 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yutaka Koyama | -- | 1426 | 2023-03-07 05:40:06 | | | |
| 2 | Conner Chen | Meta information modification | 1426 | 2023-03-08 03:09:17 | | | | |
| 3 | Conner Chen | + 2 word(s) | 1428 | 2023-03-09 01:24:12 | | |
Video Upload Options
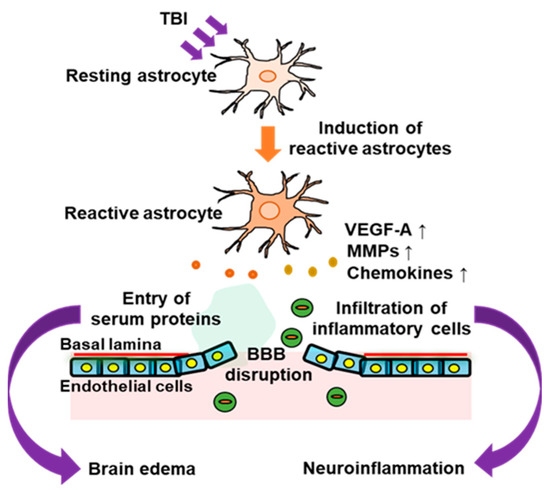
Traumatic brain injury (TBI) is an intracranial injury caused by accidents, falls, or sports. The production of endothelins (ETs) is increased in the injured brain. ET receptors are classified into distinct types, including ETA receptor (ETA-R) and ETB receptor (ETB-R). ETB-R is highly expressed in reactive astrocytes and upregulated by TBI. Activation of astrocytic ETB-R promotes conversion to reactive astrocytes and the production of astrocyte-derived bioactive factors, including vascular permeability regulators and cytokines, which cause blood–brain barrier (BBB) disruption, brain edema, and neuroinflammation in the acute phase of TBI. ETB-R antagonists alleviate BBB disruption and brain edema in animal models of TBI. The activation of astrocytic ETB receptors also enhances the production of various neurotrophic factors. These astrocyte-derived neurotrophic factors promote the repair of the damaged nervous system in the recovery phase of patients with TBI.
1. Introduction
2. Pathophysiological Responses of Astrocytes to TBI
References
- Jarrahi, A.; Braun, M.; Ahluwalia, M.; Gupta, R.V.; Wilson, M.; Munie, S.; Ahluwalia, P.; Vender, J.R.; Vale, F.L.; Dhandapani, K.M.; et al. Revisiting traumatic brain injury: From molecular mechanisms to therapeutic interventions. Biomedicines 2020, 8, 389.
- Thapa, K.; Khan, H.; Singh, T.G.; Kaur, A. Traumatic brain injury: Mechanistic insight on pathophysiology and potential therapeutic targets. J. Mol. Neurosci. 2021, 9, 1725–1742.
- Maas, A.I.; Roozenbeek, B.; Manley, G.T. Clinical trials in traumatic brain injury: Past experience and current developments. Neurotherapeutics 2010, 7, 115–126.
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology 2019, 145, 230–246.
- Lerouet, D.; Marchand-Leroux, C.; Besson, V.C. Neuropharmacology in traumatic brain injury: From preclinical to clinical neuroprotection? Fundam. Clin. Pharmacol. 2021, 35, 524–538.
- Maugeri, G.; D’Agata, V.; Trovato, B.; Roggio, F.; Castorina, A.; Vecchio, M.; Di Rosa, M.; Musumeci, G. The role of exercise on peripheral nerve regeneration: From animal model to clinical application. Heliyon 2021, 7, e08281.
- Theisen, C.C.; Sachdeva, R.; Austin, S.; Kulich, D.; Kranz, V.; Houle, J.D. Exercise and peripheral nerve grafts as a strategy to promote regeneration after acute or chronic spinal cord injury. J. Neurotrauma 2017, 34, 1909–1914.
- Sachdeva, R.; Theisen, C.C.; Ninan, V.; Twiss, J.L.; Houlé, J.D. Exercise dependent increase in axon regeneration into peripheral nerve grafts by propriospinal but not sensory neurons after spinal cord injury is associated with modulation of regeneration-associated genes. Exp. Neurol. 2016, 276, 72–82.
- Lee, H.G.; Wheeler, M.A.; Quintana, F.J. Function and therapeutic value of astrocytes in neurological diseases. Nat. Rev. Drug Discov. 2022, 21, 339–358.
- Gorshkov, K.; Aguisanda, F.; Thorne, N.; Zheng, W. Astrocytes as targets for drug discovery. Drug Discov. Today 2018, 23, 673–680.
- Zhou, Y.; Shao, A.; Yao, Y.; Tu, S.; Deng, Y.; Zhang, J. Dual roles of astrocytes in plasticity and reconstruction after traumatic brain injury. Cell Commun. Signal. 2020, 18, 62.
- Koyama, Y. Endothelin ETB receptor-mediated astrocytic activation: Pathological roles in brain disorders. Int. J. Mol. Sci. 2021, 22, 4333.
- Castejón, O.J. Morphological astrocytic changes in complicated human brain trauma. A light and electron microscopic study. Brain Inj. 1998, 12, 409–427.
- Michinaga, S.; Kimura, A.; Hatanaka, S.; Minami, S.; Asano, A.; Ikushima, Y.; Matsui, S.; Toriyama, Y.; Fujii, M.; Koyama, Y. Delayed administration of BQ788, an ETB antagonist, after experimental traumatic brain injury promotes recovery of blood-brain barrier function and a reduction of cerebral edema in mice. J. Neurotrauma 2018, 35, 1481–1494.
- Dunn, C.; Sturdivant, N.; Venier, S.; Ali, S.; Wolchok, J.; Balachandran, K. Blood-brain barrier breakdown and astrocyte reactivity evident in the absence of behavioral changes after repeated traumatic brain injury. Neurotrauma Rep. 2021, 2, 399–410.
- Zhai, Y.; Ye, S.Y.; Wang, Q.S.; Xiong, R.P.; Fu, S.Y.; Du, H.; Xu, Y.W.; Peng, Y.; Huang, Z.Z.; Yang, N.; et al. Overexpressed ski efficiently promotes neurorestoration, increases neuronal regeneration, and reduces astrogliosis after traumatic brain injury. Gene Ther. 2022.
- Prabhakar, N.K.; Khan, H.; Grewal, A.K.; Singh, T.G. Intervention of neuroinflammation in the traumatic brain injury trajectory: In vivo and clinical approaches. Int. Immunopharmacol. 2022, 108, 108902.
- Goodman, J.C.; Van, M.; Gopinath, S.P.; Robertson, C.S. Pro-inflammatory and proapoptotic elements of the neuroinflammatory response are activated in traumatic brain injury. Acta Neurochir. Suppl. 2008, 102, 437–439.
- Pedrazzi, M.; Patrone, M.; Passalacqua, M.; Ranzato, E.; Colamassaro, D.; Sparatore, B.; Pontremoli, S.; Melloni, E. Selective proinflammatory activation of astrocytes by high-mobility group box 1 protein signaling. J. Immunol. 2007, 179, 8525–8532.
- Gorina, R.; Font-Nieves, M.; Márquez-Kisinousky, L.; Santalucia, T.; Planas, A.M. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia 2011, 59, 242–255.
- Koyama, Y.; Kotani, M.; Sawamura, T.; Kuribayashi, M.; Konishi, R.; Michinaga, S. Different actions of endothelin-1 on chemokine production in rat cultured astrocytes: Reduction of CX3CL1/fractalkine and an increase in CCL2/MCP-1 and CXCL1/CINC-1. J. Neuroinflamm. 2013, 10, 51.
- Wicher, G.; Wallenquist, U.; Lei, Y.; Enoksson, M.; Li, X.; Fuchs, B.; Abu Hamdeh, S.; Marklund, N.; Hillered, L.; Nilsson, G.; et al. Interleukin-33 promotes recruitment of microglia/macrophages in response to traumatic brain injury. J. Neurotrauma 2017, 34, 3173–3182.
- Xue, J.; Zhang, Y.; Zhang, J.; Zhu, Z.; Lv, Q.; Su, J. Astrocyte-derived CCL7 promotes microglia-mediated inflammation following traumatic brain injury. Int. Immunopharmacol. 2021, 99, 107975.
- Ding, J.Y.; Kreipke, C.W.; Schafer, P.; Schafer, S.; Speirs, S.L.; Rafols, J.A. Synapse loss regulated by matrix metalloproteinases in traumatic brain injury is associated with hypoxia inducible factor-1alpha expression. Brain Res. 2009, 1268, 125–1234.
- Suehiro, E.; Fujisawa, H.; Akimura, T.; Ishihara, H.; Kajiwara, K.; Kato, S.; Fujii, M.; Yamashita, S.; Maekawa, T.; Suzuki, M. Increased matrix metalloproteinase-9 in blood in association with activation of interleukin-6 after traumatic brain injury: Influence of hypothermic therapy. J. Neurotrauma 2004, 21, 1706–1711.
- Gao, W.; Zhao, Z.; Yu, G.; Zhou, Z.; Zhou, Y.; Hu, T.; Jiang, R.; Zhang, J. VEGI attenuates the inflammatory injury and disruption of blood-brain barrier partly by suppressing the TLR4/NF-κB signaling pathway in experimental traumatic brain injury. Brain Res. 2015, 1622, 230–239.
- Michinaga, S.; Onishi, K.; Shimizu, K.; Mizuguchi, H.; Hishinuma, S. Pharmacological inhibition of transient receptor potential vanilloid 4 reduces vasogenic edema after traumatic brain injury in mice. Biol. Pharm. Bull. 2021, 44, 1759–1766.
- Hu, X.; Li, S.; Shi, Z.; Lin, W.J.; Yang, Y.; Li, Y.; Li, H.; Xu, Y.; Zhou, M.; Tang, Y. Partial ablation of astrocytes exacerbates cerebral infiltration of monocytes and neuronal loss after brain stab injury in mice. Cell Mol. Neurobio. 2022.
- Gao, X.; Li, W.; Syed, F.; Yuan, F.; Li, P.; Yu, Q. PD-L1 signaling in reactive astrocytes counteracts neuroinflammation and ameliorates neuronal damage after traumatic brain injury. J. Neuroinflamm. 2022, 19, 43.
- Zhang, W.; Hong, J.; Zhang, H.; Zheng, W.; Yang, Y. Astrocyte-derived exosomes protect hippocampal neurons after traumatic brain injury by suppressing mitochondrial oxidative stress and apoptosis. Aging (Albany NY) 2021, 13, 21642–21658.
- Michinaga, S.; Tanabe, A.; Nakaya, R.; Fukutome, C.; Inoue, A.; Iwane, A.; Minato, Y.; Tujiuchi, Y.; Miyake, D.; Mizuguchi, H.; et al. Angiopoietin-1/Tie-2 signal after focal traumatic brain injury is potentiated by BQ788, an ETB receptor antagonist, in the mouse cerebrum: Involvement in recovery of blood-brain barrier function. J. Neurochem. 2020, 154, 330–348.
- Michinaga, S.; Inoue, A.; Sonoda, K.; Mizuguchi, H.; Koyama, Y. Down-regulation of astrocytic sonic hedgehog by activation of endothelin ETB receptors: Involvement in traumatic brain injury-induced disruption of blood brain barrier in a mouse model. Neurochem. Int. 2021, 146, 105042.
- Jamjoom, A.A.B.; Rhodes, J.; Andrews, P.J.D.; Grant, S.G.N. The synapse in traumatic brain injury. Brain 2021, 144, 18–31.
- Yu, T.S.; Tensaouti, Y.; Stephanz, E.P.; Chintamen, S.; Rafikian, E.E.; Yang, M.; Kernie, S.G. Astrocytic ApoE underlies maturation of hippocampal neurons and cognitive recovery after traumatic brain injury in mice. Commun. Biol. 2021, 4, 1303.
- Baecker, J.; Wartchow, K.; Sehm, T.; Ghoochani, A.; Buchfelder, M.; Kleindienst, A. Treatment with the neurotrophic protein S100B increases synaptogenesis after traumatic brain injury. J. Neurotrauma 2020, 37, 1097–1107.
- Christopherson, K.S.; Ullian, E.M.; Stokes, C.C.; Mullowney, C.E.; Hell, J.W.; Agah, A.; Lawler, J.; Mosher, D.F.; Bornstein, P.; Barres, B.A. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 2005, 120, 421–433.
- Lu, Z.; Kipnis, J. Thrombospondin 1—A key astrocyte-derived neurogenic factor. FASEB J. 2010, 24, 1925–1934.
- Cheng, C.; Yu, Z.; Zhao, S.; Liao, Z.; Xing, C.; Jiang, Y.; Yang, Y.G.; Whalen, M.J.; Lo, E.H.; Sun, X.; et al. Thrombospondin-1 gene deficiency worsens the neurological outcomes of traumatic brain injury in mice. Int. J. Med. Sci. 2017, 14, 927–936.
- Lin, P.H.; Kuo, L.T.; Luh, H.T. The roles of neurotrophins in traumatic brain injury. Life 2021, 12, 26.
- Goss, J.R.; O’Malley, M.E.; Zou, L.; Styren, S.D.; Kochanek, P.M.; DeKosky, S.T. Astrocytes are the major source of nerve growth factor upregulation following traumatic brain injury in the rat. Exp. Neurol. 1998, 149, 301–309.
- Dixon, C.E.; Flinn, P.; Bao, J.; Venya, R.; Hayes, R.L. Nerve growth factor attenuates cholinergic deficits following traumatic brain injury in rats. Exp. Neurol. 1997, 146, 479–490.
- Furtado, A.B.V.; Gonçalves, D.F.; Hartmann, D.D.; Courtes, A.A.; Cassol, G.; Nunez-Figueredo, Y.; Argolo, D.S.; do Nascimento, R.P.; Costa, S.L.; da Silva, V.D.A.; et al. JM-20 treatment after mild traumatic brain injury reduces glial cell pro-inflammatory signaling and behavioral and cognitive deficits by increasing neurotrophin expression. Mol. Neurobiol. 2021, 58, 4615–4627.
- Zhao, J.; Qu, D.; Xi, Z.; Huan, Y.; Zhang, K.; Yu, C.; Yang, D.; Kang, J.; Lin, W.; Wu, S.; et al. Mitochondria transplantation protects traumatic brain injury via promoting neuronal survival and astrocytic BDNF. Transl. Res. 2021, 235, 102–114.
- Hao, P.; Duan, H.; Hao, F.; Chen, L.; Sun, M.; Fan, K.S.; Sun, Y.E.; Williams, D.; Yang, Z.; Li, X. Neural repair by NT3-chitosan via enhancement of endogenous neurogenesis after adult focal aspiration brain injury. Biomaterials 2017, 140, 88–102.


