 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Donata Santarsiero | -- | 3622 | 2023-03-09 15:14:18 | | | |
| 2 | Lindsay Dong | Meta information modification | 3622 | 2023-03-10 06:52:31 | | |
Video Upload Options
Kidney transplantation is the therapy of choice for patients who suffer from end-stage renal diseases. Despite improvements in surgical techniques and immunosuppressive treatments, long-term graft survival remains a challenge. The complement cascade, a part of the innate immune system, plays a crucial role in the deleterious inflammatory reactions that occur during the transplantation process, such as brain or cardiac death of the donor and ischaemia/reperfusion injury. In addition, the complement system also modulates the responses of T cells and B cells to alloantigens, thus playing a crucial role in cellular as well as humoral responses to the allograft, which lead to damage to the transplanted kidney.
1. Complement System
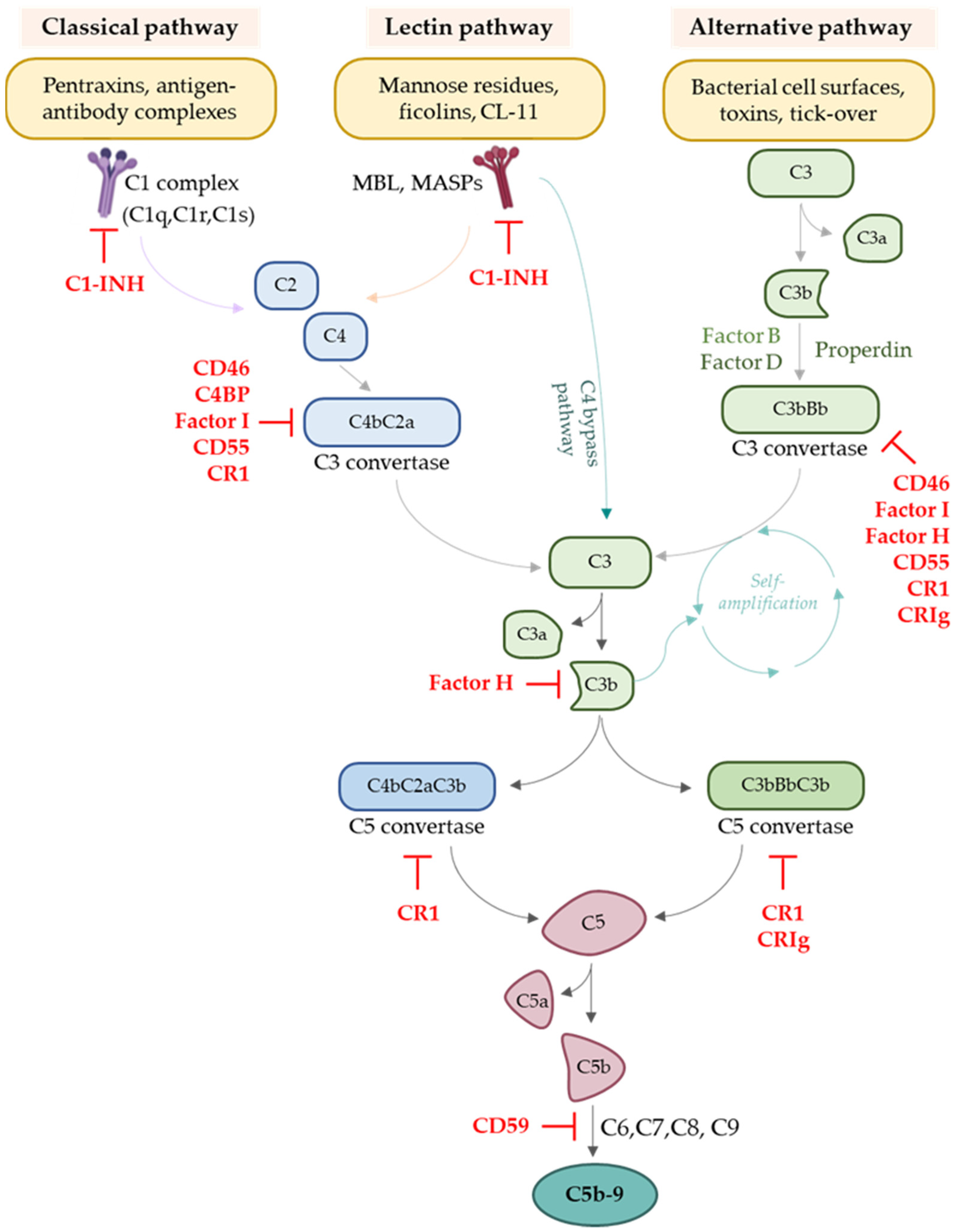
2. Involvement of the Complement System in Transplantation
During the whole process of transplantation, several events crucially impact graft function and survival and can potentially undermine the overall outcome. Even the steps that must be taken before surgical kidney implantation play a significant role. Indeed, the initial condition of both the donor and the recipient, as well as the organ preservation techniques used, are closely associated with graft quality and outcome. Firstly, the different types of donors, namely living or deceased donors—depending on whether donation occurs after brain death or cardiac death—result in differing organ quality. Regarding kidney transplant recipients, their medical condition and pharmacological treatment until a suitable transplant becomes available also significantly influence graft outcomes. Before surgical implantation, the time and method of graft preservation are crucial and delicate steps in the process of kidney transplantation. Once the organ is implanted in the recipient, the graft finally undergoes reperfusion. From this moment onwards, the graft encounters and inevitably activates the recipient innate and adaptive immune systems, which can potentially induce graft injury and rejection of the donor organ.
2.1. Complement Activation in Donor Kidneys
2.2. Complement Activation in Transplant Candidates
2.3. Ischaemia/Reperfusion Injury (IRI) and Complement Activation
2.4. Complement Activation Modulates the Adaptive Immune Response against the Graft
2.4.1. The Role of Complement in Regulating T Cell Responses
2.4.2. The Role of Complement in Antibody-Mediated Rejection
3. Complement as a Therapeutic Target in Kidney Transplantation
4. Blocking Complement to Help Pro-Tolerogenic Cell Therapies
Casiraghi et al. recently investigated how to render MSCs a therapeutic option for recipients of organs from deceased donors as well. Based on evidence that human MSCs express C3aR and C5aR [83], and that the chemoattractant effects of anaphylatoxins could guide MSCs toward injured tissues [83][84], the scholars evaluated the effect of antagonising complement receptors on MSCs given on day +2 post-transplant in preventing their recruitment into the graft and in prolonging graft survival [85]. Post-transplant MSC infusion combined with a short course of C3aR or C5aR antagonist, or the administration of MSCs pre-treated with C3aR and C5aR antagonists, prevented intragraft recruitment of MSCs [85]. Notably, antagonising C3aR or C5aR allowed MSCs to home the secondary lymphoid organs, and led to diminished C3 deposition and neutrophil recruitment, with a subsequent reduction in graft inflammation [85].
References
- Luo, S.; Hu, D.; Wang, M.; Zipfel, P.F.; Hu, Y. Complement in Hemolysis- and Thrombosis- Related Diseases. Front. Immunol. 2020, 11, 1212.
- Arbore, G.; Kemper, C.; Kolev, M. Intracellular complement − the complosome − in immune cell regulation. Mol. Immunol. 2017, 89, 2–9.
- Merle, N.S.; Noé, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257.
- Noris, M.; Remuzzi, G. Overview of Complement Activation and Regulation. Semin. Nephrol. 2013, 33, 479–492.
- De la O Becerra, K.I.; Oosterheert, W.; Bos, R.M.V.D.; Xenaki, K.T.; Lorent, J.H.; Ruyken, M.; Schouten, A.; Rooijakkers, S.H.M.; Henegouwen, P.M.P.V.B.E.; Gros, P. Multifaceted Activities of Seven Nanobodies against Complement C4b. J. Immunol. 2022, 208, 2207–2219.
- Zhou, T.; Li, Y.; Li, X.; Zeng, F.; Rao, Y.; He, Y.; Wang, Y.; Liu, M.; Li, D.; Xu, Z.; et al. Microglial debris is cleared by astrocytes via C4b-facilitated phagocytosis and degraded via RUBICON-dependent noncanonical autophagy in mice. Nat. Commun. 2022, 13, 1–22.
- Walport, M.J. Complement. New Engl. J. Med. 2001, 344, 1140–1144.
- Tew, J.G.; Kosco, M.H.; Burton, G.F.; Szakal, A.K. Follicular Dendritic Cells as Accessory Cells. Immunol. Rev. 1990, 117, 185–211.
- Thieblemont, N.; Haeffner-Cavaillon, N.; Haeffner, A.; Weiss, L.; Kazatchkine, M.D. Triggering of Complement Receptors CR1 (CD35) and CR3 (CD11b/CD18) Induces Nuclear Translocation of NF-Kappa B (P50/P65) in Human Monocytes and Enhances Viral Replication in HIV-Infected Monocytic Cells. J. Immunol. 1950, 155, 4861–4867.
- Phan, T.; Grigorova, I.; Okada, T.; Cyster, J.G. Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat. Immunol. 2007, 8, 992–1000.
- Kopf, M.; Abel, B.; Gallimore, A.; Carroll, M.; Bachmann, M.F. Complement component C3 promotes T-cell priming and lung migration to control acute influenza virus infection. Nat. Med. 2002, 8, 373–378.
- Soteros, B.M.; Sia, G.M. Complement and microglia dependent synapse elimination in brain development. Wiley Interdiscip. Rev. Syst. Biol. Med. 2021, 14, e1545.
- Mevorach, D. Clearance of dying cells and systemic lupus erythematosus: The role of C1q and the complement system. Apoptosis 2010, 15, 1114–1123.
- Defendi, F.; Thielens, N.M.; Clavarino, G.; Cesbron, J.-Y.; Dumestre-Pérard, C. The Immunopathology of Complement Proteins and Innate Immunity in Autoimmune Disease. Clin. Rev. Allergy Immunol. 2019, 58, 229–251.
- Franzin, R.; Stasi, A.; Fiorentino, M.; Stallone, G.; Cantaluppi, V.; Gesualdo, L.; Castellano, G. Inflammaging and Complement System: A Link Between Acute Kidney Injury and Chronic Graft Damage. Front. Immunol. 2020, 11, 734.
- Nauser, C.L.; Farrar, C.A.; Sacks, S.H. Complement Recognition Pathways in Renal Transplantation. J. Am. Soc. Nephrol. 2017, 28, 2571–2578.
- Ricklin, D.; Reis, E.S.; Lambris, J.D. Complement in disease: A defence system turning offensive. Nat. Rev. Nephrol. 2016, 12, 383–401.
- Grafals, M.; Thurman, J.M. The Role of Complement in Organ Transplantation. Front. Immunol. 2019, 10, 2380.
- Moore, S.R.; Menon, S.S.; Cortes, C.; Ferreira, V.P. Hijacking Factor H for Complement Immune Evasion. Front. Immunol. 2021, 12, 602277.
- Lesher, A.M.; Nilsson, B.; Song, W.-C. Properdin in complement activation and tissue injury. Mol. Immunol. 2013, 56, 191–198.
- Blaum, B.; Hannan, J.; Herbert, A.P.; Kavanagh, D.; Uhrín, D.; Stehle, T. Structural basis for sialic acid–mediated self-recognition by complement factor H. Nat. Chem. Biol. 2014, 11, 77–82.
- Harboe, M.; Ulvund, G.; Vien, L.; Fung, M.; Mollnes, T.E. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin. Exp. Immunol. 2004, 138, 439–446.
- Harboe, M.; Garred, P.; Karlstrøm, E.; Lindstad, J.K.; Stahl, G.L.; Mollnes, T.E. The down-stream effects of mannan-induced lectin complement pathway activation depend quantitatively on alternative pathway amplification. Mol. Immunol. 2009, 47, 373–380.
- Morgan, B.P.; Walters, D.; Serna, M.; Bubeck, D. Terminal complexes of the complement system: New structural insights and their relevance to function. Immunol. Rev. 2016, 274, 141–151.
- Qi, R.; Qin, W. Role of Complement System in Kidney Transplantation: Stepping From Animal Models to Clinical Application. Front. Immunol. 2022, 13, 811696.
- Khera, R.; Das, N. Complement Receptor 1: Disease associations and therapeutic implications. Mol. Immunol. 2009, 46, 761–772.
- Roozendaal, R.; Carroll, M.C. Complement receptors CD21 and CD35 in humoral immunity. Immunol. Rev. 2007, 219, 157–166.
- Vorup-Jensen, T.; Jensen, R.K. Structural Immunology of Complement Receptors 3 and 4. Front. Immunol. 2018, 9, 2716.
- Wagner, C.; Hänsch, G.M.; Stegmaier, S.; Denefleh, B.; Hug, F.; Schoels, M. The complement receptor 3, CR3 (CD11b/CD18), on T lymphocytes: Activation-dependent up-regulation and regulatory function. Eur. J. Immunol. 2001, 31, 1173–1180.
- Fayyazi, A.; Scheel, O.; Werfel, T.; Schweyer, S.; Oppermann, M.; Götze, O.; Radzun, H.J.; Zwirner, J. The C5a receptor is expressed in normal renal proximal tubular but not in normal pulmonary or hepatic epithelial cells. Immunology 2000, 99, 38–45.
- Thurman, J.M.; Nester, C.M. All Things Complement. Clin. J. Am. Soc. Nephrol. 2016, 11, 1856–1866.
- Aiello, S.; Gastoldi, S.; Galbusera, M.; Ruggenenti, P.L.; Portalupi, V.; Rota, S.; Rubis, N.; Liguori, L.; Conti, S.; Tironi, M.; et al. C5a and C5aR1 are key drivers of microvascular platelet aggregation in clinical entities spanning from aHUS to COVID-19. Blood Adv. 2022, 6, 866–881.
- Damman, J.; Bloks, V.W.; Daha, M.R.; Van Der Most, P.J.; Sanjabi, B.; Van Der Vlies, P.; Snieder, H.; Ploeg, R.J.; Krikke, C.; Leuvenink, H.G.D.; et al. Hypoxia and Complement-and-Coagulation Pathways in the Deceased Organ Donor as the Major Target for Intervention to Improve Renal Allograft Outcome. Transplantation 2015, 99, 1293–1300.
- Damman, J.; Nijboer, W.N.; Schuurs, T.A.; Leuvenink, H.G.; Morariu, A.M.; Tullius, S.G.; Van Goor, H.; Ploeg, R.J.; Seelen, M.A. Local renal complement C3 induction by donor brain death is associated with reduced renal allograft function after transplantation. Nephrol. Dial. Transplant. 2011, 26, 2345–2354.
- Poppelaars, F.; Seelen, M.A. Complement-mediated inflammation and injury in brain dead organ donors. Mol. Immunol. 2017, 84, 77–83.
- Mizuno, M.; Suzuki, Y.; Ito, Y. Complement regulation and kidney diseases: Recent knowledge of the double-edged roles of complement activation in nephrology. Clin. Exp. Nephrol. 2017, 22, 3–14.
- Jiang, S.; Jiao, Y.; Zou, G.; Gao, H.; Zhuo, L.; Li, W. Activation of Complement Pathways in Kidney Tissue May Mediate Tubulointerstitial Injury in Diabetic Nephropathy. Front. Med. 2022, 9, 845679.
- Ekdahl, K.N.; Soveri, I.; Hilborn, J.; Fellström, B.; Nilsson, B. Cardiovascular disease in haemodialysis: Role of the intravascular innate immune system. Nat. Rev. Nephrol. 2017, 13, 285–296.
- Wang, Y.; Gao, L. Inflammation and Cardiovascular Disease Associated With Hemodialysis for End-Stage Renal Disease. Front. Pharmacol. 2022, 13, 800950.
- Malek, M.; Nematbakhsh, M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Ren. Inj. Prev. 2015, 4, 20–27.
- Heylen, L.; Pirenne, J.; Naesens, M.; Sprangers, B.; Jochmans, I. “Time is tissue”—A minireview on the importance of donor nephrectomy, donor hepatectomy, and implantation times in kidney and liver transplantation. Am. J. Transplant. 2021, 21, 2653–2661.
- Sim, E.; Sim, R.B. Enzymic assay of C3b receptor on intact cells and solubilized cells. Biochem. J. 1983, 210, 567–576.
- Zheng, X.; Feng, B.; Chen, G.; Zhang, X.; Li, M.; Sun, H.; Liu, W.; Vladau, C.; Liu, R.; Jevnikar, A.M.; et al. Preventing Renal IschemiaReperfusion Injury Using Small Interfering RNA by Targeting Complement 3 Gene. Am. J. Transplant. 2006, 6, 2099–2108.
- Zheng, X.; Zhang, X.; Sun, H.; Feng, B.; Li, M.; Chen, G.; Vladau, C.; Chen, D.; Suzuki, M.; Min, L.; et al. Protection of Renal Ischemia Injury using Combination Gene Silencing of Complement 3 and Caspase 3 Genes. Transplantation 2006, 82, 1781–1786.
- Farrar, C.A.; Zhou, W.; Lin, T.; Sacks, S.H. Local extravascular pool of C3 is a determinant of postischemic acute renal failure. FASEB J. 2006, 20, 217–226.
- Peng, Q.; Li, K.; Patel, H.; Sacks, S.H.; Zhou, W. Dendritic Cell Synthesis of C3 Is Required for Full T Cell Activation and Development of a Th1 Phenotype. J. Immunol. 2006, 176, 3330–3341.
- Pratt, J.R.; Basheer, S.A.; Sacks, S.H. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat. Med. 2002, 8, 582–587.
- Liszewski, M.K.; Kolev, M.; Le Friec, G.; Leung, M.; Bertram, P.G.; Fara, A.F.; Subias, M.; Pickering, M.C.; Drouet, C.; Meri, S.; et al. Intracellular Complement Activation Sustains T Cell Homeostasis and Mediates Effector Differentiation. Immunity 2013, 39, 1143–1157.
- Kolev, M.; Dimeloe, S.; Le Friec, G.; Navarini, A.; Arbore, G.; Povoleri, G.A.; Fischer, M.; Belle, R.; Loeliger, J.; Develioglu, L.; et al. Complement Regulates Nutrient Influx and Metabolic Reprogramming during Th1 Cell Responses. Immunity 2015, 42, 1033–1047.
- Arbore, G.; West, E.E.; Rahman, J.; Le Friec, G.; Niyonzima, N.; Pirooznia, M.; Tunc, I.; Pavlidis, P.; Powell, N.; Li, Y.; et al. Complement receptor CD46 co-stimulates optimal human CD8+ T cell effector function via fatty acid metabolism. Nat. Commun. 2018, 9, 1–15.
- Einecke, G.; Sis, B.; Reeve, J.; Mengel, M.; Campbell, P.M.; Hidalgo, L.G.; Kaplan, B.; Halloran, P.F. Antibody-Mediated Microcirculation Injury Is the Major Cause of Late Kidney Transplant Failure. Am. J. Transplant. 2009, 9, 2520–2531.
- Halloran, P.F.; Chang, J.; Famulski, K.; Hidalgo, L.G.; Salazar, I.D.; Lopez, M.M.; Matas, A.; Picton, M.; de Freitas, D.; Bromberg, J.; et al. Disappearance of T Cell-Mediated Rejection Despite Continued Antibody-Mediated Rejection in Late Kidney Transplant Recipients. J. Am. Soc. Nephrol. 2015, 26, 1711–1720.
- Biglarnia, A.-R.; Huber-Lang, M.; Mohlin, C.; Ekdahl, K.N.; Nilsson, B. The multifaceted role of complement in kidney transplantation. Nat. Rev. Nephrol. 2018, 14, 767–781.
- Ugurlar, D.; Howes, S.C.; de Kreuk, B.-J.; Koning, R.I.; de Jong, R.N.; Beurskens, F.J.; Schuurman, J.; Koster, A.J.; Sharp, T.H.; Parren, P.W.H.I.; et al. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science 2018, 359, 794–797.
- De Clippel, D.; Baeten, M.; Torfs, A.; Emonds, M.-P.; Feys, H.B.; Compernolle, V.; Vandekerckhove, P. Screening for HLA antibodies in plateletpheresis donors with a history of transfusion or pregnancy. Transfusion 2014, 54, 3036–3042.
- Lederer, S.R.; Schneeberger, H.; Albert, E.; Johnson, J.P.; Gruber, R.; Land, W.; Burkhardt, K.; Hillebrand, G.; Feucht, H.E. Early renal graft dysfunction: The Role of Preformed Antibodies to DR-Typed Lymphoblastoid Cell Lines: 1. Transplantation 1996, 61, 313–319.
- Bentall, A.; Cornell, L.D.; Gloor, J.M.; Park, W.D.; Gandhi, M.J.; Winters, J.L.; Chedid, M.F.; Dean, P.G.; Stegall, M.D. Five-Year Outcomes in Living Donor Kidney Transplants With a Positive Crossmatch. Am. J. Transplant. 2012, 13, 76–85.
- Frémeaux-Bacchi, V.; Legendre, C.M. The emerging role of complement inhibitors in transplantation. Kidney Int. 2015, 88, 967–973.
- Hillmen, P.; Young, N.S.; Schubert, J.; Brodsky, R.A.; Socié, G.; Muus, P.; Röth, A.; Szer, J.; Elebute, M.O.; Nakamura, R.; et al. The Complement Inhibitor Eculizumab in Paroxysmal Nocturnal Hemoglobinuria. N. Engl. J. Med. 2006, 355, 1233–1243.
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian, C.; Bingham, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; et al. Terminal Complement Inhibitor Eculizumab in Atypical Hemolytic–Uremic Syndrome. N. Engl. J. Med. 2013, 368, 2169–2181.
- Noris, M.; Galbusera, M.; Gastoldi, S.; Macor, P.; Banterla, F.; Bresin, E.; Tripodo, C.; Bettoni, S.; Donadelli, R.; Valoti, E.; et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood 2014, 124, 1715–1726.
- Fakhouri, F.; Schwotzer, N.; Golshayan, D.; Frémeaux-Bacchi, V. The Rational Use of Complement Inhibitors in Kidney Diseases. Kidney Int. Rep. 2022, 7, 1165–1178.
- Weitz, M.; Amon, O.; Bassler, D.; Koenigsrainer, A.; Nadalin, S. Prophylactic eculizumab prior to kidney transplantation for atypical hemolytic uremic syndrome. Pediatr. Nephrol. 2011, 26, 1325–1329.
- Siedlecki, A.M.; Isbel, N.; Walle, J.V.; Eggleston, J.J.; Cohen, D.J.; Licht, C.; Frémeaux-Bacchi, V.; Ariceta, G.; Ardissino, G.; Fakhouri, F.; et al. Eculizumab Use for Kidney Transplantation in Patients With a Diagnosis of Atypical Hemolytic Uremic Syndrome. Kidney Int. Rep. 2018, 4, 434–446.
- Davis, A.E.; Mejia, P.; Lu, F. Biological activities of C1 inhibitor. Mol. Immunol. 2008, 45, 4057–4063.
- Huang, E.; Vo, A.; Choi, J.; Ammerman, N.; Lim, K.; Sethi, S.; Kim, I.; Kumar, S.; Najjar, R.; Peng, A.; et al. Three-Year Outcomes of a Randomized, Double-Blind, Placebo-Controlled Study Assessing Safety and Efficacy of C1 Esterase Inhibitor for Prevention of Delayed Graft Function in Deceased Donor Kidney Transplant Recipients. Clin. J. Am. Soc. Nephrol. 2019, 15, 109–116.
- MD, S.J. A Phase I/II, Double-Blind, Placebo-Controlled Study: Assessing Safety and Efficacy of Preoperative Renal Allograft Infusions of C1 Inhibitor (Berinert®) (Human) (C1INH) vs. Placebo Administration in Recipients of a Renal Allograft From Deceased High Risk Donors and Its Impact on Delayed Graft Function (DGF) and Ischemia/Reperfusion Injury (IRI). 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04696146 (accessed on 26 October 2022).
- University of Wisconsin, Madison A Phase I/II, Single Center, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Feasibility of Using Human Recombinant C1 Inhibitor(RUCONEST®) as a Therapeutic Strategy to Reduce the Incidence of Delayed Graft Function in Recipients of Kidneys From Donation After Cardio-Circulatory Death. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03791476 (accessed on 26 October 2022).
- University of Wisconsin, Madison A Phase I, Single Center, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate Tolerability of C1 Inhibitor (CINRYZE) as a Donor Pre-Treatment Strategy in Brain Dead Donors Who Meet a Kidney Donor Risk Index (KDRI) Above 60%. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT02435732 (accessed on 26 October 2022).
- Jordan, S.C.; Lorant, T.; Choi, J.; Kjellman, C.; Winstedt, L.; Bengtsson, M.; Zhang, X.; Eich, T.; Toyoda, M.; Eriksson, B.-M.; et al. IgG Endopeptidase in Highly Sensitized Patients Undergoing Transplantation. N. Engl. J. Med. 2017, 377, 442–453.
- Hansa Biopharma, A.B. A Phase II Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Efficacy of Intravenous IdeS After Administration of Ascending Doses in Chronic Kidney Disease Patients. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT02224820 (accessed on 26 October 2022).
- Hansa Biopharma, A.B. A Phase II Study to Evaluate the Safety, Tolerability, Efficacy and Pharmacokinetics of Intravenous Ascending Doses of IdeS in Kidney Transplantation. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT02475551 (accessed on 26 October 2022).
- MD, S.J. A Phase I/II Trial to Evaluate the Safety and Tolerability of Ides® (IgG Endopeptidase) to Eliminate Donor Specific HLA Antibodies (DSAs) and Prevent Antibody-Mediated Rejection Post-Transplant in Highly-HLA Sensitized Patients. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT02426684 (accessed on 26 October 2022).
- Perico, N.; Casiraghi, F.; Introna, M.; Gotti, E.; Todeschini, M.; Cavinato, R.A.; Capelli, C.; Rambaldi, A.; Cassis, P.; Rizzo, P.; et al. Autologous Mesenchymal Stromal Cells and Kidney Transplantation. Clin. J. Am. Soc. Nephrol. 2011, 6, 412–422.
- Perico, N.; Casiraghi, F.; Gotti, E.; Introna, M.; Todeschini, M.; Cavinato, R.A.; Capelli, C.; Rambaldi, A.; Cassis, P.; Rizzo, P.; et al. Mesenchymal stromal cells and kidney transplantation: Pretransplant infusion protects from graft dysfunction while fostering immunoregulation. Transpl. Int. 2013, 26, 867–878.
- Casiraghi, F.; Perico, N.; Cortinovis, F.C.M.; Remuzzi, G. Mesenchymal stromal cells in renal transplantation: Opportunities and challenges. Nat. Rev. Nephrol. 2016, 12, 241–253.
- Tan, J.; Wu, W.; Xu, X.; Liao, L.; Zheng, F.; Messinger, S.; Sun, X.; Chen, J.; Yang, S.; Cai, J.; et al. Induction Therapy With Autologous Mesenchymal Stem Cells in Living-Related Kidney Transplants. JAMA 2012, 307, 1169–1177.
- Peng, Y.; Ke, M.; Xu, L.; Liu, L.; Chen, X.; Xia, W.; Li, X.; Chen, Z.; Ma, J.; Liao, D.; et al. Donor-Derived Mesenchymal Stem Cells Combined With Low-Dose Tacrolimus Prevent Acute Rejection After Renal Transplantation. Transplantation 2013, 95, 161–168.
- Reinders, M.; Van Kooten, C.; Rabelink, T.; De Fijter, J.W. Mesenchymal Stromal Cell Therapy for Solid Organ Transplantation. Transplantation 2018, 102, 35–43.
- Casiraghi, F.; Azzollini, N.; Todeschini, M.; Cavinato, R.A.; Cassis, P.; Solini, S.; Rota, C.; Morigi, M.; Introna, M.; Maranta, R.; et al. Localization of Mesenchymal Stromal Cells Dictates Their Immune or Proinflammatory Effects in Kidney Transplantation. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2012, 12, 2373–2383.
- Herrera, M.; Bussolati, B.; Bruno, S.; Morando, L.; Mauriello-Romanazzi, G.; Sanavio, F.; Stamenkovic, I.; Biancone, L.; Camussi, G. Exogenous mesenchymal stem cells localize to the kidney by means of CD44 following acute tubular injury. Kidney Int. 2007, 72, 430–441.
- Dong, F.; Harvey, J.; Finan, A.; Weber, K.; Agarwal, U.; Penn, M.S. Myocardial CXCR4 Expression Is Required for Mesenchymal Stem Cell Mediated Repair Following Acute Myocardial Infarction. Circulation 2012, 126, 314–324.
- Ingrid U. Schraufstatter, Richard G. DiScipio, Ming Zhao, Sophia K. Khaldoyanidi; C3a and C5a Are Chemotactic Factors for Human Mesenchymal Stem Cells, Which Cause Prolonged ERK1/2 Phosphorylation1. J Immunol 2009, 182 (6): 3827–3836.
- Hengartner, N.-E.; Fiedler, J.; Schrezenmeier, H.; Huber-Lang, M.; Brenner, R.E. Crucial Role of IL1beta and C3a in the In Vitro-Response of Multipotent Mesenchymal Stromal Cells to Inflammatory Mediators of Polytrauma. PLoS ONE 2015, 10, e0116772.
- Casiraghi, F.; Todeschini, M.; Azzollini, N.; Cravedi, P.; Cassis, P.; Solini, S.; Fiori, S.; Rota, C.; Karachi, A.; Carrara, C.; et al. Effect of Timing and Complement Receptor Antagonism on Intragraft Recruitment and Protolerogenic Effects of Mesenchy-mal Stromal Cells in Murine Kidney Transplantation. Transplantation 2019, 103, 1121–1130.

