Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rehab Fouad Abdelhamid | -- | 3341 | 2023-03-08 06:20:08 | | | |
| 2 | Camila Xu | Meta information modification | 3341 | 2023-03-08 09:38:19 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Abdelhamid, R.F.; Nagano, S. Oxidative Stress and Aging in Neurodegeneration Disorders. Encyclopedia. Available online: https://encyclopedia.pub/entry/41958 (accessed on 23 July 2026).
Abdelhamid RF, Nagano S. Oxidative Stress and Aging in Neurodegeneration Disorders. Encyclopedia. Available at: https://encyclopedia.pub/entry/41958. Accessed July 23, 2026.
Abdelhamid, Rehab F., Seiichi Nagano. "Oxidative Stress and Aging in Neurodegeneration Disorders" Encyclopedia, https://encyclopedia.pub/entry/41958 (accessed July 23, 2026).
Abdelhamid, R.F., & Nagano, S. (2023, March 08). Oxidative Stress and Aging in Neurodegeneration Disorders. In Encyclopedia. https://encyclopedia.pub/entry/41958
Abdelhamid, Rehab F. and Seiichi Nagano. "Oxidative Stress and Aging in Neurodegeneration Disorders." Encyclopedia. Web. 08 March, 2023.
Copy Citation
Healthy aging is characterized by a gradual breakdown of physiological systems leading to a reduction in cognitive functions and brain health, but the timing and duration of this decline vary in individule. Oxidative stress (OS) is a crucial factor in the aging process that can cause direct damage to the brain’s cellular organization, causing neurodegenerative disease.
oxidative stress
free radical
aging
neurodegenerative diseases
1. Introduction
Healthy aging is characterized by a gradual breakdown of physiological systems leading to a reduction in cognitive functions and brain health, but the timing and extent of this decline vary among older people. Oxidative stress (OS) is a crucial factor in the aging process that can cause direct damage to the brain’s cellular architecture, causing neurodegenerative disease.
Aging is the main lead factor for many diseases, including cancer, metabolic, cardiac and neurodegenerative diseases. Aging is linked to a loss of homeostasis, involving degradation of structural components, reduced cellular maintenance and a decrease in overall physiological function/metabolism. There are two major aging theories: the free radical theory, which postulates chronological accumulation of defects in gene expression and environmental damage. According to the free radical theory of aging by Denham Harman, 1956 [1], alterations in normal metabolic and mitochondrial function are induced by production of free radicals, which cause damage, aging, and associated aging illnesses.
The second and more recent theory is the mitochondrial theory of aging by J. Miquel and colleagues in 1980 [2]. This theory posits that the loss of balance between free radical production and repair mechanisms is responsible for aging.
The redox network is also essential in antioxidant defense. Mitochondria serve a crucial function by turning stored energy into adenosine triphosphate (ATP) through oxidative phosphorylation and phospholipid synthesis, buffering calcium, and coordinating programmed cell death [3]. Free radicals such as reactive oxygen species (ROS), reactive nitrogen species (RNS), and reactive sulfur species (RSS), which are present in all cells, but which are restricted by the antioxidant systems that neutralize them, are primarily produced by oxidative phosphorylation [4]. Free radicals are produced naturally and modulate cellular processes such as inflammation, cell survival, and stress responses, as well as numerous illnesses such as cardiovascular problems, muscular dysfunction, allergies, and malignancies [5].
Free radicals are most likely formed due to processes involving molecular oxygen that are catalyzed in the cell by oxidative enzymes and in connective tissues by trace metals such as iron, cobalt, and manganese.
The primary distinction between healthy aging and accelerated aging is the balance between free radical elevation and the body’s ability to guard and fight against RNS and ROS (Figure 1).
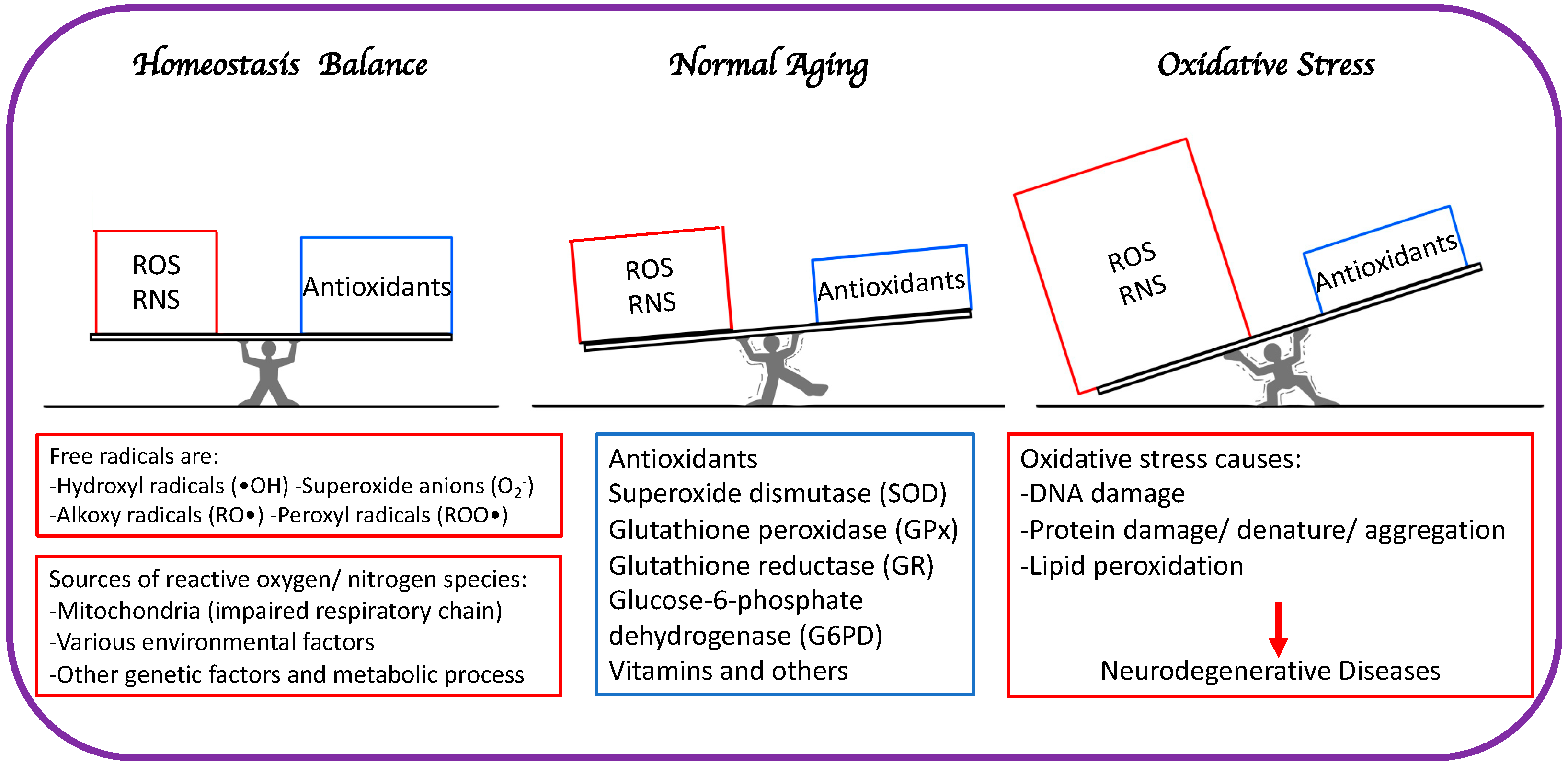
Figure 1. The primary distinction between healthy aging and accelerated aging is the balance between free radical generation (RNS and ROS) and antioxidation to minimize oxidative stress.
In the event of loss of homeostasis between free radical production and detoxification, ROS production may overpower antioxidant defenses, resulting in a noxious state known as OS and general degradation of normal cellular functions. This has been documented in numerous clinical studies involving mitochondrial malfunction and aging [6].
The brain is especially prone to oxidative damage due to its high oxygen consumption, limited antioxidant defenses, and high concentration of polyunsaturated fatty acids that are easily oxidized [7].
Lipid peroxides (LPO) are highly reactive molecules that include malondialdehyde (MDA), 4-hydroxy-2-nonenal (HNE), acrolein, isoprostanes (IsoPs), and neuroprostanes (NeuroPs). They can disrupt proteins and DNA structure and functions [8][9][10]. Increased MDA, IsoPs, and HNE have been observed in brain tissues of Tg2576 Alzheimer’s disease (AD) model mice and post-mortem AD brains [11][12]. Oxidative damage to DNA results in formation of oxidized base adducts, including 8-hydroxyguanosine (8-OHG) and 5,6-diamino-5-formamidopyridine in brains of mild cognitive impairment (MCI) patients. Despite the brain’s great potential for ROS formation, its defense system against OS remains restricted and diminishes with age, owing to low amounts of endogenous antioxidants such as catalase (CAT), glutathione (GSH), glutathione peroxidase (GPx), and vitamin E, compared to other tissues, such as liver [13][14]. The limited regenerative ability of postmitotic neurons renders OS more damaging to the brain, where the damage accumulates over time, compared to other organs [15].
2. Other Sources of Free Radicals Contribute to OS
Even though mitochondrial dysfunction is the primary source of free radicals, there are several other sources, such as genetic (endogenous), environmental, and lifestyle causes (exogenous) [16].
2.1. Environmental Factors: Radiation/UV Rays and Pollution
Environmental factors, including smoking, UV radiation, heavy metal ions, ozone, allergens, drugs or toxins, pollutants, and pesticides, may all contribute to elevated ROS production in cells [17][18]. Ionizing radiation transforms hydroxyl radicals, superoxides, and organic radicals into organic hydroperoxides and hydrogen peroxide. Subsequently, peroxides react with metal ions, particularly Fe and Cu through redox reactions, with further oxidative activity at the cellular level. Several studies have demonstrated that exposure of fibroblasts to alpha particles leads to an intracellular increase in oxygen and an acceleration of peroxide formation [19][20]. Ultraviolet radiation (UVA) induces oxidative processes by stimulating riboflavin, porphyrins, and NADPH-oxidase, resulting in synthesis of 8-oxo-guanine and a drop in intracellular GSH levels, with a return to normal after exposure cessation [21]. Heavy metals are crucial to the formation of free radicals [22].
Nickel, arsenic, iron, copper, cadmium and lead can produce free radicals by Fenton or Haber-Weiss reactions [23][24], as well as through direct reactions between metal ions and cellular constituents with similar effects, such as generation of thiol-type radicals. In brain tissue, lead causes lipid peroxidation and raises GPx concentrations. By attaching to sulfhydryl groups, arsenic generates peroxides, superoxides, and nitric oxide, and inhibits antioxidant enzymes such as glutathione-S-transferase (GST), GPx, and glutathione reductase (GR) [25]. Free radicals produced by these reactions can cause substitutions of DNA bases, including guanine for cytosine, guanine for thymine, and cytosine for thymine [26]. Even in healthy individuals, ozone exposure can impair lung function by increasing inflammatory infiltration in the respiratory epithelium [27].
2.2. Lifestyle Related Factors
Lifestyle factors such as smoking, drinking alcohol, diet, exercise, and frequency of exercise all contribute to OS [28]. Some research has shown that ROS are present at the skeletal muscle level and help to control how muscle works. Muscle fibers always produce small quantities of reactive oxygen radicals, which are elevated by muscle contraction [29]. These oxygen radicals have many direct and indirect effects on muscle activity (contractility, excitability, metabolism, and calcium homeostasis) and are involved in skeletal muscle fatigue during hard exercise [30].
Long, exhausting exercise and overcoming limits as a phase of overtraining cause a massive response to OS. Endogenous antioxidant status is improved by moderate exercise, low-intensity training, and training for a long time.
ROS are an essential part of how cells talk to each other and how antioxidant genes are turned on and off. Nuclear factor kappa B and mitogen-activated protein kinase are up-regulated by physical activity [31][32]. These activate gene expression of various enzymes and proteins that maintain oxidative/antioxidant intracellular homeostasis [33].
Physical exercise is the main non-drug therapy for treating chronic diseases, especially heart diseases, and lifestyle changes [34]. Relevant studies [35] have shown that autophagy, a process that breaks down and recycles cellular organs and nutrients, is important for cardiovascular benefits of training.
Cigarette smoke has oxidants, free radicals, and organic components such as nitric oxide and nitric superoxide. These cause neutrophils and macrophages to gather in lung tissues, increasing the production of oxidants in the area [36][37].
2.3. Genetic Factors
Enzymes that are responsible for fighting damage caused by OS and these enzymes and pathways are prone to free radical damage [38][39][40][41][42]. Oligodendrocytes are susceptible to oxidative damage due to their function in maintenance and creation of myelin and their limited repair mechanisms, suggesting that white matter may be more susceptible to oxidative activity than gray matter. Antioxidant defense enzymes in the brain, such as superoxide dismutase (SOD), CAT, GPx, and GST are essential for neutralizing toxic byproducts of oxidative phosphorylation. Allelic variants of polymorphisms encoding these antioxidants are associated with anomalies in SOD, CAT, GPx, and GST activity in the central nervous system [38][43][44].
3. Mechanistic Evolution of Neurodegenerative Diseases Caused by OS
Neurodegenerative disease is an irreversible condition in which neuronal function declines over time, leading to neuronal death. The incidence of neurodegenerative disease is increasing every year, especially in countries with aging populations. Common neurodegenerative diseases include Alzheimer’s (AD) and Parkinson’s (PD) diseases. Mitochondria are the primary source of ROS and are the main cause of neurodegenerative diseases (Figure 2).
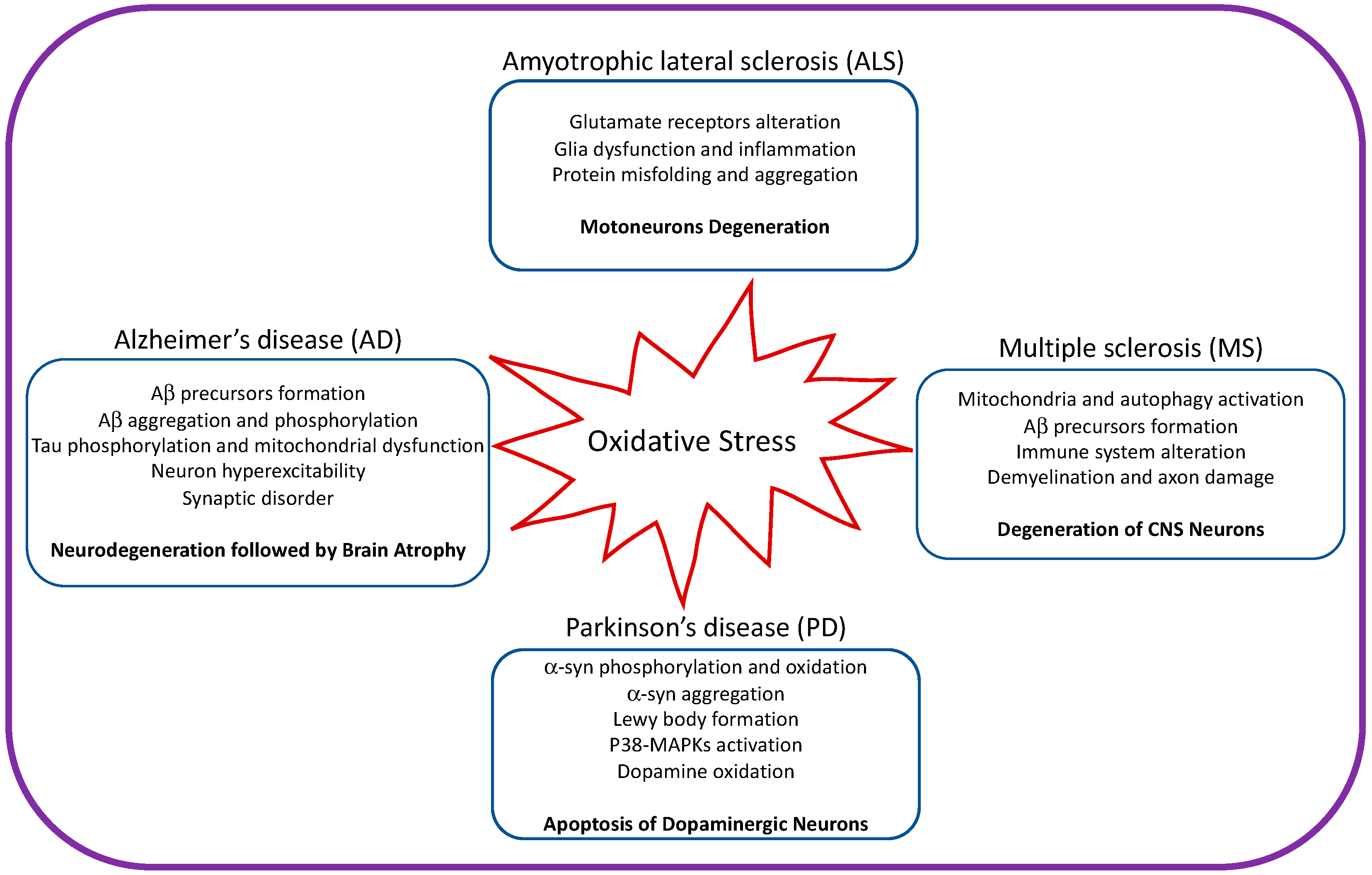
Figure 2. Illustration showing the impact of oxidative stress on amyotrophic lateral sclerosis, Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis.
Mitochondrial synthesis of ATP through oxidative phosphorylation, has the substantial disadvantage of producing unpaired electrons [45][46]. The electron transport chain comprises five multiprotein complexes that mediate interaction of these electrons with oxygen, resulting in ROS hydrogen peroxide (H2O2), superoxide anions (O2−), and hydroxyl radicals (•OH) [14][47]. Electron transfer from NADH to ubiquinone is catalyzed by mitochondrial complex I (reduced nicotinamide adenine dinucleotide [NADH] coenzyme Q reductase) (coenzyme Q). Complex II also provides electrons to ubiquinone (succinate dehydrogenase). Electrons from reduced ubiquinone are donated to complex III (cytochrome bc1), and subsequently, to cytochrome c (CytC). Complex IV (CytC oxidase) is involved in interactions between molecular oxygen and electrons extracted from CytC, resulting in water production [48]. Complexes I, II, and III are most often linked to premature electron leakage to oxygen and are the primary source of ROS generation [49].
Additionally, elevated ROS levels trigger formation of additional reactive species, such as RNS, when O2 reacts with other molecules, such as nitric oxide, to generate peroxynitrite (ONOO−). Furthermore, in addition to ROS and RNS, mitochondria generate RSSs, which are very reactive. Free oxygen radicals gradually damage proteins, lipids, and nucleic acids, resulting in inefficient or aberrant cellular functions, inflammation, and cell death [50].
Mitochondrial internal components and mitochondrial DNA (mtDNA) in particular, are very vulnerable to OS-induced damage, resulting in impaired mitochondrial bioenergetics, increased ROS generation, and OS [51]. Enzymatic and non-enzymatic mechanisms safeguard antioxidant systems. SOD, CAT, GPx, GR, and thioredoxin (TRX) are the major enzymes involved in catalytic ROS elimination. Non-enzymatic complexes include vitamins A, C, and E, GSH, and proteins such as albumin and ceruloplasmin [52][53].
4. Major Degenerative Disorders Caused by OS
4.1. OS in Amyotrophic Lateral Sclerosis (ALS)
Despite substantial investigation, the genesis of amyotrophic lateral sclerosis (ALS) is still not fully clear. Ninety percent of ALS cases are sporadic and appear to lack a genetic foundation, whereas 10% of patients have familial ALS, mostly an autosomal dominant [54]. ALS has been linked to various occupational and environmental variables, such as exposure to various chemicals, metals, and pesticides, electromagnetic fields (EMFs), and lifestyle choices, including smoking and excessive exercise [55][56][57][58].
A recognized but poorly understood pathogenic ALS feature is abnormally high free radical levels and inadequate antioxidation. Undoubtedly, OS is essential for motor neuron death, but we do not know exactly when oxidative damage occurs [59]. OS biomarkers have been identified in brain tissue of ALS patients, cerebrospinal fluid (CSF), blood, and urine [60]. It is challenging to track OS biomarkers over a long period due to the short lifespans of ALS patients. Further, because of OS’s random start and the present lack of tools to forecast its progression, it is difficult to be certain whether OS is a cause of ALS-associated neurodegeneration or a result of other underlying etiologic variables [57]. Investigations using a murine ALS model have revealed altered mitochondrial structures and nuclear factor erythroid 2-related factor 2 (Nrf2) pathway activation during early ALS stages, indicating OS involvement in the disease’s early stages. OS damage is typically caused by stimulating formation of intracellular antioxidant molecules [61][62].
However, these studies used the murine mutant SOD1 ALS model, and SOD1 mutations only account for 20% of human familial ALS cases. Twenty individuals with sporadic ALS had significantly higher levels of lipid peroxidation and lower levels of the antioxidant enzymes CAT, GR, GSH, and glucose-6-phosphate dehydrogenase (G6PD) in their erythrocytes [63]. Progression of alterations parallel the pathophysiology of ALS, supporting OS participation in ALS development. Additionally, the aforementioned environmental and endoplasmic reticulum stress factors work together to promote pro-oxidative states that may ultimately harm motor neurons [64].
The intermembrane space (IMS) of mitochondria contains a protein known as Coiled-Coil-Helix-Coiled-Coil-Helix Domain-Containing Protein 10 (CHCHD10), which has no known function [65]. However, it is believed to be involved in maintaining mitochondrial crista morphology and proper oxidative phosphorylation. In particular, in multiprotein complexes I, II, III, and IV, overexpression of mutant CHCHD10 harboring an allele linked to ALS alters mitochondrial structure and impairs electron transport chain function [66][67]. In addition, fibroblasts from an ALS patient with a CHCHD10 mutation displayed mitochondrial ultrastructural damage and mitochondrial network fragmentation [66][67].
4.2. OS in Alzheimer’s Disease (AD)
The most common neurodegenerative condition, AD, is characterized by a steady decline in behavior, cognition, and functioning that profoundly affect day-to-day activities. AD brains have higher protein, DNA, and lipid oxidation rates, as well as redox-active metals [51].
Pathological indicators of AD include extracellular senile plaques (SP) and intracellular neurofibrillary tangles (NFT). Protein aggregation is sporadic, and its molecular mechanism is poorly understood. Several investigations have revealed that AD brains display elevated OS, which is crucial to disease development [68]. Amyloid fibril precursors are neurotoxic owing to OS generated by toxic conformer of amyloid oligomers and additional neurotoxic effects such as neuronal membrane rupture, microglia and astrocyte activation, and Ca2+ dyshomeostasis [69][70][71].
Tau protein aggregation is another hallmark of AD although in most cases, it is of late onset. Several reports have indicated that neurotoxicity caused by beta amyloid alteration is the main trigger of tau alteration to form tangles. NFTs in the early stages of AD are intracellular deposits; however, these progress to extracellular deposits in later stages. NFTs are composed of paired helical filaments (PHFs) in which the major component is tau protein [72][73][74][75][76].
When tau proteins are hyperphosphorylated, they self-assemble into NFTs and may be detected in neurons [77][78]. According to recent data, amyloid deposition occurs 15–20 years before dementia manifests itself, and tau pathology appears thereafter [79][80][81][82][83].
Using multiphoton imaging, researchers identified a clear link between free radical generation and amyloid plaques in AD mouse models and human AD brain tissues, where fluorogenic free radical markers decreased following administration of a synthetic antioxidant [84].
4.3. OS in Parkinson’s Disease (PD)
Unlike AD, PD is clinically recognized by four cardinal motor symptoms: bradykinesia, stiffness, resting tremors, and trouble with posture and walking [83][84][85]. In PD brains, the substantia nigra pars compacta and to a lesser extent, the globus pallidus, putamen, and caudate nucleus, exhibit selective dopaminergic neuronal loss. The nigrostriatal pathway’s degenerating neurons release less of the neurotransmitter dopamine [86]. Lewy bodies are clusters of aberrant proteins seen in neurons of individuals with Parkinson’s disease. They are components of α-synuclein (α-syn) protein, broadly disseminated in the neurological system, but activities of which are not well understood [87]. α-syn fibrillation creates clumps that take up significant space within cells and ultimately result in neuronal death [88]. As in AD, pathophysiological pathways underlying PD are strongly related to OS [89]. Evidence of OS involvement may be seen in the substantia nigra of PD patients, where oxidized lipids, proteins, and DNA can be found [90][91]. Additionally, the monoamine oxidase (MAO) that breaks down dopamine creates hydrogen peroxide, whereas dopamine on its own produces superoxide anion and reactive quinones. These reactive substances cause other nearby neurons, as well as dopaminergic neurons, to become cytotoxic [92][93].
4.4. OS in Multiple Sclerosis (MS)
Multiple sclerosis (MS) is a multifactorial autoimmune disease of the central nervous system (CNS), characterized by chronic inflammation, demyelination, and axon and neuron loss. Depending on the location of the demyelinating lesions, MS patients can develop almost any neurological sign or symptom, including motor, sensory, and cognitive impairment [94][95].
OS is heavily involved in several MS pathological hallmarks such as myelin destruction, axonal degeneration, and inflammation [96]. In MS as in other neurodegenerative diseases OS triggers activation of autophagy and microglia as well as of the neuroimmune system. Neurons, astrocytes, and oligodendrocytes produce chemicals that bind to microglial receptors in healthy settings, suppressing their activated states [97][98][99][100]. When certain molecules (such as myelin CD47) are expressed less, microglial activation is increased, which may cause myelin debris to be phagocytosed and to provide neurotrophic factors [101][102]. Microglial physiological functions are transformed into harmful inflammatory insults by long-term damage, systemic inflammation, proinflammatory cytokine release, and ROS signaling [103].
Due to a combination of circumstances, including high levels of iron and polyunsaturated fatty acids, high iron requirements and mitochondrial activity, and restricted cell regeneration, the CNS is particularly susceptible to OS. Oxidative damage also affects the immune response that is developing in the periphery and controls MS illness outside of the CNS. First, by lowering its electrical resistance and thus changing its permeability, elevated ROS levels harm the brain endothelium [94][104][105][106].
5. Significance of Non-Coding RNAs in OS
OS readily damages RNA due to its single-stranded structure and high concentration close to mitochondria, where most intracellular ROS are generated [107][108]. An overabundance of ROS can cause chemical modification or even separation of RNA bases and breakage of RNA strands [91][109]. Oxidatively damaged RNA accumulates in cells, leading to decreased protein synthesis, erroneous protein generation, and ultimately cell death [110][111].
Non-coding RNAs (ncRNAs), which do not encode proteins, make up the majority of RNAs in human cells [112][113]. As a category of ncRNAs, regulatory ncRNAs, including long ncRNAs (lncRNAs) and small ncRNAs, which includes microRNAs (miRNAs), circular RNAs (circRNAs), and PIWI-interacting RNAs (piRNAs), are involved in regulating gene expression [114]. Compared to mRNAs, these regulatory ncRNAs persist relatively longer. Hence, oxidative damage can cause prolonged effects. ncRNAs of several types have been linked to various neurogenerative diseases [115][116][117][118]. However, the direct relationship between ncRNA oxidation and neuronal diseases is still unclear, except in PD, in which the interaction between OS and regulatory ncRNAs has been well studied [119]. In contrast, regulatory small ncRNAs such as miRNAs and lncRNAs help to regulate ROS production [118]. Their interactions are involved in the pathophysiology of PD [120].
RNA oxidation is not only a hallmark of PD but also a crucial first step in the development of the disorder [112][121]. 8-oxo-7, 8-dihydroguanosine (8-oxoG) is one of many oxidative marks on RNA, and it is possibly the most common and well studied [122]. Guanine’s vulnerability to free radicals means that it can form this base adduct, leading to mismatched bases [109].
Neuronal apoptosis and autophagy are influenced by lncRNAs; thus, α-syn accumulation and degradation that restricts their function would have deleterious effects on cellular homeostasis [123]. For instance, free radical damage to miRNAs can cause them to misidentify their target mRNAs, which can increase expression of specific proteins [124]. A reduction in α-syn expression was mediated by two miRNAs in an experiment performed by Je and Kim [123]. High levels of α-syn and subsequent development of PD resulted from stress-induced oxidative loss of translational inhibition by these two miRNAs.
Furthermore, Chen et al. found that OS triggered modification of circRNA N6-methyladenosine (m6A). m6A-modified circRNAs can modulate expression of stress response genes (UBC and PPP2CA), which may constitute the mechanistic basis for OS-induced neurodegenerative disorders [125].
The regulatory role of miRNAs in OS is closely linked to α-syn, which is responsible for inducing OS. Both microRNA-141-3p and microRNA-9-5p target the 3′ UTR of SIRT1 mRNA. Since SIRT1 inhibits formation of α-syn aggregates, knockdown of microRNA-141-3p and microRNA-9-5p may alleviate OS and boost the viability of in vitro PD model [126][127].
SOD, CAT, and GPx are responsible for detoxifying oxidants and repairing oxidative damage. MicroRNA-137 and microRNA-494-3p aggravate OS by reducing SOD and GPx activity in PD rats treated with these miRNAs. MicroRNA-335 suppresses expression of FTH1, thereby promoting release of Fe2+ ions and generation of free radicals [128]. Downregulation of microRNA-410 expression in PD is associated with elevated ROS production [129].
Regulatory lncRNAs and OS are hallmarks of PD. In PD, mitochondrial dysfunction is linked to ROS overproduction. α-syn aggregate formation can exacerbate OS by decreasing complex I activity or by activating microglia. Upregulation of beta-amyloid-cleaving enzyme-antisense (BACE1-AS), a lncRNA, was also associated with increasing levels of α-syn in PD [130]. In addition, the lncRNA microRNA-17-92a-1 cluster host gene (MIR17HG) promotes α-syn expression. MicroRNA-153-5p cannot inhibit α-syn expression because MIR17HG acts as a sponge for microRNA-153-5p [131]. Inhibition of autophagy by GSK3 promotes α-syn buildup and therefore aggregation. MicroRNA-15b-5p suppressed GSK3 expression, but binding of SNHG1 to microRNA-15b-5p rescued GSK3 expression [132].
References
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300.
- Miquel, J.; Economos, A.C.; Fleming, J.; Johnson, J.E., Jr. Mitochondrial Role in Cell Aging. Exp. Gerontol. 1980, 15, 575–591.
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The Role of Mitochondria in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 710, 132933.
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Opportunities. Antioxid. Redox Signal. 2019, 30, 1450–1499.
- Zuo, L.; Koozechian, M.S.; Chen, L.L. Characterization of Reactive Nitrogen Species in Allergic Asthma. Ann. Allergy Asthma Immunol. 2014, 112, 18–22.
- Zuo, L.; Zhou, T.; Pannell, B.K.; Ziegler, A.C.; Best, T.M. Biological and Physiological Role of Reactive Oxygen Species—The Good, the Bad and the Ugly. Acta Physiol. 2015, 214, 329–348.
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274.
- Taso, O.V.; Philippou, A.; Moustogiannis, A.; Zevolis, E.; Koutsilieris, M. Lipid Peroxidation Products and Their Role in Neurodegenerative Diseases. Ann. Res. Hosp. 2019, 3, 2.
- Bradley-Whitman, M.A.; Lovell, M.A. Biomarkers of Lipid Peroxidation in Alzheimer Disease (AD): An Update. Arch. Toxicol. 2015, 89, 1035–1044.
- Ciamporcero, E.; Daga, M.; Pizzimenti, S.; Roetto, A.; Dianzani, C.; Compagnone, A.; Palmieri, A.; Ullio, C.; Cangemi, L.; Pili, R.; et al. Crosstalk between Nrf2 and YAP Contributes to Maintaining the Antioxidant Potential and Chemoresistance in Bladder Cancer. Free Radic. Biol. Med. 2018, 115, 447–457.
- Perluigi, M.; Coccia, R.; Butterfield, D.A. 4-Hydroxy-2-Nonenal, a Reactive Product of Lipid Peroxidation, and Neurodegenerative Diseases: A Toxic Combination Illuminated by Redox Proteomics Studies. Antioxid. Redox Signal. 2012, 17, 1590–1609.
- Kao, Y.-C.; Ho, P.-C.; Tu, Y.-K.; Jou, I.-M.; Tsai, K.-J. Lipids and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1505.
- Cheng, K.C.; Cahill, D.S.; Kasai, H.; Nishimura, S.; Loeb, L.A. 8-Hydroxyguanine, an Abundant Form of Oxidative DNA Damage, Causes G—T and A—C Substitutions. J. Biol. Chem. 1992, 267, 166–172.
- Cadenas, S. ROS and Redox Signaling in Myocardial Ischemia-Reperfusion Injury and Cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89.
- Maynard, S.; Fang, E.F.; Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. DNA Damage, DNA Repair, Aging, and Neurodegeneration. Cold Spring Harb. Perspect. Med. 2015, 5, a025130.
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694.
- Antunes dos Santos, A.; Ferrer, B.; Marques Gonçalves, F.; Tsatsakis, A.M.; Renieri, E.A.; Skalny, A.V.; Farina, M.; Rocha, J.B.T.; Aschner, M. Oxidative Stress in Methylmercury-Induced Cell Toxicity. Toxics 2018, 6, 47.
- Mahajan, A.; Taliun, D.; Thurner, M.; Robertson, N.R.; Torres, J.M.; Rayner, N.W.; Payne, A.J.; Steinthorsdottir, V.; Scott, R.A.; Grarup, N.; et al. Fine-Mapping Type 2 Diabetes Loci to Single-Variant Resolution Using High-Density Imputation and Islet-Specific Epigenome Maps. Nat. Genet. 2018, 50, 1505–1513.
- Spitz, D.R.; Azzam, E.I.; Jian Li, J.; Gius, D. Metabolic Oxidation/Reduction Reactions and Cellular Responses to Ionizing Radiation: A Unifying Concept in Stress Response Biology. Cancer Metastasis Rev. 2004, 23, 311–322.
- Spitz, D.R.; Hauer-Jensen, M. Ionizing Radiation-Induced Responses: Where Free Radical Chemistry Meets Redox Biology and Medicine. Antioxid. Redox Signal. 2014, 20, 1407–1409.
- Marchitti, S.A.; Chen, Y.; Thompson, D.C.; Vasiliou, V. Ultraviolet Radiation: Cellular Antioxidant Response and the Role of Ocular Aldehyde Dehydrogenase Enzymes. Eye Contact Lens 2011, 37, 206–213.
- Ściskalska, M.; Zalewska, M.; Grzelak, A.; Milnerowicz, H. The Influence of the Occupational Exposure to Heavy Metals and Tobacco Smoke on the Selected Oxidative Stress Markers in Smelters. Biol. Trace Elem. Res. 2014, 159, 59–68.
- Koppenol, W.H. The Haber-Weiss Cycle—70 Years Later. Redox Rep. 2001, 6, 229–234.
- Das, T.K.; Wati, M.R.; Fatima-Shad, K. Oxidative Stress Gated by Fenton and Haber Weiss Reactions and Its Association with Alzheimer’s Disease. Arch. Neurosci. 2015, 2, e60038.
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19.
- Jan, A.T.; Azam, M.; Siddiqui, K.; Ali, A.; Choi, I.; Haq, Q.M.R. Heavy Metals and Human Health: Mechanistic Insight into Toxicity and Counter Defense System of Antioxidants. Int. J. Mol. Sci. 2015, 16, 29592–29630.
- Li, J.; Zou, B.; Yeo, Y.H.; Feng, Y.; Xie, X.; Lee, D.H.; Fujii, H.; Wu, Y.; Kam, L.Y.; Ji, F.; et al. Prevalence, Incidence, and Outcome of Non-Alcoholic Fatty Liver Disease in Asia, 1999–2019: A Systematic Review and Meta-Analysis. Lancet Gastroenterol. Hepatol. 2019, 4, 389–398.
- Fisher-Wellman, K.; Bloomer, R.J. Acute Exercise and Oxidative Stress: A 30 Year History. Dyn. Med. 2009, 8, 1.
- He, F.; Li, J.; Liu, Z.; Chuang, C.-C.; Yang, W.; Zuo, L. Redox Mechanism of Reactive Oxygen Species in Exercise. Front. Physiol. 2016, 7, 486.
- Pingitore, A.; Lima, G.P.P.; Mastorci, F.; Quinones, A.; Iervasi, G.; Vassalle, C. Exercise and Oxidative Stress: Potential Effects of Antioxidant Dietary Strategies in Sports. Nutrition 2015, 31, 916–922.
- Adams, V.; Späte, U.; Kränkel, N.; Christian, S.P.; Linke, A.; Schuler, G.; Hambrecht, R. Nuclear Factor-Kappa B Activation in Skeletal Muscle of Patients with Chronic Heart Failure: Correlation with the Expression of Inducible Nitric Oxide Synthase. Eur. J. Cardiovasc. Prev. Rehabil. 2003, 10, 273–277.
- Ji, L.L.; Gomez-Cabrera, M.-C.; Vina, J. Role of Nuclear Factor kappaB and Mitogen-Activated Protein Kinase Signaling in Exercise-Induced Antioxidant Enzyme Adaptation. Appl. Physiol. Nutr. Metab. 2007, 32, 930–935.
- Antonioni, A.; Fantini, C.; Dimauro, I.; Caporossi, D. Redox Homeostasis in Sport: Do Athletes Really Need Antioxidant Support? Res. Sport. Med. 2019, 27, 147–165.
- Ren, J.; Taegtmeyer, H. Too Much or Not Enough of a Good Thing—The Janus Faces of Autophagy in Cardiac Fuel and Protein Homeostasis. J. Mol. Cell. Cardiol. 2015, 84, 223–226.
- Wu, N.N.; Tian, H.; Chen, P.; Wang, D.; Ren, J.; Zhang, Y. Physical Exercise and Selective Autophagy: Benefit and Risk on Cardiovascular Health. Cells 2019, 8, 1436.
- Rahman, I.; Biswas, S.K.; Kode, A. Oxidant and Antioxidant Balance in the Airways and Airway Diseases. Eur. J. Pharmacol. 2006, 533, 222–239.
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-Hydroxy-2′-Deoxyguanosine (8-OHdG): A Critical Biomarker of Oxidative Stress and Carcinogenesis. J. Environ. Sci. Health Part C 2009, 27, 120–139.
- Salminen, L.E.; Paul, R.H. Oxidative Stress and Genetic Markers of Suboptimal Antioxidant Defense in the Aging Brain: A Theoretical Review. Rev. Neurosci. 2014, 25, 805–819.
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, e8416763.
- Ighodaro, O.M.; Akinloye, O.A. First Line Defence Antioxidants-Superoxide Dismutase (SOD), Catalase (CAT) and Glutathione Peroxidase (GPX): Their Fundamental Role in the Entire Antioxidant Defence Grid. Alex. J. Med. 2018, 54, 287–293.
- Górny, M.; Bilska-Wilkosz, A.; Iciek, M.; Hereta, M.; Kamińska, K.; Kamińska, A.; Chwatko, G.; Rogóż, Z.; Lorenc-Koci, E. Alterations in the Antioxidant Enzyme Activities in the Neurodevelopmental Rat Model of Schizophrenia Induced by Glutathione Deficiency during Early Postnatal Life. Antioxidants 2020, 9, 538.
- Gusti, A.M.T.; Qusti, S.Y.; Alshammari, E.M.; Toraih, E.A.; Fawzy, M.S. Antioxidants-Related Superoxide Dismutase (SOD), Catalase (CAT), Glutathione Peroxidase (GPX), Glutathione-S-Transferase (GST), and Nitric Oxide Synthase (NOS) Gene Variants Analysis in an Obese Population: A Preliminary Case-Control Study. Antioxidants 2021, 10, 595.
- Chiras, D.; Kitsos, G.; Petersen, M.B.; Skalidakis, I.; Kroupis, C. Oxidative Stress in Dry Age-Related Macular Degeneration and Exfoliation Syndrome. Crit. Rev. Clin. Lab. Sci. 2015, 52, 12–27.
- Kowalska, M.; Wize, K.; Prendecki, M.; Lianeri, M.; Kozubski, W.; Dorszewska, J. Genetic Variants and Oxidative Stress in Alzheimer’s Disease. Curr. Alzheimer Res. 2020, 17, 208–223.
- Mailloux, R.J. Teaching the Fundamentals of Electron Transfer Reactions in Mitochondria and the Production and Detection of Reactive Oxygen Species. Redox Biol. 2015, 4, 381–398.
- Glantzounis, G.K.; Salacinski, H.J.; Yang, W.; Davidson, B.R.; Seifalian, A.M. The Contemporary Role of Antioxidant Therapy in Attenuating Liver Ischemia-Reperfusion Injury: A Review. Liver Transplant. 2005, 11, 1031–1047.
- Panov, A.V.; Dikalov, S.I. Cardiolipin, Perhydroxyl Radicals, and Lipid Peroxidation in Mitochondrial Dysfunctions and Aging. Oxid. Med. Cell. Longev. 2020, 2020, 1323028.
- Srinivasan, S.; Avadhani, N.G. Cytochrome c Oxidase Dysfunction in Oxidative Stress. Free Radic. Biol. Med. 2012, 53, 1252–1263.
- Turrens, J.F. Mitochondrial Formation of Reactive Oxygen Species. J. Physiol. 2003, 552, 335–344.
- Ghosh, N.; Das, A.; Chaffee, S.; Roy, S.; Sen, C.K. Reactive Oxygen Species, Oxidative Damage and Cell Death. In Immunity and Inflammation in Health and Disease; Chatterjee, S., Jungraithmayr, W., Bagchi, D., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 45–55.
- Jellinger, K.A. Basic Mechanisms of Neurodegeneration: A Critical Update. J. Cell. Mol. Med. 2010, 14, 457–487.
- Halliwell, B.; Gutteridge, J.M.C. The Antioxidants of Human Extracellular Fluids. Arch. Biochem. Biophys. 1990, 280, 1–8.
- Aruoma, O.I.; Neergheen, V.S.; Bahorun, T.; Jen, L.-S. Free Radicals, Antioxidants and Diabetes: Embryopathy, Retinopathy, Neuropathy, Nephropathy and Cardiovascular Complications. Neuroembryol. Aging 2006, 4, 117–137.
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310.
- Ingre, C.; Roos, P.M.; Piehl, F.; Kamel, F.; Fang, F. Risk Factors for Amyotrophic Lateral Sclerosis. Clin. Epidemiol. 2015, 7, 181–193.
- Motataianu, A.; Serban, G.; Barcutean, L.; Balasa, R. Oxidative Stress in Amyotrophic Lateral Sclerosis: Synergy of Genetic and Environmental Factors. Int. J. Mol. Sci. 2022, 23, 9339.
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxid. Med. Cell. Longev. 2020, 2020, 5021694.
- Carrì, M.T.; Valle, C.; Bozzo, F.; Cozzolino, M. Oxidative Stress and Mitochondrial Damage: Importance in Non-SOD1 ALS. Front. Cell. Neurosci. 2015, 9, 41.
- Agar, J.; Durham, H. Relevance of Oxidative Injury in the Pathogenesis of Motor Neuron Diseases. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2003, 4, 232–242.
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583.
- Kraft, A.D.; Resch, J.M.; Johnson, D.A.; Johnson, J.A. Activation of the Nrf2–ARE Pathway in Muscle and Spinal Cord during ALS-like Pathology in Mice Expressing Mutant SOD1. Exp. Neurol. 2007, 207, 107–117.
- Velde, C.V.; McDonald, K.K.; Boukhedimi, Y.; McAlonis-Downes, M.; Lobsiger, C.S.; Hadj, S.B.; Zandona, A.; Julien, J.-P.; Shah, S.B.; Cleveland, D.W. Misfolded SOD1 Associated with Motor Neuron Mitochondria Alters Mitochondrial Shape and Distribution Prior to Clinical Onset. PLoS ONE 2011, 6, e22031.
- Babu, G.N.; Kumar, A.; Chandra, R.; Puri, S.K.; Singh, R.L.; Kalita, J.; Misra, U.K. Oxidant–Antioxidant Imbalance in the Erythrocytes of Sporadic Amyotrophic Lateral Sclerosis Patients Correlates with the Progression of Disease. Neurochem. Int. 2008, 52, 1284–1289.
- Keon, M.; Musrie, B.; Dinger, M.; Brennan, S.E.; Santos, J.; Saksena, N.K. Destination Amyotrophic Lateral Sclerosis. Front. Neurol. 2021, 12, 596006.
- Anderson, C.J.; Bredvik, K.; Burstein, S.R.; Davis, C.; Meadows, S.M.; Dash, J.; Case, L.; Milner, T.A.; Kawamata, H.; Zuberi, A.; et al. ALS/FTD Mutant CHCHD10 Mice Reveal a Tissue-Specific Toxic Gain-of-Function and Mitochondrial Stress Response. Acta Neuropathol. 2019, 138, 103–121.
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A Mitochondrial Origin for Frontotemporal Dementia and Amyotrophic Lateral Sclerosis through CHCHD10 Involvement. Brain 2014, 137, 2329–2345.
- Genin, E.C.; Bannwarth, S.; Ropert, B.; Lespinasse, F.; Mauri-Crouzet, A.; Augé, G.; Fragaki, K.; Cochaud, C.; Donnarumma, E.; Lacas-Gervais, S.; et al. CHCHD10 and SLP2 Control the Stability of the PHB Complex: A Key Factor for Motor Neuron Viability. Brain 2022, 145, 3415–3430.
- Fontana, I.C.; Zimmer, A.R.; Rocha, A.S.; Gosmann, G.; Souza, D.O.; Lourenco, M.V.; Ferreira, S.T.; Zimmer, E.R. Amyloid-β Oligomers in Cellular Models of Alzheimer’s Disease. J. Neurochem. 2020, 155, 348–369.
- Fernandez-Perez, E.J.; Peters, C.; Aguayo, L.G. Membrane Damage Induced by Amyloid Beta and a Potential Link with Neuroinflammation. Curr. Pharm. Des. 2016, 22, 1295–1304.
- Meraz-Ríos, M.A.; Lira-De León, K.I.; Campos-Peña, V.; De Anda-Hernández, M.A.; Mena-López, R. Tau Oligomers and Aggregation in Alzheimer’s Disease. J. Neurochem. 2010, 112, 1353–1367.
- Calvo-Rodriguez, M.; Bacskai, B.J. Mitochondria and Calcium in Alzheimer’s Disease: From Cell Signaling to Neuronal Cell Death. Trends Neurosci. 2021, 44, 136–151.
- Liu, Z.; Li, T.; Li, P.; Wei, N.; Zhao, Z.; Liang, H.; Ji, X.; Chen, W.; Xue, M.; Wei, J. The Ambiguous Relationship of Oxidative Stress, Tau Hyperphosphorylation, and Autophagy Dysfunction in Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2015, 2015, e352723.
- Rojas Quijano, F.A.; Morrow, D.; Wise, B.M.; Brancia, F.L.; Goux, W.J. Prediction of Nucleating Sequences from Amyloidogenic Propensities of Tau-Related Peptides. Biochemistry 2006, 45, 4638–4652.
- Goedert, M. Tau Protein and the Neurofibrillary Pathology of Alzheimer’s Disease. Trends Neurosci. 1993, 16, 460–465.
- Weaver, C.L.; Espinoza, M.; Kress, Y.; Davies, P. Conformational Change as One of the Earliest Alterations of Tau in Alzheimer’s Disease. Neurobiol. Aging 2000, 21, 719–727.
- Grabowski, T.J.; Cho, H.S.; Vonsattel, J.P.G.; Rebeck, G.W.; Greenberg, S.M. Novel Amyloid Precursor Protein Mutation in an Iowa Family with Dementia and Severe Cerebral Amyloid Angiopathy. Ann. Neurol. 2001, 49, 697–705.
- Hansson, O. Biomarkers for Neurodegenerative Diseases. Nat. Med. 2021, 27, 954–963.
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the Pathologic Process in Alzheimer Disease: Age Categories from 1 to 100 Years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969.
- Fleisher, A.S.; Chen, K.; Quiroz, Y.T.; Jakimovich, L.J.; Gomez, M.G.; Langois, C.M.; Langbaum, J.B.; Ayutyanont, N.; Roontiva, A.; Thiyyagura, P. Florbetapir PET Analysis of Amyloid-β Deposition in the Presenilin 1 E280A Autosomal Dominant Alzheimer’s Disease Kindred: A Cross-Sectional Study. Lancet Neurol. 2012, 11, 1057–1065.
- O’Brien, J.T.; Herholz, K. Amyloid Imaging for Dementia in Clinical Practice. BMC Med. 2015, 13, 163.
- Gunasingh Masilamoni, J.; Philip Jesudason, E.; Dhandayuthapani, S.; Ashok, B.S.; Vignesh, S.; Jebaraj, W.C.E.; Paul, S.F.; Jayakumar, R. The Neuroprotective Role of Melatonin against Amyloid β Peptide Injected Mice. Free Radic. Res. 2008, 42, 661–673.
- Smith, D.G.; Cappai, R.; Barnham, K.J. The Redox Chemistry of the Alzheimer’s Disease Amyloid Beta Peptide. Biochim. Biophys. Acta 2007, 1768, 1976–1990.
- Jankovic, J. Parkinson’s Disease: Clinical Features and Diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376.
- Blandini, F.; Nappi, G.; Tassorelli, C.; Martignoni, E. Functional Changes of the Basal Ganglia Circuitry in Parkinson’s Disease. Prog. Neurobiol. 2000, 62, 63–88.
- Agosta, F.; Weiler, M.; Filippi, M. Propagation of Pathology through Brain Networks in Neurodegenerative Diseases: From Molecules to Clinical Phenotypes. CNS Neurosci. Ther. 2015, 21, 754–767.
- Jansen van Rensburg, Z.; Abrahams, S.; Bardien, S.; Kenyon, C. Toxic Feedback Loop Involving Iron, Reactive Oxygen Species, α-Synuclein and Neuromelanin in Parkinson’s Disease and Intervention with Turmeric. Mol. Neurobiol. 2021, 58, 5920–5936. Available online: https://link.springer.com/article/10.1007/s12035-021-02516-5 (accessed on 3 January 2023).
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325.
- Jenner, P. Oxidative Stress in Parkinson’s Disease. Ann. Neurol. 2003, 53, S26–S38.
- Fahn, S.; Cohen, G. The Oxidant Stress Hypothesis in Parkinson’s Disease: Evidence Supporting It. Ann. Neurol. 1992, 32, 804–812.
- Delcambre, S.; Nonnenmacher, Y.; Hiller, K. Dopamine Metabolism and Reactive Oxygen Species Production. In Mitochondrial Mechanisms of Degeneration and Repair in Parkinson’s Disease; Buhlman, L.M., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 25–47. ISBN 978-3-319-42139-1.
- Nunomura, A.; Moreira, P.I.; Castellani, R.J.; Lee, H.; Zhu, X.; Smith, M.A.; Perry, G. Oxidative Damage to RNA in Aging and Neurodegenerative Disorders. Neurotox. Res. 2012, 22, 231–248.
- Li, Z.; Chen, X.; Liu, Z.; Ye, W.; Li, L.; Qian, L.; Ding, H.; Li, P.; Aung, L.H.H. Recent Advances: Molecular Mechanism of RNA Oxidation and Its Role in Various Diseases. Front. Mol. Biosci. 2020, 7, 184.
- Jellinger, K.A. Recent Advances in Our Understanding of Neurodegeneration. J. Neural Transm. 2009, 116, 1111–1162.
- Pegoretti, V.; Swanson, K.A.; Bethea, J.R.; Probert, L.; Eisel, U.L.M.; Fischer, R. Inflammation and Oxidative Stress in Multiple Sclerosis: Consequences for Therapy Development. Oxid. Med. Cell. Longev. 2020, 2020, e7191080.
- Compston, A. Genetic Epidemiology of Multiple Sclerosis. J. Neurol. Neurosurg. Psychiatry 1997, 62, 553–561.
- Adamczyk-Sowa, M.; Galiniak, S.; Żyracka, E.; Grzesik, M.; Naparło, K.; Sowa, P.; Bartosz, G.; Sadowska-Bartosz, I. Oxidative Modification of Blood Serum Proteins in Multiple Sclerosis after Interferon Beta and Melatonin Treatment. Oxid. Med. Cell. Longev. 2017, 2017, e7905148.
- Hernangómez, M.; Carrillo-Salinas, F.J.; Mecha, M.; Correa, F.; Mestre, L.; Loría, F.; Feliú, A.; Docagne, F.; Guaza, C. Brain Innate Immunity in the Regulation of Neuroinflammation: Therapeutic Strategies by Modulating CD200-CD200R Interaction Involve the Cannabinoid System. Curr. Pharm. Des. 2014, 20, 4707–4722.
- Nair, A.; Frederick, T.J.; Miller, S.D. Astrocytes in Multiple Sclerosis: A Product of Their Environment. Cell. Mol. Life Sci. 2008, 65, 2702–2720.
- Kawabori, M.; Yenari, M.A. Inflammatory Responses in Brain Ischemia. Curr. Med. Chem. 2015, 22, 1258–1277.
- Degan, D.; Ornello, R.; Tiseo, C.; Carolei, A.; Sacco, S.; Pistoia, F. The Role of Inflammation in Neurological Disorders. Curr. Pharm. Des. 2018, 24, 1485–1501.
- Kasschau, M.; Sherman, K.; Haider, L.; Frontario, A.; Shaw, M.; Datta, A.; Bikson, M.; Charvet, L. A Protocol for the Use of Remotely-Supervised Transcranial Direct Current Stimulation (TDCS) in Multiple Sclerosis (MS). J. Vis. Exp. 2015, 106, e53542.
- Gitik, M.; Liraz-Zaltsman, S.; Oldenborg, P.-A.; Reichert, F.; Rotshenker, S. Myelin Down-Regulates Myelin Phagocytosis by Microglia and Macrophages through Interactions between CD47 on Myelin and SIRPα (Signal Regulatory Protein-α) on Phagocytes. J. Neuroinflamm. 2011, 8, 24.
- Perry, V.H. Contribution of Systemic Inflammation to Chronic Neurodegeneration. Acta Neuropathol. 2010, 120, 277–286.
- Rodgers, J.M.; Miller, S.D. Cytokine Control of Inflammation and Repair in the Pathology of Multiple Sclerosis. Yale J. Biol. Med. 2012, 85, 447.
- Carson, M.J.; Doose, J.M.; Melchior, B.; Schmid, C.D.; Ploix, C.C. CNS Immune Privilege: Hiding in Plain Sight. Immunol. Rev. 2006, 213, 48–65.
- Holman, D.W.; Klein, R.S.; Ransohoff, R.M. The Blood–Brain Barrier, Chemokines and Multiple Sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 220–230.
- Tanaka, M.; Song, H.; Küpfer, P.A.; Leumann, C.J.; Sonntag, W.E. An Assay for RNA Oxidation Induced Abasic Sites Using the Aldehyde Reactive Probe. Free Radic. Res. 2011, 45, 237–247.
- Pieper, A.A.; Verma, A.; Zhang, J.; Snyder, S.H. Poly (ADP-Ribose) Polymerase, Nitric Oxide and Cell Death. Trends Pharmacol. Sci. 1999, 20, 171–181.
- Nunomura, A.; Perry, G. RNA and Oxidative Stress in Alzheimer’s Disease: Focus on MicroRNAs. Oxid. Med. Cell. Longev. 2020, 2020, e2638130.
- Moreira, P.I.; Nunomura, A.; Nakamura, M.; Takeda, A.; Shenk, J.C.; Aliev, G.; Smith, M.A.; Perry, G. Nucleic Acid Oxidation in Alzheimer Disease. Free Radic. Biol. Med. 2008, 44, 1493–1505.
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of Transcription in Human Cells. Nature 2012, 489, 101–108.
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and Their Integrated Networks. J. Integr. Bioinform. 2019, 16, 20190027.
- Johnson, R.; Noble, W.; Tartaglia, G.G.; Buckley, N.J. Neurodegeneration as an RNA Disorder. Prog. Neurobiol. 2012, 99, 293–315.
- Salta, E.; De Strooper, B. Noncoding RNAs in Neurodegeneration. Nat. Rev. Neurosci. 2017, 18, 627–640.
- Abdelhamid, R.F.; Ogawa, K.; Beck, G.; Ikenaka, K.; Takeuchi, E.; Yasumizu, Y.; Jinno, J.; Kimura, Y.; Baba, K.; Nagai, Y.; et al. PiRNA/PIWI Protein Complex as a Potential Biomarker in Sporadic Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2022, 59, 1693–1705.
- Tai, Y.; Chen, J.; Tao, Z.; Ren, J. Non-Coding RNAs: New Players in Mitophagy and Neurodegeneration. Neurochem. Int. 2022, 152, 105253.
- Zhang, H.; Liu, X.; Liu, Y.; Liu, J.; Gong, X.; Li, G.; Tang, M. Crosstalk between Regulatory Non-Coding RNAs and Oxidative Stress in Parkinson’s Disease. Front. Aging Neurosci. 2022, 14, 975248.
- Konovalova, J.; Gerasymchuk, D.; Parkkinen, I.; Chmielarz, P.; Domanskyi, A. Interplay between MicroRNAs and Oxidative Stress in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 6055.
- Cervinkova, B.; Krcmova, L.K.; Sestakova, V.; Solichova, D.; Solich, P. A Fully Validated Bioanalytical Method Using an UHPLC–MS/MS System for Quantification of DNA and RNA Oxidative Stress Biomarkers. Anal Bioanal. Chem. 2017, 409, 3611–3621.
- Lushchak, V.I.; Duszenko, M.; Gospodaryov, D.V.; Garaschuk, O. Oxidative Stress and Energy Metabolism in the Brain: Midlife as a Turning Point. Antioxidants 2021, 10, 1715.
- Zhang, X.; Li, L. The Significance of 8-OxoGsn in Aging-Related Diseases. Aging Dis. 2020, 11, 1329–1338.
- Yan, L.L.; Simms, C.L.; McLoughlin, F.; Vierstra, R.D.; Zaher, H.S. Oxidation and Alkylation Stresses Activate Ribosome-Quality Control. Nat. Commun. 2019, 10, 5611.
- Je, G.; Kim, Y.-S. Mitochondrial ROS-Mediated Post-Transcriptional Regulation of α-Synuclein through MiR-7 and MiR-153. Neurosci. Lett. 2017, 661, 132–136.
- Chen, N.; Tang, J.; Su, Q.; Chou, W.-C.; Zheng, F.; Guo, Z.; Yu, G.; Shao, W.; Li, H.; Wu, S. Paraquat-Induced Oxidative Stress Regulates N6-Methyladenosine (M6A) Modification of Circular RNAs. Environ. Pollut. 2021, 290, 117816.
- Zheng, Y.; Dong, L.; Liu, N.; Luo, X.; He, Z. Mir-141-3p Regulates Apoptosis and Mitochondrial Membrane Potential via Targeting Sirtuin1 in a 1-Methyl-4-Phenylpyridinium In Vitro Model of Parkinson’s Disease. BioMed Res. Int. 2020, 2020, e7239895.
- Ge, H.; Yan, Z.; Zhu, H.; Zhao, H. MiR-410 Exerts Neuroprotective Effects in a Cellular Model of Parkinson’s Disease Induced by 6-Hydroxydopamine via Inhibiting the PTEN/AKT/MTOR Signaling Pathway. Exp. Mol. Pathol. 2019, 109, 16–24.
- Li, X.; Si, W.; Li, Z.; Tian, Y.; Liu, X.; Ye, S.; Huang, Z.; Ji, Y.; Zhao, C.; Hao, X.; et al. MiR-335 Promotes Ferroptosis by Targeting Ferritin Heavy Chain 1 in In Vivo and In Vitro Models of Parkinson’s Disease. Int. J. Mol. Med. 2021, 47, 61.
- Li, Y.; Fang, J.; Zhou, Z.; Zhou, Q.; Sun, S.; Jin, Z.; Xi, Z.; Wei, J. Downregulation of LncRNA BACE1-AS Improves Dopamine-Dependent Oxidative Stress in Rats with Parkinson’s Disease by Upregulating MicroRNA-34b-5p and Downregulating BACE1. Cell Cycle 2020, 19, 1158–1171.
- Zhang, J.; Yang, Y.; Zhou, C.; Zhu, R.; Xiao, X.; Zhou, B.; Wan, D. LncRNA MiR-17-92a-1 Cluster Host Gene (MIR17HG) Promotes Neuronal Damage and Microglial Activation by Targeting the MicroRNA-153-3p/Alpha-Synuclein Axis in Parkinson’s Disease. Bioengineered 2022, 13, 4493–4516.
- Xie, N.; Qi, J.; Li, S.; Deng, J.; Chen, Y.; Lian, Y. Upregulated LncRNA Small Nucleolar RNA Host Gene 1 Promotes 1-Methyl-4-Phenylpyridinium Ion-Induced Cytotoxicity and Reactive Oxygen Species Production through MiR-15b-5p/GSK3β Axis in Human Dopaminergic SH-SY5Y Cells. J. Cell. Biochem. 2019, 120, 5790–5801.
- Rodrigo, R.; Libuy, M.; Feliú, F.; Hasson, D. Oxidative Stress-Related Biomarkers in Essential Hypertension and Ischemia-Reperfusion Myocardial Damage. Dis. Mark. 2013, 35, 773–790.
- Cherubini, A.; Ruggiero, C.; Polidori, M.C.; Mecocci, P. Potential Markers of Oxidative Stress in Stroke. Free Radic. Biol. Med. 2005, 39, 841–852.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
939
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
08 Mar 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No