 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Takeharu Sakamoto | -- | 2329 | 2023-03-14 12:05:45 | | | |
| 2 | Lindsay Dong | Meta information modification | 2329 | 2023-03-15 09:08:41 | | |
Video Upload Options
Hypoxia-inducible factor-1α (HIF-1α) is a transcription factor that plays a crucial role in cells adapting to a low-oxygen environment by facilitating a switch from oxygen-dependent ATP production to glycolysis. Mediated by membrane type-1 matrix metalloproteinase (MT1-MMP) expression, Munc-18-1 interacting protein 3 (Mint3) binds to the factor inhibiting HIF-1 (FIH-1) and inhibits its suppressive effect, leading to HIF-1α activation. Defects in Mint3 generally lead to improved acute inflammation, which is regulated by HIF-1α and subsequent glycolysis, as well as the suppression of the proliferation and metastasis of cancer cells directly through its expression in cancer cells and indirectly through its expression in macrophages or fibroblasts associated with cancer. Mint3 in inflammatory monocytes enhances the chemotaxis into metastatic sites and the production of vascular endothelial growth factors, which leads to the expression of E-selectin at the metastatic sites and the extravasation of cancer cells. Fibroblasts express L1 cell adhesion molecules in a Mint3-dependent manner and enhance integrin-mediated cancer progression. In pancreatic cancer cells, Mint3 directly promotes cancer progression. Naphthofluorescein, a Mint3 inhibitor, can disrupt the interaction between FIH-1 and Mint3 and potently suppress Mint3-mediated inflammation, cancer progression, and metastasis without causing marked adverse effects.
1. Introduction
2. HIF Proteins
3. Mechanisms of Mint3-Mediated HIF-1 Activation
3.1. Mint3 Indirectly Upregulates HIF-1α Activity through Its Interaction with FIH-1
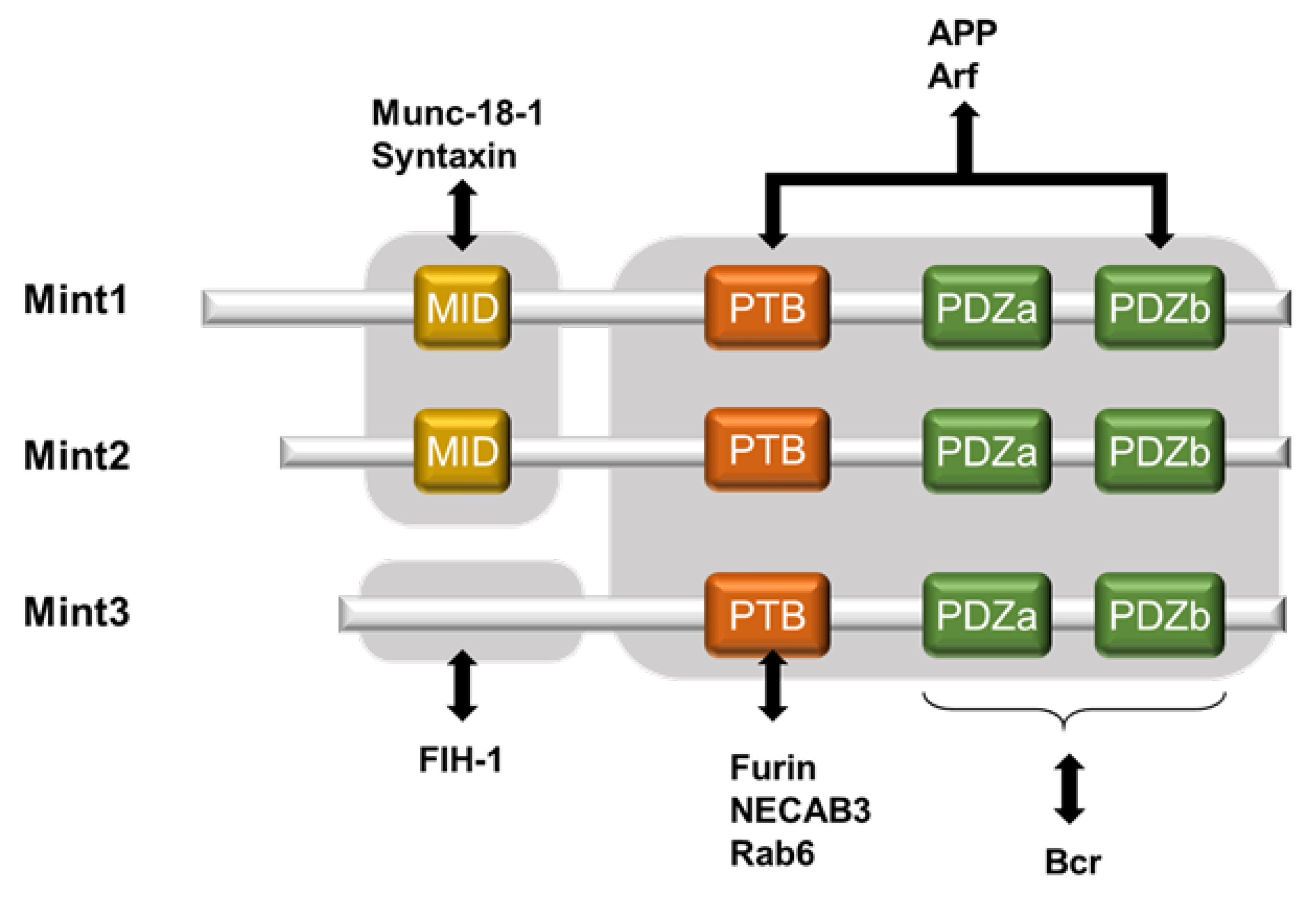
3.2. Factors That Support Mint3-Mediated HIF-1α Activation
4. Mint3 Mediates Inflammatory Responses
5. Role of Mint3 in Cancer Progression
The effects of Mint3 on the innate immune system may be applicable in cancers because Mint3 has the potential to increase cancer malignancy and metastatic features through its expression in inflammatory monocytes (IMs), which are defined with the following markers: Gr-1/Ly6C+, CD11b/CD115+ [46]. In addition, a relationship between Mint3 activity in cancer cells and cancer-associated fibroblasts (CAFs) has been previously reported [47][48][49]. Because Mint3 has key roles in enhancing cancer progression and metastasis by upregulating HIF-1α, which is also related to the tumor microenvironments, it could be an attractive target for cancer treatment, as tumor-microenvironments-related factors are gaining more attraction, which is described as one of the important hallmarks of cancers [50].
5.1. Impact of Mint3 Activity in Cancer Cells
5.2. Metastatic Ability of Cancer Cells Achieved through Mint3 Expression in IM
5.3. Supportive Effect of Mint3 Expression in CAFs on Cancer Progression
6. Therapeutic Efficacy of Targeting Mint3-Related Environments
References
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314.
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344.
- Luo, Z.; Tian, M.; Yang, G.; Tan, Q.; Chen, Y.; Li, G.; Zhang, Q.; Li, Y.; Wan, P.; Wu, J. Hypoxia signaling in human health and diseases: Implications and prospects for therapeutics. Signal Transduct. Target. Ther. 2022, 7, 218.
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Forster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 2003, 112, 645–657.
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162.
- Rezvani, H.R.; Ali, N.; Serrano-Sanchez, M.; Dubus, P.; Varon, C.; Ged, C.; Pain, C.; Cario-Andre, M.; Seneschal, J.; Taieb, A.; et al. Loss of epidermal hypoxia-inducible factor-1alpha accelerates epidermal aging and affects re-epithelialization in human and mouse. J. Cell Sci. 2011, 124, 4172–4183.
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M.; et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 2010, 7, 391–402.
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237.
- Wang, G.L.; Semenza, G.L. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc. Natl. Acad. Sci. USA 1993, 90, 4304–4308.
- Jiang, B.H.; Semenza, G.L.; Bauer, C.; Marti, H.H. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am. J. Physiol. 1996, 271, C1172–C1180.
- Pugh, C.W.; O’Rourke, J.F.; Nagao, M.; Gleadle, J.M.; Ratcliffe, P.J. Activation of hypoxia-inducible factor-1; definition of regulatory domains within the alpha subunit. J. Biol. Chem. 1997, 272, 11205–11214.
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82.
- Rankin, E.B.; Biju, M.P.; Liu, Q.; Unger, T.L.; Rha, J.; Johnson, R.S.; Simon, M.C.; Keith, B.; Haase, V.H. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J. Clin. Investig. 2007, 117, 1068–1077.
- Renassia, C.; Peyssonnaux, C. New insights into the links between hypoxia and iron homeostasis. Curr. Opin. Hematol. 2019, 26, 125–130.
- Wang, V.; Davis, D.A.; Haque, M.; Huang, L.E.; Yarchoan, R. Differential gene up-regulation by hypoxia-inducible factor-1alpha and hypoxia-inducible factor-2alpha in HEK293T cells. Cancer Res. 2005, 65, 3299–3306.
- Hatanaka, M.; Shimba, S.; Sakaue, M.; Kondo, Y.; Kagechika, H.; Kokame, K.; Miyata, T.; Hara, S. Hypoxia-inducible factor-3alpha functions as an accelerator of 3T3-L1 adipose differentiation. Biol. Pharm. Bull. 2009, 32, 1166–1172.
- Sakamoto, T.; Seiki, M. Mint3 enhances the activity of hypoxia-inducible factor-1 (HIF-1) in macrophages by suppressing the activity of factor inhibiting HIF-1. J. Biol. Chem. 2009, 284, 30350–30359.
- Okamoto, M.; Sudhof, T.C. Mints, Munc18-interacting proteins in synaptic vesicle exocytosis. J. Biol. Chem. 1997, 272, 31459–31464.
- Okamoto, M.; Südhof, T.C. Mint 3: A ubiquitous mint isoform that does not bind to munc18-1 or -2. Eur. J. Cell Biol. 1998, 77, 161–165.
- Han, J.; Wang, Y.; Wang, S.; Chi, C. Interaction of Mint3 with Furin regulates the localization of Furin in the trans-Golgi network. J. Cell Sci. 2008, 121, 2217–2223.
- Borg, J.P.; Ooi, J.; Levy, E.; Margolis, B. The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol. Cell Biol. 1996, 16, 6229–6241.
- Tanahashi, H.; Tabira, T. X11L2, a new member of the X11 protein family, interacts with Alzheimer’s beta-amyloid precursor protein. Biochem. Biophys. Res. Commun. 1999, 255, 663–667.
- Teber, I.; Nagano, F.; Kremerskothen, J.; Bilbilis, K.; Goud, B.; Barnekow, A. Rab6 interacts with the mint3 adaptor protein. Biol. Chem. 2005, 386, 671–677.
- Nakaoka, H.J.; Hara, T.; Yoshino, S.; Kanamori, A.; Matsui, Y.; Shimamura, T.; Sato, H.; Murakami, Y.; Seiki, M.; Sakamoto, T. NECAB3 Promotes Activation of Hypoxia-inducible factor-1 during Normoxia and Enhances Tumourigenicity of Cancer Cells. Sci. Rep. 2016, 6, 22784.
- Ten, T.; Nagatoishi, S.; Maeda, R.; Hoshino, M.; Nakayama, Y.; Seiki, M.; Sakamoto, T.; Tsumoto, K. Structural and thermodynamical insights into the binding and inhibition of FIH-1 by the N-terminal disordered region of Mint3. J. Biol. Chem. 2021, 297, 101304.
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67.
- Sakamoto, T.; Seiki, M. Integrated functions of membrane-type 1 matrix metalloproteinase in regulating cancer malignancy: Beyond a proteinase. Cancer Sci. 2017, 108, 1095–1100.
- Yana, I.; Weiss, S.J. Regulation of membrane type-1 matrix metalloproteinase activation by proprotein convertases. Mol. Biol. Cell 2000, 11, 2387–2401.
- Sato, H.; Takino, T.; Okada, Y.; Cao, J.; Shinagawa, A.; Yamamoto, E.; Seiki, M. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 1994, 370, 61–65.
- Ohuchi, E.; Imai, K.; Fujii, Y.; Sato, H.; Seiki, M.; Okada, Y. Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J. Biol. Chem. 1997, 272, 2446–2451.
- Barbolina, M.V.; Stack, M.S. Membrane type 1-matrix metalloproteinase: Substrate diversity in pericellular proteolysis. Semin. Cell Dev. Biol. 2008, 19, 24–33.
- Sakamoto, T.; Seiki, M. Cytoplasmic tail of MT1-MMP regulates macrophage motility independently from its protease activity. Genes Cells 2009, 14, 617–626.
- Sakamoto, T.; Seiki, M. A membrane protease regulates energy production in macrophages by activating hypoxia-inducible factor-1 via a non-proteolytic mechanism. J. Biol. Chem. 2010, 285, 29951–29964.
- Michl, J.; Ohlbaum, D.J.; Silverstein, S.C. 2-Deoxyglucose selectively inhibits Fc and complement receptor-mediated phagocytosis in mouse peritoneal macrophages. I. Description of the inhibitory effect. J. Exp. Med. 1976, 144, 1465–1483.
- Riedemann, N.C.; Guo, R.F.; Ward, P.A. Novel strategies for the treatment of sepsis. Nat. Med. 2003, 9, 517–524.
- Hara, T.; Mimura, K.; Abe, T.; Shioi, G.; Seiki, M.; Sakamoto, T. Deletion of the Mint3/Apba3 gene in mice abrogates macrophage functions and increases resistance to lipopolysaccharide-induced septic shock. J. Biol. Chem. 2011, 286, 32542–32551.
- Uematsu, T.; Fujita, T.; Nakaoka, H.J.; Hara, T.; Kobayashi, N.; Murakami, Y.; Seiki, M.; Sakamoto, T. Mint3/Apba3 depletion ameliorates severe murine influenza pneumonia and macrophage cytokine production in response to the influenza virus. Sci. Rep. 2016, 6, 37815.
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262.
- Salminen, A.; Hyttinen, J.M.; Kaarniranta, K. AMP-activated protein kinase inhibits NF-kappaB signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med. (Berl.) 2011, 89, 667–676.
- Friedlander, A.M. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J. Biol. Chem. 1986, 261, 7123–7126.
- D’Souza, C.A.; Heitman, J. Dismantling the Cryptococcus coat. Trends Microbiol. 2001, 9, 112–113.
- Kimura, A.; Abe, H.; Tsuruta, S.; Chiba, S.; Fujii-Kuriyama, Y.; Sekiya, T.; Morita, R.; Yoshimura, A. Aryl hydrocarbon receptor protects against bacterial infection by promoting macrophage survival and reactive oxygen species production. Int. Immunol. 2014, 26, 209–220.
- Wang, Y.; Shi, P.; Chen, Q.; Huang, Z.; Zou, D.; Zhang, J.; Gao, X.; Lin, Z. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J. Mol. Cell Biol. 2019, 11, 1069–1082.
- Boockvar, K.S.; Granger, D.L.; Poston, R.M.; Maybodi, M.; Washington, M.K.; Hibbs, J.B., Jr.; Kurlander, R.L. Nitric oxide produced during murine listeriosis is protective. Infect. Immun. 1994, 62, 1089–1100.
- Uematsu, T.; Tsuchiya, K.; Kobayashi, N.; Seiki, M.; Inoue, J.I.; Kaneko, S.; Sakamoto, T. Mint3 depletion-mediated glycolytic and oxidative alterations promote pyroptosis and prevent the spread of Listeria monocytogenes infection in macrophages. Cell Death Dis. 2021, 12, 404.
- Hara, T.; Nakaoka, H.J.; Hayashi, T.; Mimura, K.; Hoshino, D.; Inoue, M.; Nagamura, F.; Murakami, Y.; Seiki, M.; Sakamoto, T. Control of metastatic niche formation by targeting APBA3/Mint3 in inflammatory monocytes. Proc. Natl. Acad. Sci. USA 2017, 114, E4416–E4424.
- Nakaoka, H.J.; Tanei, Z.; Hara, T.; Weng, J.S.; Kanamori, A.; Hayashi, T.; Sato, H.; Orimo, A.; Otsuji, K.; Tada, K.; et al. Mint3-mediated L1CAM expression in fibroblasts promotes cancer cell proliferation via integrin alpha5beta1 and tumour growth. Oncogenesis 2017, 6, e334.
- Kanamori, A.; Matsubara, D.; Saitoh, Y.; Fukui, Y.; Gotoh, N.; Kaneko, S.; Seiki, M.; Murakami, Y.; Inoue, J.I.; Sakamoto, T. Mint3 depletion restricts tumor malignancy of pancreatic cancer cells by decreasing SKP2 expression via HIF-1. Oncogene 2020, 39, 6218–6230.
- Ikeda, J.; Ohe, C.; Tanaka, N.; Yoshida, T.; Saito, R.; Atsumi, N.; Kobayashi, T.; Kinoshita, H.; Tsuta, K.; Sakamoto, T. Hypoxia inducible factor-1 activator munc-18-interacting protein 3 promotes tumour progression in urothelial carcinoma. Clin. Transl. Discov. 2023, 3, e158.
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46.
- Sakamoto, T.; Fukui, Y.; Kondoh, Y.; Honda, K.; Shimizu, T.; Hara, T.; Hayashi, T.; Saitoh, Y.; Murakami, Y.; Inoue, J.I.; et al. Pharmacological inhibition of Mint3 attenuates tumour growth, metastasis, and endotoxic shock. Commun. Biol. 2021, 4, 1165.
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225.
- Hara, T.; Murakami, Y.; Seiki, M.; Sakamoto, T. Mint3 in bone marrow-derived cells promotes lung metastasis in breast cancer model mice. Biochem. Biophys. Res. Commun. 2017, 490, 688–692.
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1alpha and HIF2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22.
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186.
- Kiefel, H.; Bondong, S.; Hazin, J.; Ridinger, J.; Schirmer, U.; Riedle, S.; Altevogt, P. L1CAM: A major driver for tumor cell invasion and motility. Cell Adhes. Migr. 2012, 6, 374–384.
- Kiefel, H.; Pfeifer, M.; Bondong, S.; Hazin, J.; Altevogt, P. Linking L1CAM-mediated signaling to NF-kappaB activation. Trends Mol. Med. 2011, 17, 178–187.
- Chen, D.L.; Zeng, Z.L.; Yang, J.; Ren, C.; Wang, D.S.; Wu, W.J.; Xu, R.H. L1cam promotes tumor progression and metastasis and is an independent unfavorable prognostic factor in gastric cancer. J. Hematol. Oncol. 2013, 6, 43.


