Ca2+) regulator and an inducer of apoptosis [1][2].
Mitochondrial damage can lead to impaired adenosine triphosphate (ATP) production, an increase in oxidative stress products, and an increase in the level of Ca2+ in the cytosol. These undesirable consequences can be prevented through the processes of mitophagy. Mitophagy is a specific type of autophagy in which the maintenance of cellular structure and function is carried out via elimination of unhealthy mitochondria [3][4]. The term mitophagy first appeared in 2005; since then, the popularity of this topic has grown exponentially [5][6]. Due to the role played by mitochondria in the cell, the topic of mitophagy has attracted researchers in anti-aging medicine [7][8], cardiology [9][10], endocrinology [11][12][13], nephrology [14][15], and neuroscience [16][17][18][19].
2. Role of Mitochondria in the Synapse
Mitochondria are localized in all synaptic compartments: in the presynaptic terminal, at the base of the dendritic spine, in the astrocytic body and processes, and in microglial cells (when considered a part of the synapse). In the presynaptic terminal, mitochondria provide energy (ATP) for the formation of synaptic vesicles, the release of neurotransmitters into the synaptic cleft, and reuptake of the mediator. In dendrites, mitochondria are located at the base of the most functionally loaded dendritic spines, where they are responsible for providing energy for the main stages of synaptic plasticity: phosphorylation, externalization and synaptic recruitment of receptors, and changes in the structure of the dendritic spine. The quality of the mitochondrial state at the base of the dendritic spine, including membrane potential, also determines Ca2+ homeostasis, its global and local gradients [1][2][20]. In astrocytic processes, mitochondria support the operation of glutamate transporter-1 (GLT-1), excitatory amino acid transporter (EAAT) 1 and 2, as well as many other functions [21][22][23].
If the quality of the mitochondria is reduced, then the energy supply of all processes will be disrupted. For example, astrocytic mitochondria dysfunction induced with a subsequent decrease in the energy supply for the uptake of glutamate from the synaptic cleft can lead to the process of glutamate excitotoxicity [21]. In the dendritic spine, disruption of the mitochondria can lead to a decrease in the ability to maintain the function of the spine. This violation may be caused both by a decrease in energy supply and by a deficiency in the local Ca2+ signaling [24].
Damaged mitochondria contribute to the production of reactive oxygen species that disrupt the structure of macromolecules such as DNA, lipids, and proteins, which ultimately leads to necrosis and/or apoptotic death of the nerve cell [25][26].
The number of processes that depend on mitochondria in synapse is equal to all currently known molecular mechanisms in this area and requires a separate review. Researchers have previously reported in more detail on the role of mitochondria in this area [1]. Thus, a functional deficiency of synaptic mitochondria leads to negative changes in this area (Figure 1). That is why mitochondria and the processes of maintaining their quality have become such an important target for therapeutic intervention.
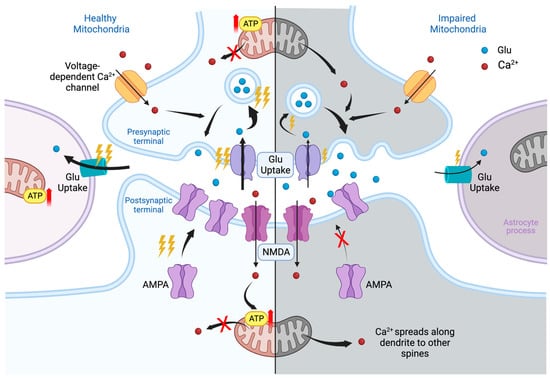
Figure 1. Glutamatergic synapse in healthy (left) and impaired (right) mitochondria. The synapse consists of three parts: on top—the presynaptic terminal, on the bottom—the postsynaptic region, on the side—a process of astroglia. In the presynaptic terminal, the mitochondrion provides energy for the reuptake of glutamate (Glu), previously released into synaptic cleft, the formation of vesicles containing Glu, and, finally, the release of neurotransmitters into the postsynaptic terminal, as shown in the figure. Ca2+ enters the presynaptic terminal through voltage-dependent ion channels; binds to synaptotagmin, a membrane-trafficking protein holding two calcium-binding domains; and thereby participates in the release of Glu. Healthy mitochondria regulate intracellular Ca2+. In case of disruption of the mitochondria, the energy supply of all presynaptic processes deteriorates and the levels of neurotransmitters decreases. Nevertheless, dysregulation of Ca2+ homeostasis at the initial stage increases the probability of transmitter release, thereby elevating its overall concentration at the synaptic cleft. Astrocytic leaflet provides the reuptake of Glu and regulates perisynaptic potassium levels. Impairment of the mitochondria in astrocyte processes violates reuptake of neurotransmitters. Its excess in synaptic cleft creates conditions for excessive excitation of the postsynaptic membrane and the effect of excitotoxicity. The main events of synaptic plasticity occur at the postsynaptic terminal. The largest and most active dendritic spines of mushroom type are supported by nearby shaft mitochondria clusters. The initial plasticity events are believed to be triggered by Ca2+ entry via N-methyl-d-aspartate (NMDA) channels. Elevated intra-spine Ca2+ levels activate the buffer protein calmodulin and corresponding kinases. Kinases phosphorylate existing α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors; promote the recruitment of extra-synaptic ones, which enhances the signal transduction; and further trigger long-term synaptic modifications. Calcium influx is also required to activate ATP production by local mitochondria. Leakage of calcium from mitochondria may be associated with its spread towards the neighboring spines and decrease in local ATP supply. Ultimately, the lack of energy support will weaken the synapse, reduce its plastic potential and create the conditions for loss of its head volume and mushroom shape, degradation, and/or pruning. Thunder symbol indicates level of consuming of ATP. Red arrow means increase of ATP production.
3. Role of Mitophagy in Neurodegeneration
To understand how various compounds can determine the course of neurodegenerative pathologies through mitophagy, it is necessary to understand the role of mitophagy in neurodegeneration. If the violation of mitophagy occurs because of pathological processes of neurodegenerative pathology, then these compounds are cofactors that can change the course of pathology through mitophagy. If the disruption of mitophagy is primary in relation to neurodegenerative processes, then these compounds are toxins that may receive a status of etiological factors. This provision will significantly change the understanding of neurodegenerative pathologies and the role of surrounding compounds.
Alzheimer’s disease is one of the most common neurodegenerative diseases characterized by a progressive impairment in cognitive functions and episodic memory, changes in behavior and personality, followed by social and everyday maladaptation. Genetic variants of AD occur only in 1–2% of cases and are associated with mutations in the APP, PS1, and PS2 genes [27][28]. At the tissue and cellular level, the disease is characterized by extracellular deposits of Amyloid beta (Aβ) plaques and intraneuronal accumulation of pTau [28][29]. Mitochondria are involved in the pathogenesis of AD, and their damage contributes to the progression of AD. On the one hand, the work of mitochondria is disrupted under the influence of the pTau protein and Aβ. Aβ and pTau contribute to damage to mitochondrial DNA (mtDNA), the respiratory chain, a decrease in cytochrome oxidase activity, damage to proteins responsible for the transportation of mitochondria along the neuron, and proteins responsible for normal fission–fusion of mitochondria. As a result, the mitochondrion ceases to provide the cell with a sufficient supply of ATP, the production of reactive oxygen species (ROS) increases, the balance of fission and fusion of mitochondria is disturbed in favor of excessive fission, and the process of elimination of damaged mitochondria in the soma is disturbed. Disruption of the mitochondrial permeability transition pore (mPTP) through the effect of Aβ on cyclophilin D (CypD) leads to severe mitochondrial stress and apoptosis [27][30]. On the other hand, disruption of mitochondrial function promotes an increase in the production of pTau and Aß. For example, in AD models, the disruption of complexes I and III with rotenone and antimycin A increased the level of ROS, leading to the accumulation of Aβ [31]. In addition, mitochondrial dysfunction leads to hyperphosphorylation of Tau [29][32]. The involvement of mitochondria in AD pathogenesis is multifaceted and complex. Stimulation of mitophagy in various AD models contributed to the leveling of AD manifestations [33][34][35].
Parkinson’s disease (PD) is characterized by typical motor disturbances, including bradykinesia, tremor, rigidity, and postural unsteadiness, as well as a range of non-motor symptoms. The pathological picture of PD is associated with progressive loss of dopaminergic neurons and aberrant accumulation of α-synuclein (α-syn) in the form of Lewy bodies in the substantia nigra compactus (SNpc). Mitophagy disorders have been found in patients suffering from PD and examined postmortem [17][36]. α-syn can lead to a deficiency of complex I, decreased ATP production and increased ROS levels, membrane depolarization, and the release of cytochrome c into the cytosol, leading to apoptosis. α-syn binds to endoplasmic reticulum (ER) or mitochondria-associated membrane (MAM) membranes, reducing the interaction between mitochondrion and ER, promoting mitochondrial fragmentation and disruption of mitophagy. This fragmentation may also be associated with disruption of the mitochondrial fission factors (Drp1, Opa1) [36]. At the same time, mutations in the genes of proteins involved in mitophagy PARK6 (coding for PINK1) and PARK2 (coding for Parkin) are associated with autosomal recessive PD; however, whether PD is caused by a violation of mitophagy in this case remains to be seen. It should be considered that there are compensatory pathways of mitophagy; therefore, the development of PD may be associated with other functions of the PINK1 and Parkin proteins [36][37].
The main problem of neurodegenerative diseases is a lack of mitophagy, but in some pathologies, excessive mitophagy can be a negative pathophysiological link. Neurological pathologies with excessive mitophagy include stroke [38][39] and multiple sclerosis (MS) [40][41]. The cell needs to maintain a balance between the number of mitochondria and mitochondrial health in order to provide enough energy on the one hand, and on the other hand to maintain other homeostatic parameters.
Thus, mitophagy in neurodegenerative processes is an important link in the self-sustaining feedback loop of neurodegeneration, which is why the search for and study of compounds that contribute to the proper course of mitophagy is so important.
4. Mechanisms of Mitophagy
Mitophagy is provided by mitochondria-specific mechanisms for the elimination of damaged mitochondria. The purpose of mitophagy is to isolate the cell from damaged mitochondria and to create space for new healthy ones. This process is carried out with the help of phagophore formation and utilization by lysosomal enzymes. There are different signaling pathways that enable the formation of phagophores. Traditionally, these pathways are divided into those that are Parkin-dependent and Parkin-independent. The Parkin-dependent mitophagy signaling pathway initiates the appearance of PTEN-induced putative kinase 1 (PINK1) on the membrane. PINK1 stabilizes on the outer mitochondrial membrane in response to stress, then autophosphorylates, leading to the accumulation of Parkin on the mitochondrial surface. Parkin promotes ubiquitination of the surface of membrane proteins. Ubiquitins promote attachment of mitochondria to the autophagosome membrane via p62 protein and microtubule-associated proteins with 1A/1B-light chain 3 II (LC3II) [42][43]. Appearance and stabilization of PINK1 on the membrane surface is associated with a decrease in mitochondrial membrane potential [44][45]. Another important link in the induction of the PINK1/Parkin pathway of mitophagy may be ataxia-telangiectasia mutated protein kinase (ATM), which is thought to phosphorylate PINK1. Knockdown of the genes responsible for ATM synthesis leads to disruption of mitophagy stimulated by lead (Pb) or spermidine [46][47].
Parkin-independent mitophagy pathways are activated in response to hypoxia and are associated with the stabilization of BNIP3, Nix, and FUNDC1 proteins on the outer mitochondrial membrane. These proteins, in turn, can bind to LC3II, promoting autophagosome organization [16][48].
Dynamin-related protein 1 (Drp1) plays an important role in mitochondrial division. In the region of the mitochondrial fission ring, Drp1 attaches to fission 1 (Fis1), mitochondrial fission factor (Mff), and mitochondrial dynamic 49/51 kDa protein MiD49/51 proteins, contributing to further division processes [49][50]. Drp1 is involved in Bnip3-dependent apoptosis by controlling the permeabilization of the outer mitochondrial membrane [50][51].
The mechanisms of mitophagy are not isolated from the mechanisms of other processes that may be associated with common autophagy pathways, mitochondrial biogenesis processes, and mitochondrial fusion and division processes. All of the above are controlled by higher order signaling pathways. The main molecules of such signaling pathways are mammalian target of rapamycin (mTOR), adenosine monophosphate (AMP), activated protein kinase (AMPK), and transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2). Changes in glucose and ATP levels affect the functioning of mTOR and AMPK. A decrease in the level of ATP and an increase in AMP contribute to the activation of autophagy and mitophagy. AMP-activated protein kinase (AMPK) is a highly conserved sensor of low levels of intracellular ATP that is rapidly activated after almost all mitochondrial stresses, even those that do not disrupt the mitochondrial membrane potential [52][53]. In turn, AMPK activates unc-51-like kinase 1 (ULK1), which phosphorylates various proteins necessary for normal mitophagy, such as Parkin, TANK-binding kinase 1 (TBK1), and Mff, Beclin-1. Phosphorylated TBK1 itself phosphorylates other proteins regulating mitophagy. Beclin-1 is an important protein for autophagosome formation and recruitment of Parkin [54][55][56]. In contrast to AMPK, mTOR is a major inhibitor of autophagy and mitophagy. It is activated in response to various stimuli, including those caused by an increase in the amount of nutrients and insulin levels, performs its functions as part of the mTORC1 complex, and inhibits ULK1 [12][57]. Nrf2 regulates the expression of many enzymes with antioxidant and detoxifying functions. Nrf2 does not have an inhibitory or activating effect on mitophagy, but participates as a mitophagy regulator, stimulating mitochondrial biogenesis factors (Nrf1, PGC-1α) on the one hand, and mitophagy factors (p62, PINK1) on the other hand [58][59][60][61]. The summary of different possible signaling pathways of mitophagy are presented in Figure 2.
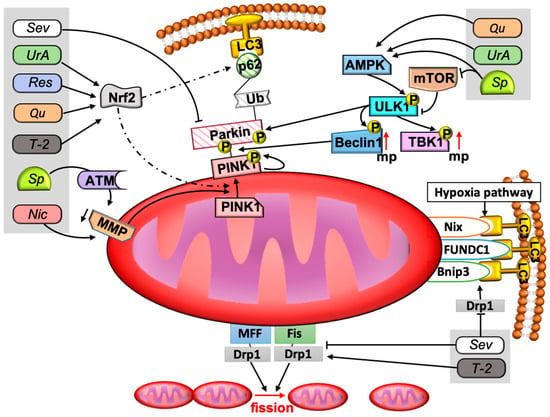
Figure 2. The signaling pathways of mitophagy and examples of participation of different active compounds. The figure shows the signaling pathways of mitophagy and the place of described compounds in these signaling pathways. Mitophagy is provided in several ways. The main pathway is the Parkin-dependent pathway. A decrease in the membrane potential contributes to the stabilization of PINK1 on the outer mitochondrial membrane. PINK1 phosphorylates Parkin. Parkin is recruited to the outer membrane and provides attachment to the phagophore via ubiquitin chains, p62 protein, and LC3II. Parkin-independent pathways are activated by hypoxia and are provided by Nix, FUNDC1, and Bnip3 molecules. Mitophagy is regulated by AMPK, mTOR, and Nrf2. AMPK and mTOR are an activator and inhibitor of ULK1 kinase, respectively. ULK1 phosphorylates Parkin and other proteins involved in the proper course of mitophagy, such as TBK1 and Beclin1. Nrf2 is a transcription factor that can promote mitophagy under certain circumstances and mitochondrial biogenesis in others. At the lower pole of the mitochondria, the role of Mff, Fis1, and Drp1 has been demonstrated. These factors are very important for proper fission of mitochondria. Drp1 also promotes Bnip3-mediated mitophagy. Resveratrol, urolithin A, quercetin, and T-2 toxin contribute to the regulation of the mitophagy process through the Nrf2 factor. Niclosamide and spermidine reduce the membrane potential of the mitochondria, and spermidine has this effect indirectly, through ATM. Urolithin A, spermidine, and quercetin can also stimulate AMPK, and spermidine blocks mTOR. Sevoflurane reduces p62 and Parkin, and sevoflurane is also able to inhibit mitochondrial division and the Bnip3-mediated mitophagy pathway by reducing Drp1. Abbreviations: AMPK—AMP kinase, ATM—ataxia-telangiectasia mutated protein kinase (ATM), Drp1—Dynamin-related protein 1, LC3—1A/1B-light chain 3 II (LC3II) protein, MMP—mitochondria membrane potential, mTOR—mammalian target of rapamycin, MFF—mitochondrial fission factor, Fis—fission 1, mp—mitophagy, Nic—niclosamide, Nrf2—the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), p in yellow circles means phosphorylation of protein, PINK1—PTEN-induced putative kinase 1, Qu—quercetin, Res—resveratrol, Sev—sevoflurane, Sp—spermidine, TBK1—TANK-binding kinase 1, T-2—T-2 toxin, Ub—ubiquitin, ULK1—unc-51-like kinase 1, UrA—urolithin A. Solid lines with arrowheads correspond to activating or promoting effects and inhibiting effects in the case of lines without arrowheads. Dotted line means regulation of expression.
References
- Kushnireva, L.; Korkotian, E. Mitochondria-Endoplasmic Reticulum Interaction in Central Neurons. In Updates on Endoplasmic Reticulum; IntechOpen: London, UK, 2022.
- Cowan, K.; Anichtchik, O.; Luo, S. Mitochondrial integrity in neurodegeneration. CNS Neurosci. Ther. 2019, 25, 825–836.
- Okamoto, K. Organellophagy: Eliminating cellular building blocks via selective autophagy. J. Cell Biol. 2014, 205, 435–445.
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705.
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5.
- Priault, M.; Salin, B.; Schaeffer, J.; Vallette, F.D.; Di Rago, J.P.; Martinou, J.C. Impairing the bioenergetic status and the biogenesis of mitochondria triggers mitophagy in yeast. Cell Death Differ. 2005, 12, 1613–1621.
- Markaki, M.; Palikaras, K.; Tavernarakis, N. Novel insights into the anti-aging role of mitophagy. Int. Rev. Cell Mol. Biol. 2018, 340, 169–208.
- Lin, C.L. Mitophagy and mitohormetics: Promising antiaging strategy. In Anti-Aging Drug Discovery on the Basis of Hallmarks of Aging; Academic Press: Cambridge, MA, USA, 2022; pp. 279–289.
- Ajoolabady, A.; Aslkhodapasandhokmabad, H.; Aghanejad, A.; Zhang, Y.; Ren, J. Mitophagy receptors and mediators: Therapeutic targets in the management of cardiovascular ageing. Ageing Res. Rev. 2020, 62, 101129.
- Morciano, G.; Patergnani, S.; Bonora, M.; Pedriali, G.; Tarocco, A.; Bouhamida, E.; Marchi, S.; Ancora, G.; Anania, G.; Wieckowski, M.R.; et al. Mitophagy in cardiovascular diseases. J. Clin. Med. 2020, 9, 892.
- Bhansali, S.; Bhansali, A.; Dutta, P.; Walia, R.; Dhawan, V. Metformin upregulates mitophagy in patients with T2DM: A randomized placebo-controlled study. J. Cell. Mol. Med. 2020, 24, 2832–2846.
- Saxena, S.; Mathur, A.; Kakkar, P. Critical role of mitochondrial dysfunction and impaired mitophagy in diabetic nephropathy. J. Cell. Physiol. 2019, 234, 19223–19236.
- Sidarala, V.; Pearson, G.L.; Parekh, V.S.; Thompson, B.; Christen, L.; Gingerich, M.A.; Zhu, J.; Stromer, T.; Ren, J.; Reck, E.C.; et al. Mitophagy protects β cells from inflammatory damage in diabetes. JCI Insight 2020, 5, e141138.
- Dagar, N.; Kale, A.; Steiger, S.; Anders, H.J.; Gaikwad, A.B. Receptor-Mediated Mitophagy: An Emerging Therapeutic Target in Acute Kidney Injury. Mitochondrion 2022, 66, 82–91.
- Chen, K.; Feng, L.; Hu, W.; Chen, J.; Wang, X.; Wang, L.; He, Y. Optineurin inhibits NLRP3 inflammasome activation by enhancing mitophagy of renal tubular cells in diabetic nephropathy. FASEB J. 2019, 33, 4571–4585.
- Chu, C.T. Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol. Dis. 2019, 122, 23–34.
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s disease: From pathogenesis to treatment. Cells 2019, 8, 712.
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial dysfunction and mitophagy in Parkinson’s disease: From mechanism to therapy. Trends Biochem. Sci. 2021, 46, 329–343.
- Clark, E.H.; de la Torre, A.V.; Hoshikawa, T.; Briston, T. Targeting mitophagy in Parkinson’s disease. J. Biol. Chem. 2021, 296, 100209.
- Korkotian, E.; Meshcheriakova, A.; Segal, M. Presenilin 1 regulates i and mitochondria/ER interaction in cultured rat hippocampal neurons. Oxidative Med. Cell. Longev. 2019, 2019, 7284967.
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells 2019, 8, 184.
- Peterson, A.R.; Binder, D.K. Astrocyte glutamate uptake and signaling as novel targets for antiepileptogenic therapy. Front. Neurol. 2020, 11, 1006.
- Bantle, C.M.; Hirst, W.D.; Weihofen, A.; Shlevkov, E. Mitochondrial dysfunction in astrocytes: A role in Parkinson’s disease? Front. Cell Dev. Biol. 2021, 8, 608026.
- Kushnireva, L.; Basnayake, K.; Holcman, D.; Segal, M.; Korkotian, E. Dynamic regulation of mitochondrial in hippocampal neurons. Int. J. Mol. Sci. 2022, 23, 12321.
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583.
- Collin, F. Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 2407.
- Pradeepkiran, J.A.; Reddy, P.H. Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101191.
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s disease: Cellular and molecular mechanisms. Trends Neurosci. 2017, 40, 151–166.
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional mitochondria and mitophagy as drivers of Alzheimer’s disease pathogenesis. Front. Aging Neurosci. 2019, 11, 311.
- Albensi, B.C. Dysfunction of mitochondria: Implications for Alzheimer’s disease. Int. Rev. Neurobiol. 2019, 145, 13–27.
- Leuner, K.; Schütt, T.; Kurz, C.; Eckert, S.H.; Schiller, C.; Occhipinti, A.; Mai, S.; Haass, C.; Reichert, A.S.; Müller, W.E. Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid. Redox Signal. 2012, 16, 1421–1433.
- Neddens, J.; Temmel, M.; Flunkert, S.; Kerschbaumer, B.; Hoeller, C.; Loeffler, T.; Niederkofler, V.; Daum, G.; Attems, J.; Hutter-Paier, B. Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 52.
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412.
- Lautrup, S.; Lou, G.; Aman, Y.; Nilse, H.; Tao, J.; Fang, E.F. Microglial mitophagy mitigates neuroinflammation in Alzheimer’s disease. Neurochem. Int. 2019, 129, 104469.
- Lee, H.J.; Jung, Y.H.; Choi, G.E.; Kim, J.S.; Chae, C.W.; Lim, J.R.; Seo, Y.K.; Yoon, J.H.; Cho, J.H.; Lee, S.J.; et al. Urolithin A suppresses high glucose-induced neuronal amyloidogenesis by modulating TGM2-dependent ER-mitochondria contacts and calcium homeostasis. Cell Death Differ. 2021, 28, 184–202.
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s disease and other age-related neurodegenerative diseases. Cells 2020, 9, 150.
- Chu, C.T. Multiple pathways for mitophagy: A neurodegenerative conundrum for Parkinson’s disease. Neurosci. Lett. 2019, 697, 66–71.
- Guan, R.; Zou, W.; Dai, X.; Yu, X.; Liu, H.; Chen, Q.; Teng, W. Mitophagy, a potential therapeutic target for stroke. J. Biomed. Sci. 2018, 25, 87.
- Song, M.; Zhou, Y.; Fan, X. Mitochondrial quality and quantity control: Mitophagy is a potential therapeutic target for ischemic stroke. Mol. Neurobiol. 2022, 59, 3110–3123.
- Patergnani, S.; Fossati, V.; Bonora, M.; Giorgi, C.; Marchi, S.; Missiroli, S.; Rusielewicz, T.; Wieckowski, M.R.; Pinton, P. Mitochondria in multiple sclerosis: Molecular mechanisms of pathogenesis. Int. Rev. Cell Mol. Biol. 2017, 328, 49–103.
- Patergnani, S.; Bonora, M.; Ingusci, S.; Previati, M.; Marchi, S.; Zucchini, S.; Perrone, M.; Wieckowski, M.R.; Castellazzi, M.; Pugliatti, M.; et al. Antipsychotic drugs counteract autophagy and mitophagy in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2020078118.
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624.
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. In Autophagosome and Phagosome; Humana Press: Totowa, NJ, USA, 2008; pp. 77–88.
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221.
- McWilliams, T.G.; Muqit, M.M. PINK1 and Parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91.
- Qi, Y.; Qiu, Q.; Gu, X.; Tian, Y.; Zhang, Y. ATM mediates spermidine-induced mitophagy via PINK1 and Parkin regulation in human fibroblasts. Sci. Rep. 2016, 6, 24700.
- Gu, X.; Qi, Y.; Feng, Z.; Ma, L.; Gao, K.; Zhang, Y. Lead (Pb) induced ATM-dependent mitophagy via PINK1/Parkin pathway. Toxicol. Lett. 2018, 291, 92–100.
- Pradeepkiran, J.A.; Hindle, A.; Kshirsagar, S.; Reddy, P.H. Are mitophagy enhancers therapeutic targets for Alzheimer’s disease? Biomed. Pharmacother. 2022, 149, 112918.
- Pernaute, B.; Pérez-Montero, S.; Nieto, J.M.S.; Di Gregorio, A.; Lima, A.; Lawlor, K.; Bowling, S.; Liccardi, G.; Tomás, A.; Meier, P.; et al. DRP1 levels determine the apoptotic threshold during embryonic differentiation through a mitophagy-dependent mechanism. Dev. Cell 2022, 57, 1316–1330.e7.
- Tong, M.; Zablocki, D.; Sadoshima, J. The role of Drp1 in mitophagy and cell death in the heart. J. Mol. Cell. Cardiol. 2020, 142, 138–145.
- Lee, Y.; Lee, H.Y.; Hanna, R.A.; Gustafsson, Å.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, H1924–H1931.
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135.
- Iorio, R.; Celenza, G.; Petricca, S. Mitophagy: Molecular mechanisms, new concepts on parkin activation and the emerging role of AMPK/ULK1 Axis. Cells 2021, 11, 30.
- Nazarko, V.Y.; Zhong, Q. ULK1 targets Beclin-1 in autophagy. Nat. Cell Biol. 2013, 15, 727–728.
- Park, J.M.; Seo, M.; Jung, C.H.; Grunwald, D.; Stone, M.; Otto, N.M.; Toso, E.; Ahn, Y.; Kyba, M.; Griffin, T.J.; et al. ULK1 phosphorylates Ser30 of BECN1 in association with ATG14 to stimulate autophagy induction. Autophagy 2018, 14, 584–597.
- Kumar, A.; Shaha, C. SESN2 facilitates mitophagy by helping Parkin translocation through ULK1 mediated Beclin1 phosphorylation. Sci. Rep. 2018, 8, 615.
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295.
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496.
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free. Radic. Biol. Med. 2015, 88, 179–188.
- Gureev, A.P.; Sadovnikova, I.S.; Starkova, N.N.; Starkov, A.A.; Popov, V.N. p62-Nrf2-p62 mitophagy regulatory loop as a target for preventive therapy of neurodegenerative diseases. Brain Sci. 2020, 10, 847.
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
918
Entry Collection:
Neurodegeneration
Revision:
1 time
(View History)
Update Date:
15 Mar 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No