 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Aaron Reigh Robart | -- | 5642 | 2023-03-06 17:00:51 | | | |
| 2 | Camila Xu | Meta information modification | 5642 | 2023-03-07 01:39:02 | | |
Video Upload Options
Approaches for crystallization of RNA and how they are used in practice.
1. Introduction
2. RNA Purification and Folding
2.1. Producing Homogeneous Transcripts: Hammerhead Ribozyme
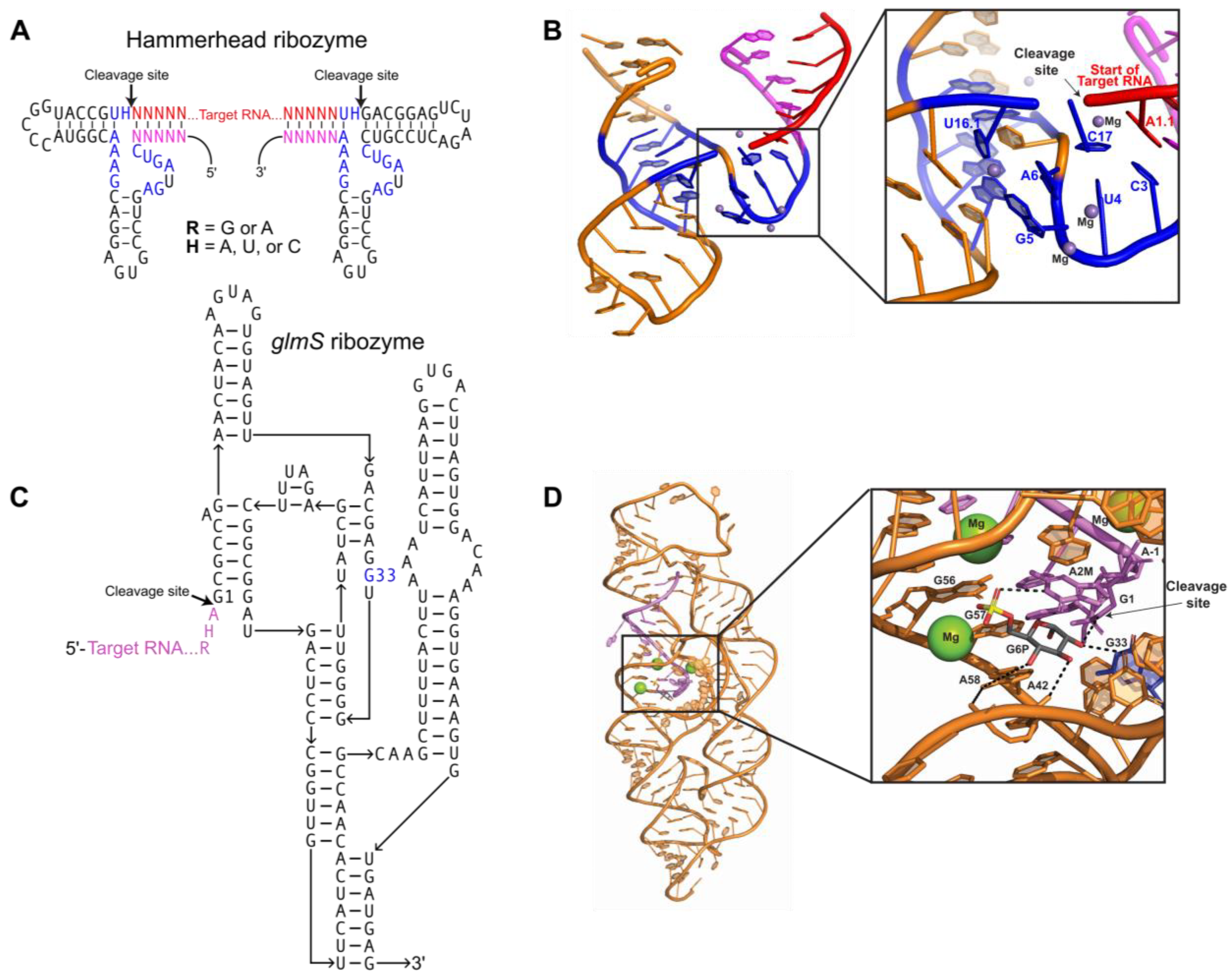
2.2. Producing Homogeneous Transcripts: glmS Ribozyme
2.3. Purification of Transcribed RNA
3. RNA-Driven Crystallization Modules
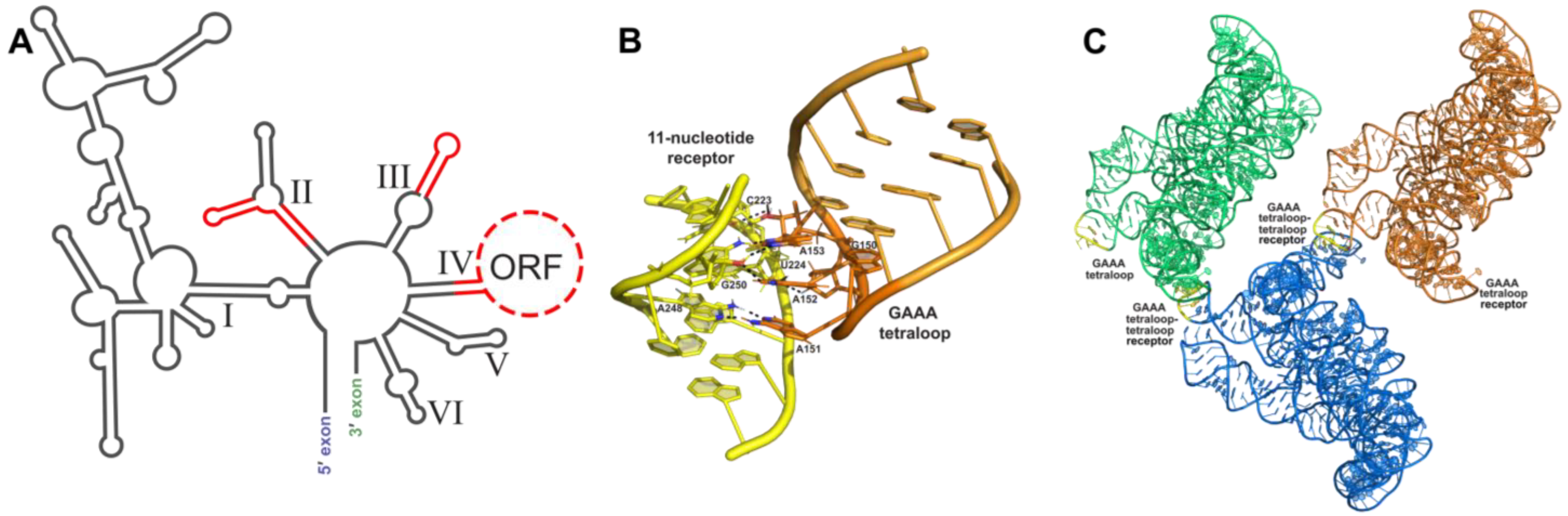
3.1. Tetraloop Interactions as RNA Crystallization Modules
3.2. Loop–Loop Interactions as RNA Crystallization Modules
4. Protein-Assisted RNA Crystallography
4.1. U1A Protein Module
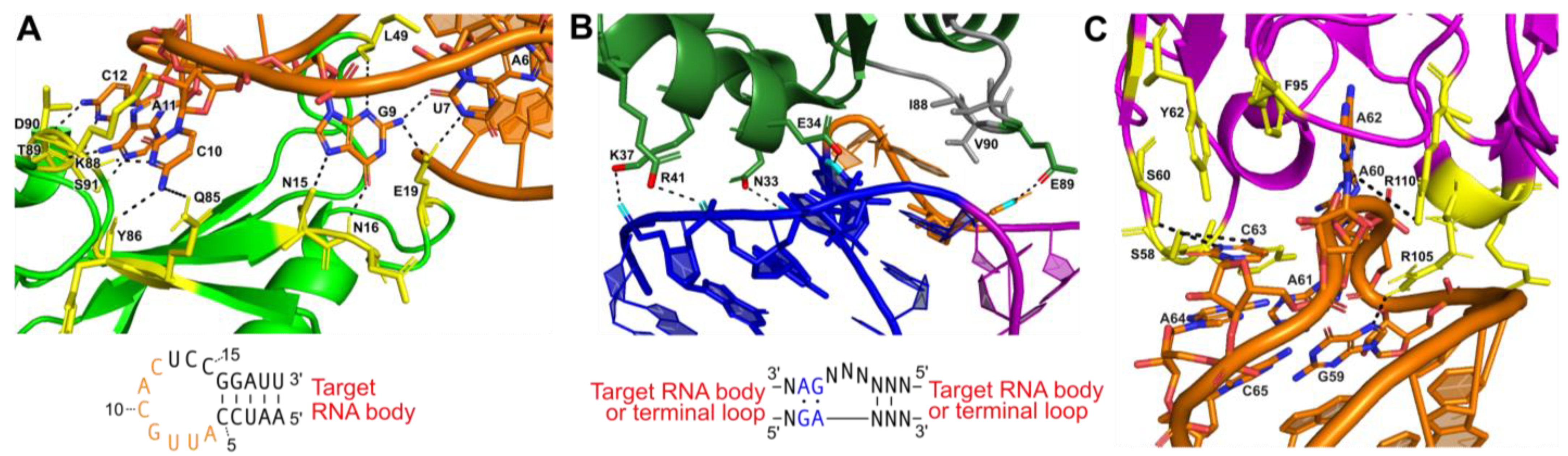
4.2. Kink-Turn Module
4.3. Antibody Fragment Module
5. Solving the ‘Phase Problem’ for RNA Crystals
Formation of electron density maps of crystallized macromolecules requires the amplitude and phase of each diffracted wave [105]. X-ray diffraction datasets collected from a crystal use predetermined X-ray energies, and the intensities of diffracted waves—or “diffraction spots”—are used to determine amplitude [105]. However, this information is essentially useless without a means to determine the phase of each wave. Phase information is necessary to offset the scattered waves when they are added together during reconstruction of the electron density map; consequently, they are critical for building structures from diffraction data [106]. Unfortunately, unlike amplitudes that can be directly measured as intensities on an X-ray detector, information regarding the phase is lost and cannot be directly observed without specific additional experimental considerations [105][106]. This inherent block between crystal diffraction data and a solved structure is referred to as the “phase problem” in crystallography. Several strategies for solving the phase problem have been developed, such as molecular replacement, various methods of isomorphous replacement, and anomalous diffraction [105][106][107]. In this section, molecular replacement models for RNA and RNA–protein complexes, as well as isomorphous replacement with multiple different heavy metals used to support anomalous diffraction methods will be discussed.
5.1. Molecular Replacement Methods
Molecular replacement (MR) is one commonly used method for solving the phase problem, especially for protein crystallography [105][106][107]. This method applies the phases of a structurally similar model to the experimental diffraction data of the target crystal in order to obtain preliminary electron density maps [105][106][107]. Although MR is applicable for estimating phases for any type of macromolecule, it is often better suited for proteins or nucleic acid–protein complexes, as nucleic acid structures make up only 1.8% of the total number of structures in the PDB [108]. This dearth of solved nucleic acid structures can make finding a suitable model for molecular replacement of an all-nucleic acid target quite difficult. MR search models should have high structural similarity to the target molecule [107]. If possible, the input of weak experimental phases determined by anomalous scattering into the search for a model will enhance the chances of success [109]. For a comprehensive review on finding MR models for RNA, see [108].
If no structures are available, a search model can also be designed by homology modeling of the target molecule, or by de novo structure predictions [110][111]. Computational structural biology powered by artificial intelligence (AI) has been revolutionary, providing powerful tools to model macromolecular structures and predict their functions. Alphafold2 is an AI system that uses deep learning algorithms to predict protein structures with astonishing accuracy and is a promising prospect for MR phasing [112]. RNA-based prediction algorithms are also being developed [113][114]: the Rosetta framework FARFAR2 (fragment assembly of RNA with full-atom refinement) uses small RNA fragments that are mended together to create predictions, and it generally performs well in recovering known native-like structures of RNA [114]. Though these advanced computational methods can be limited in the size of the macromolecules that they can predict, their potential applications in phasing will enable a robust and potentially automated pipeline to solve the phase problem in crystallography.
5.2. Isomorphous Replacement and Anomalous Scattering
5.2.1. Isomorphous Replacement with Mg2+ Mimics
5.2.2. Engineering Heavy-Metal Binding Sites
Heavy metal derivatives have historically been produced by a method affectionately referred to as “soak and pray”, where the crystal is soaked in a heavy metal atom solution with the hope that the heavy metal atoms will bind to one or more specific locations within the RNA [124]. Although this method typically results in derivatized crystals, RNA containing suitable specific sites for heavy metal binding is not predictable; thus, the process becomes highly time-consuming through rounds of trial and error [130][131][132]. To address this issue, a general “directed soaking” method has been devised by Batey and Kieft that involves engineering one or more reliable, non-structure-perturbing cation-binding sites into the RNA structure and then soaking hexamine cations into resulting RNA crystals [123][124]. Their method is based on the observation that G-U wobble pairs in A-form helices create a binding site for many cations, including hexamine complexes [127][133][134][135][136]. The identity and orientation of the base pairs that flank the wobble pairs should be taken into consideration, as this can affect cation binding [124]. This method has successfully been used with both cobalt(III) and iridium(III) hexamine, but even cesium has been shown to be effective for phasing when bound to the motif [137]. Since engineering of this site only changes a few nucleotides, it can typically be performed without perturbing the fold or function of the RNA.
5.2.3. Selenium Incorporation into Nucleic Acids
X-ray crystallography of proteins has been greatly impacted by the utilization of selenium derivatization as a phasing module [138]. Selenium is a popular choice for phasing by anomalous diffraction and is in the same periodic family as oxygen and sulfur, thus selenium substitution often does not cause structural perturbations [138]. Although this method typically involves substituting selenium for sulfur atoms in methionine residues, selenium has been successfully incorporated into large nucleic acids by multiple enzymatic approaches [138][139]. Small selenium-derivatized nucleic acids (60 nt. or less) can be produced easily during oligonucleotide synthesis while larger ones can be prepared using DNA or RNA nucleotide triphosphates ((d)NTPs) incorporated by polymerase activity [138]. These Se-modified dNTP/NTPs are commercially available and can include selenium substitutions in the base, sugar, or phosphate portions. For preparation of large selenium derivatized RNAs, in vitro transcription using NTP𝛼Se analogs has been used, where one of the oxygen atoms on the alpha phosphate of the NTP is replaced with selenium [138]. These analogs perform as substrates for T7 RNA polymerase just as well as natural NTPs, but certain bases may affect activity of resulting Se-modified RNAs. When in vitro transcriptional incorporation of NTP𝛼Se analogs into the hammerhead ribozyme was tested, it was observed that ribozymes produced with UTP𝛼Se and CTP𝛼Se analogs had the same catalytic activity as wild type [138]. However, ribozymes produced with GTP𝛼Se had only 30% wild type activity and ribozymes produced with ATP𝛼Se had only very low activity [138]. This suggests that incorporation of selenium into an RNA crystallization target may require some trial-and-error optimization. Aside from direct polymerization incorporation, Se-modified RNAs may also be produced by enzymatic ligation of two or more selenium-containing fragments. This method was used in the crystallization of a rat spliceosomal U6 snRNA stem-loop motif using T4 RNA ligase [140][141].
5.2.4. Soaking with Halogens
MAD and SAD methods combined with halogen soaking have also been used as techniques for phasing, with limited success [142]. Both bromine and iodine have been utilized, where the halides incorporate into the ordered solvent shell as anomalous scatterers. The absorption edge of bromine is achievable at all synchrotron beam lines and although the absorption edges of iodine are not easily accessible, it does have a significant anomalous effect [142]. Soaking with this method can require high halide concentrations (0.2-1M), and soaks should last only a few seconds because of their fast diffusion into the crystals [142].
References
- Milligan, J.F.; Groebe, D.R.; Witherell, G.W.; Uhlenbeck, O.C. Oligoribonucleotide Synthesis Using T7 RNA Polymerase and Synthetic DNA Templates. Nucleic Acids Res. 1987, 15, 8783–8798.
- Kao, C.; Zheng, M.; Rüdisser, S. A Simple and Efficient Method to Reduce Nontemplated Nucleotide Addition at the 3 Terminus of RNAs Transcribed by T7 RNA Polymerase. RNA 1999, 5, 1268–1272.
- Macdonald, L.E.; Zhou, Y.; McAllister, W.T. Termination and Slippage by Bacteriophage T7 RNA Polymerase. J. Mol. Biol. 1993, 232, 1030–1047.
- Hutchins, C.J.; Rathjen, P.D.; Forster, A.C.; Symons, R.H. Self-Cleavage of plus and Minus RNA Transcripts of Avocado Sunblotch Viroid. Nucleic Acids Res. 1986, 14, 3627–3640.
- Buzayan, J.M.; Gerlach, W.L.; Bruening, G.; Keese, P.; Gould, A.R. Nucleotide Sequence of Satellite Tobacco Ringspot Virus RNA and Its Relationship to Multimeric Forms. Virology 1986, 151, 186–199.
- Symons, R.H. Plant Pathogenic RNAs and RNA Catalysis. Nucleic Acids Res. 1997, 25, 2683–2689.
- Pata, J.D.; King, B.R.; Steitz, T.A. Assembly, Purification and Crystallization of an Active HIV-1 Reverse Transcriptase Initiation Complex. Nucleic Acids Res. 2002, 30, 4855–4863.
- Nagai, K.; Oubridge, C.; Ito, N.; Jessen, T.H.; Avis, J.; Evans, P. Crystal Structure of the U1A Spliceosomal Protein Complexed with Its Cognate RNA Hairpin. Nucleic Acids Symp. Ser. 1995, 34, 1–2.
- Ruffner, D.E.; Stormo, G.D.; Uhlenbeck, O.C. Sequence Requirements of the Hammerhead RNA Self-Cleavage Reaction. Biochemistry 1990, 29, 10695–10702.
- Stage-Zimmermann, T.K.; Uhlenbeck, O.C. Hammerhead Ribozyme Kinetics. RNA 1998, 4, 875–889.
- Canny, M.D.; Jucker, F.M.; Kellogg, E.; Khvorova, A.; Jayasena, S.D.; Pardi, A. Fast Cleavage Kinetics of a Natural Hammerhead Ribozyme. J. Am. Chem. Soc. 2004, 126, 10848–10849.
- O’Rourke, S.M.; Scott, W.G. Structural Simplicity and Mechanistic Complexity in the Hammerhead Ribozyme. Prog. Mol. Biol. Transl. Sci. 2018, 159, 177–202.
- Price, S.R.; Ito, N.; Oubridge, C.; Avis, J.M.; Nagai, K. Crystallization of RNA-Protein Complexes. I. Methods for the Large-Scale Preparation of RNA Suitable for Crystallographic Studies. J. Mol. Biol. 1995, 249, 398–408.
- Bassi, G.S.; Murchie, A.I.; Walter, F.; Clegg, R.M.; Lilley, D.M. Ion-Induced Folding of the Hammerhead Ribozyme: A Fluorescence Resonance Energy Transfer Study. EMBO J. 1997, 16, 7481–7489.
- Hammann, C.; Lilley, D.M.J. Folding and Activity of the Hammerhead Ribozyme. Chembiochem 2002, 3, 690–700.
- Perriman, R.; Delves, A.; Gerlach, W.L. Extended Target-Site Specificity for a Hammerhead Ribozyme. Gene 1992, 113, 157–163.
- Koizumi, M.; Hayase, Y.; Iwai, S.; Kamiya, H.; Inoue, H.; Ohtsuka, E. Design of RNA Enzymes Distinguishing a Single Base Mutation in RNA. Nucleic Acids Res. 1989, 17, 7059–7071.
- Sheldon, C.C.; Symons, R.H. Mutagenesis Analysis of a Self-Cleaving RNA. Nucleic Acids Res. 1989, 17, 5679–5685.
- O’Rourke, S.M.; Estell, W.; Scott, W.G. Minimal Hammerhead Ribozymes with Uncompromised Catalytic Activity. J. Mol. Biol. 2015, 427, 2340–2347.
- Scott, W.G. Ribozymes. Curr. Opin. Struct. Biol. 2007, 17, 280–286.
- Watson, P.Y.; Fedor, M.J. The GlmS Riboswitch Integrates Signals from Activating and Inhibitory Metabolites in vivo. Nat. Struct. Mol. Biol. 2011, 18, 359–363.
- Ferré-D’Amaré, A.R.; Scott, W.G. Small Self-Cleaving Ribozymes. Cold Spring Harb. Perspect. Biol. 2010, 2, a003574.
- Pereira, M.J.B.; Behera, V.; Walter, N.G. Nondenaturing Purification of Co-Transcriptionally Folded RNA Avoids Common Folding Heterogeneity. PLoS ONE 2010, 5, e12953.
- Ke, A.; Zhou, K.; Ding, F.; Cate, J.H.D.; Doudna, J.A. A Conformational Switch Controls Hepatitis Delta Virus Ribozyme Catalysis. Nature 2004, 429, 201–205.
- Ke, A.; Doudna, J.A. Crystallization of RNA and RNA-Protein Complexes. Methods 2004, 34, 408–414.
- Batey, R.T.; Sagar, M.B.; Doudna, J.A. Structural and Energetic Analysis of RNA Recognition by a Universally Conserved Protein from the Signal Recognition Particle. J. Mol. Biol. 2001, 307, 229–246.
- Pan, J.; Woodson, S.A. Folding Intermediates of a Self-Splicing RNA: Mispairing of the Catalytic Core. J. Mol. Biol. 1998, 280, 597–609.
- Kladwang, W.; Hum, J.; Das, R. Ultraviolet Shadowing of RNA Can Cause Significant Chemical Damage in Seconds. Sci. Rep. 2012, 2, 517.
- Turner, D.H.; Sugimoto, N.; Freier, S.M. RNA Structure Prediction. Annu. Rev. Biophys. Biophys. Chem. 1988, 17, 167–192.
- Lukavsky, P.J.; Puglisi, J.D. Large-Scale Preparation and Purification of Polyacrylamide-Free RNA Oligonucleotides. RNA 2004, 10, 889–893.
- Toor, N.; Keating, K.S.; Taylor, S.D.; Pyle, A.M. Crystal Structure of a Self-Spliced Group II Intron. Science 2008, 320, 77–82.
- Chan, R.T.; Robart, A.R.; Rajashankar, K.R.; Pyle, A.M.; Toor, N. Crystal Structure of a Group II Intron in the Pre-Catalytic State. Nat. Struct. Mol. Biol. 2012, 19, 555–557.
- Nachtergaele, S.; He, C. The Emerging Biology of RNA Post-Transcriptional Modifications. RNA Biol. 2016, 14, 156–163.
- Kudrin, P.; Meierhofer, D.; Vågbø, C.B.; Ørom, U.A.V. Nuclear RNA-Acetylation Can Be Erased by the Deacetylase SIRT7. bioRxiv 2021, arXiv:2021.04.06.438707.
- Flynn, R.A.; Pedram, K.; Malaker, S.A.; Batista, P.J.; Smith, B.A.H.; Johnson, A.G.; George, B.M.; Majzoub, K.; Villalta, P.W.; Carette, J.E.; et al. Small RNAs Are Modified with N-Glycans and Displayed on the Surface of Living Cells. Cell 2021, 184, 3109–3124.e22.
- Peabody, D.S. The RNA Binding Site of Bacteriophage MS2 Coat Protein. EMBO J. 1993, 12, 595–600.
- Yoon, J.-H.; Srikantan, S.; Gorospe, M. MS2-TRAP (MS2-Tagged RNA Affinity Purification): Tagging RNA to Identify Associated MiRNAs. Methods 2012, 58, 81–87.
- Lim, F.; Peabody, D.S. RNA Recognition Site of PP7 Coat Protein. Nucleic Acids Res. 2002, 30, 4138–4144.
- Fritz, S.E.; Haque, N.; Hogg, J.R. Highly Efficient in Vitro Translation of Authentic Affinity-Purified Messenger Ribonucleoprotein Complexes. RNA 2018, 24, 982–989.
- Youngman, E.M.; Green, R. Affinity Purification of in Vivo-Assembled Ribosomes for in Vitro Biochemical Analysis. Methods 2005, 36, 305–312.
- Ferré-D’Amaré, A.R. Use of the Spliceosomal Protein U1A to Facilitate Crystallization and Structure Determination of Complex RNAs. Methods 2010, 52, 159–167.
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating Non-Coding RNAs in Complete Genomes. Nucleic Acids Res. 2005, 33, D121–D124.
- Edwards, A.L.; Garst, A.D.; Batey, R.T. Determining Structures of RNA Aptamers and Riboswitches by X-Ray Crystallography. Methods Mol. Biol. 2009, 535, 135–163.
- Woese, C.R.; Winker, S.; Gutell, R.R. Architecture of Ribosomal RNA: Constraints on the Sequence of “Tetra-Loops”. Proc. Natl. Acad. Sci. USA 1990, 87, 8467–8471.
- Hermann, T.; Patel, D.J. Stitching Together RNA Tertiary Architectures. J. Mol. Biol. 1999, 294, 829–849.
- Richardson, K.E.; Adams, M.S.; Kirkpatrick, C.C.; Gohara, D.W.; Znosko, B.M. Identification and Characterization of New RNA Tetraloop Sequence Families. Biochemistry 2019, 58, 4809–4820.
- Ferré-D’Amaré, A.R.; Zhou, K.; Doudna, J.A. A General Module for RNA Crystallization. J. Mol. Biol. 1998, 279, 621–631.
- Coonrod, L.A.; Lohman, J.R.; Berglund, J.A. Utilizing the GAAA Tetraloop/Receptor to Facilitate Crystal Packing and Determination of the Structure of a CUG RNA Helix. Biochemistry 2012, 51, 8330–8337.
- Reiter, N.J.; Osterman, A.; Torres-Larios, A.; Swinger, K.K.; Pan, T.; Mondragón, A. Structure of a Bacterial Ribonuclease P Holoenzyme in Complex with tRNA. Nature 2010, 468, 784–789.
- Robart, A.R.; Chan, R.T.; Peters, J.K.; Rajashankar, K.R.; Toor, N. Crystal Structure of a Eukaryotic Group II Intron Lariat. Nature 2014, 514, 193–197.
- Toor, N.; Rajashankar, K.; Keating, K.S.; Pyle, A.M. Structural Basis for Exon Recognition by a Group II Intron. Nat. Struct. Mol. Biol. 2008, 15, 1221–1222.
- Montange, R.K.; Batey, R.T. Structure of the S-Adenosylmethionine Riboswitch Regulatory mRNA Element. Nature 2006, 441, 1172–1175.
- Grundy, F.J.; Henkin, T.M. The S Box Regulon: A New Global Transcription Termination Control System for Methionine and Cysteine Biosynthesis Genes in Gram-Positive Bacteria. Mol. Microbiol. 1998, 30, 737–749.
- Winkler, W.C.; Nahvi, A.; Sudarsan, N.; Barrick, J.E.; Breaker, R.R. An mRNA Structure That Controls Gene Expression by Binding S-Adenosylmethionine. Nat. Struct. Biol. 2003, 10, 701–707.
- Pley, H.W.; Flaherty, K.M.; McKay, D.B. Model for an RNA Tertiary Interaction from the Structure of an Intermolecular Complex between a GAAA Tetraloop and an RNA Helix. Nature 1994, 372, 111–113.
- Pley, H.W.; Flaherty, K.M.; McKay, D.B. Three-Dimensional Structure of a Hammerhead Ribozyme. Nature 1994, 372, 68–74.
- Costa, M.; Michel, F. Frequent Use of the Same Tertiary Motif by Self-Folding RNAs. EMBO J. 1995, 14, 1276–1285.
- Costa, M.; Michel, F. Rules for RNA Recognition of GNRA Tetraloops Deduced by in Vitro Selection: Comparison with in Vivo Evolution. EMBO J. 1997, 16, 3289–3302.
- Abramovitz, D.L.; Pyle, A.M. Remarkable Morphological Variability of a Common RNA Folding Motif: The GNRA Tetraloop-Receptor Interaction. J. Mol. Biol. 1997, 266, 493–506.
- Murphy, F.L.; Cech, T.R. GAAA Tetraloop and Conserved Bulge Stabilize Tertiary Structure of a Group I Intron Domain. J. Mol. Biol. 1994, 236, 49–63.
- Cate, J.H.; Gooding, A.R.; Podell, E.; Zhou, K.; Golden, B.L.; Kundrot, C.E.; Cech, T.R.; Doudna, J.A. Crystal Structure of a Group I Ribozyme Domain: Principles of RNA Packing. Science 1996, 273, 1678–1685.
- Szewczak, A.A.; Podell, E.R.; Bevilacqua, P.C.; Cech, T.R. Thermodynamic Stability of the P4-P6 Domain RNA Tertiary Structure Measured by Temperature Gradient Gel Electrophoresis. Biochemistry 1998, 37, 11162–11170.
- Baird, N.J.; Westhof, E.; Qin, H.; Pan, T.; Sosnick, T.R. Structure of a Folding Intermediate Reveals the Interplay between Core and Peripheral Elements in RNA Folding. J. Mol. Biol. 2005, 352, 712–722.
- Chauhan, S.; Woodson, S.A. Tertiary Interactions Determine the Accuracy of RNA Folding. J. Am. Chem. Soc. 2008, 130, 1296–1303.
- Qin, H.; Sosnick, T.R.; Pan, T. Modular Construction of a Tertiary RNA Structure: The Specificity Domain of the Bacillus Subtilis RNase P RNA. Biochemistry 2001, 40, 11202–11210.
- Shcherbakova, I.; Brenowitz, M. Perturbation of the Hierarchical Folding of a Large RNA by the Destabilization of Its Scaffold’s Tertiary Structure. J. Mol. Biol. 2005, 354, 483–496.
- Treiber, D.K.; Williamson, J.R. Concerted Kinetic Folding of a Multidomain Ribozyme with a Disrupted Loop-Receptor Interaction. J. Mol. Biol. 2001, 305, 11–21.
- Pomeranz Krummel, D.A.; Oubridge, C.; Leung, A.K.W.; Li, J.; Nagai, K. Crystal Structure of Human Spliceosomal U1 SnRNP at 5.5 Å Resolution. Nature 2009, 458, 475–480.
- Quigley, G.J.; Rich, A. Structural Domains of Transfer RNA Molecules. Science 1976, 194, 796–806.
- Lehnert, V.; Jaeger, L.; Michel, F.; Westhof, E. New Loop-Loop Tertiary Interactions in Self-Splicing Introns of Subgroup IC and ID: A Complete 3D Model of the Tetrahymena Thermophila Ribozyme. Chem. Biol. 1996, 3, 993–1009.
- Batey, R.T.; Rambo, R.P.; Doudna, J.A. Tertiary Motifs in RNA Structure and Folding. Angew. Chem. Int. Ed. Engl. 1999, 38, 2326–2343.
- Gregorian, R.S.; Crothers, D.M. Determinants of RNA Hairpin Loop-Loop Complex Stability. J. Mol. Biol. 1995, 248, 968–984.
- Eisinger, J. Complex Formation between Transfer RNA’S with Complementary Anticodons. Biochem. Biophys. Res. Commun. 1971, 43, 854–861.
- Eisinger, J.; Gross, N. The Anticodon-Anticodon Complex. J. Mol. Biol. 1974, 88, 165–174.
- Grosjean, H.; Söll, D.G.; Crothers, D.M. Studies of the Complex between Transfer RNAs with Complementary Anticodons. I. Origins of Enhanced Affinity between Complementary Triplets. J. Mol. Biol. 1976, 103, 499–519.
- Labuda, D.; Grosjean, H.; Striker, G.; Pörschke, D. Codon:Anticodon and Anticodon:Anticodon Interaction: Evaluation of Equilibrium and Kinetic Parameters of Complexes Involving a G:U Wobble. Biochim. Biophys. Acta 1982, 698, 230–236.
- Houssier, C.; Grosjean, H. Temperature Jump Relaxation Studies on the Interactions between Transfer RNAs with Complementary Anticodons. The Effect of Modified Bases Adjacent to the Anticodon Triplet. J. Biomol. Struct. Dyn. 1985, 3, 387–408.
- Romby, P.; Giegé, R.; Houssier, C.; Grosjean, H. Anticodon-Anticodon Interactions in Solution. Studies of the Self-Association of Yeast or Escherichia Coli tRNAAsp and of Their Interactions with Escherichia Coli tRNAVal. J. Mol. Biol. 1985, 184, 107–118.
- Skripkin, E.; Paillart, J.C.; Marquet, R.; Ehresmann, B.; Ehresmann, C. Identification of the Primary Site of the Human Immunodeficiency Virus Type 1 RNA Dimerization in Vitro. Proc. Natl. Acad. Sci. USA 1994, 91, 4945–4949.
- Paillart, J.C.; Marquet, R.; Skripkin, E.; Ehresmann, B.; Ehresmann, C. Mutational Analysis of the Bipartite Dimer Linkage Structure of Human Immunodeficiency Virus Type 1 Genomic RNA. J. Biol. Chem. 1994, 269, 27486–27493.
- Brunel, C.; Marquet, R.; Romby, P.; Ehresmann, C. RNA Loop-Loop Interactions as Dynamic Functional Motifs. Biochimie 2002, 84, 925–944.
- Oubridge, C.; Ito, N.; Evans, P.R.; Teo, C.H.; Nagai, K. Crystal Structure at 1.92 A Resolution of the RNA-Binding Domain of the U1A Spliceosomal Protein Complexed with an RNA Hairpin. Nature 1994, 372, 432–438.
- Rupert, P.B.; Ferré-D’Amaré, A.R. Crystal Structure of a Hairpin Ribozyme-Inhibitor Complex with Implications for Catalysis. Nature 2001, 410, 780–786.
- Cochrane, J.C.; Lipchock, S.V.; Strobel, S.A. Structural Investigation of the GlmS Ribozyme Bound to Its Catalytic Cofactor. Chem. Biol. 2007, 14, 97–105.
- Adams, P.L.; Stahley, M.R.; Kosek, A.B.; Wang, J.; Strobel, S.A. Crystal Structure of a Self-Splicing Group I Intron with Both Exons. Nature 2004, 430, 45–50.
- Ferré-D’Amaré, A.R.; Zhou, K.; Doudna, J.A. Crystal Structure of a Hepatitis Delta Virus Ribozyme. Nature 1998, 395, 567–574.
- Xiao, H.; Edwards, T.E.; Ferré-D’Amaré, A.R. Structural Basis for Specific, High-Affinity Tetracycline Binding by an in Vitro Evolved Aptamer and Artificial Riboswitch. Chem. Biol. 2008, 15, 1125–1137.
- Kulshina, N.; Baird, N.J.; Ferré-D’Amaré, A.R. Recognition of the Bacterial Second Messenger Cyclic Diguanylate by Its Cognate Riboswitch. Nat. Struct. Mol. Biol. 2009, 16, 1212–1217.
- Smith, K.D.; Lipchock, S.V.; Ames, T.D.; Wang, J.; Breaker, R.R.; Strobel, S.A. Structural Basis of Ligand Binding by a C-Di-GMP Riboswitch. Nat. Struct. Mol. Biol. 2009, 16, 1218–1223.
- Shechner, D.M.; Grant, R.A.; Bagby, S.C.; Koldobskaya, Y.; Piccirilli, J.A.; Bartel, D.P. Crystal Structure of the Catalytic Core of an RNA-Polymerase Ribozyme. Science 2009, 326, 1271–1275.
- Klein, D.J.; Schmeing, T.M.; Moore, P.B.; Steitz, T.A. The Kink-Turn: A New RNA Secondary Structure Motif. EMBO J. 2001, 20, 4214–4221.
- Huang, L.; Lilley, D.M.J. The Molecular Recognition of Kink-Turn Structure by the L7Ae Class of Proteins. RNA 2013, 19, 1703–1710.
- Lilley, D.M.J. The L7Ae Proteins Mediate a Widespread and Highly Functional Protein–RNA Interaction. Biochemist 2019, 41, 40–44.
- Zhang, J.; Ferré-D’Amaré, A.R. Cocrystal Structure of a T-Box Riboswitch Stem I Domain in Complex with Its Cognate tRNA. Nature 2013, 500, 363–366.
- Zhang, J.; Ferré-D’Amaré, A.R. New Molecular Engineering Approaches for Crystallographic Studies of Large RNAs. Curr. Opin. Struct. Biol. 2014, 26, 9–15.
- Koide, S. Engineering of Recombinant Crystallization Chaperones. Curr. Opin. Struct. Biol. 2009, 19, 449–457.
- Dutzler, R.; Campbell, E.B.; Cadene, M.; Chait, B.T.; MacKinnon, R. X-Ray Structure of a ClC Chloride Channel at 3.0 A Reveals the Molecular Basis of Anion Selectivity. Nature 2002, 415, 287–294.
- Tereshko, V.; Uysal, S.; Koide, A.; Margalef, K.; Koide, S.; Kossiakoff, A.A. Toward Chaperone-Assisted Crystallography: Protein Engineering Enhancement of Crystal Packing and X-Ray Phasing Capabilities of a Camelid Single-Domain Antibody (VHH) Scaffold. Protein Sci. 2008, 17, 1175–1187.
- Iwata, S.; Ostermeier, C.; Ludwig, B.; Michel, H. Structure at 2.8 A Resolution of Cytochrome c Oxidase from Paracoccus Denitrificans. Nature 1995, 376, 660–669.
- Lieberman, R.L.; Culver, J.A.; Entzminger, K.C.; Pai, J.C.; Maynard, J.A. Crystallization Chaperone Strategies for Membrane Proteins. Methods 2011, 55, 293–302.
- Piccirilli, J.A.; Koldobskaya, Y. Crystal Structure of an RNA Polymerase Ribozyme in Complex with an Antibody Fragment. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 2918–2928.
- Fellouse, F.A.; Esaki, K.; Birtalan, S.; Raptis, D.; Cancasci, V.J.; Koide, A.; Jhurani, P.; Vasser, M.; Wiesmann, C.; Kossiakoff, A.A.; et al. High-Throughput Generation of Synthetic Antibodies from Highly Functional Minimalist Phage-Displayed Libraries. J. Mol. Biol. 2007, 373, 924–940.
- Koide, A.; Gilbreth, R.N.; Esaki, K.; Tereshko, V.; Koide, S. High-Affinity Single-Domain Binding Proteins with a Binary-Code Interface. Proc. Natl. Acad. Sci. USA 2007, 104, 6632–6637.
- Ye, J.-D.; Tereshko, V.; Frederiksen, J.K.; Koide, A.; Fellouse, F.A.; Sidhu, S.S.; Koide, S.; Kossiakoff, A.A.; Piccirilli, J.A. Synthetic Antibodies for Specific Recognition and Crystallization of Structured RNA. Proc. Natl. Acad. Sci. USA 2008, 105, 82–87.
- Rhodes, G. Crystallography Made Crystal Clear: A Guide for Users of Macromolecular Models; Elsevier Science & Technology: Burlington, VT, USA, 2006; ISBN 978-0-08-045554-9.
- Taylor, G.L. Introduction to Phasing. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 325–338.
- Evans, P.; McCoy, A. An Introduction to Molecular Replacement. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 1–10.
- Marcia, M.; Humphris-Narayanan, E.; Keating, K.S.; Somarowthu, S.; Rajashankar, K.; Pyle, A.M. Solving Nucleic Acid Structures by Molecular Replacement: Examples from Group II Intron Studies. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 2174–2185.
- Kleywegt, G.J.; Jones, T.A. Template Convolution to Enhance or Detect Structural Features in Macromolecular Electron-Density Maps. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 179–185.
- Giorgetti, A.; Raimondo, D.; Miele, A.E.; Tramontano, A. Evaluating the Usefulness of Protein Structure Models for Molecular Replacement. Bioinformatics 2005, 21 (Suppl. 2), ii72–ii76.
- Thompson, J.; Baker, D. Incorporation of Evolutionary Information into Rosetta Comparative Modeling. Proteins 2011, 79, 2380–2388.
- McCoy, A.J.; Sammito, M.D.; Read, R.J. Implications of AlphaFold2 for Crystallographic Phasing by Molecular Replacement. Acta Crystallogr. D Struct. Biol. 2022, 78, 1–13.
- Baek, M.; McHugh, R.; Anishchenko, I.; Baker, D.; DiMaio, F. Accurate Prediction of Nucleic Acid and Protein-Nucleic Acid Complexes Using RoseTTAFoldNA. bioRxiv 2022, arXiv:2022.09.09.507333.
- Watkins, A.M.; Rangan, R.; Das, R. FARFAR2: Improved De Novo Rosetta Prediction of Complex Global RNA Folds. Structure 2020, 28, 963–976.e6.
- Grigg, J.C.; Ke, A. Structural Determinants for Geometry and Information Decoding of TRNA by T Box Leader RNA. Structure 2013, 21, 2025–2032.
- Grigg, J.C.; Price, I.R.; Ke, A. TRNA Fusion to Streamline RNA Structure Determination: Case Studies in Probing Aminoacyl-TRNA Sensing Mechanisms by the T-Box Riboswitch. Crystals 2022, 12, 694.
- Xiao, H.; Murakami, H.; Suga, H.; Ferré-D’Amaré, A.R. Structural Basis of Specific tRNA Aminoacylation by a Small in Vitro Selected Ribozyme. Nature 2008, 454, 358–361.
- Ferré-D’Amaré, A.R.; Doudna, J.A. Methods to Crystallize RNA. In Current Protocols in Nucleic Acid Chemistry; Wiley & Sons: New York, NY, USA, 2001; Chapter 7, Unit 7.6.
- Rupert, P.B.; Ferré-D’Amaré, A.R. Crystallization of the Hairpin Ribozyme: Illustrative Protocols. Methods Mol. Biol. 2004, 252, 303–311.
- Pyle, A. Metal Ions in the Structure and Function of RNA. J. Biol. Inorg. Chem. 2002, 7, 679–690.
- Wedekind, J.E. Metal Ion Binding and Function in Natural and Artificial Small RNA Enzymes from a Structural Perspective. Met. Ions Life Sci. 2011, 9, 299–345.
- Jenkins, J.L.; Wedekind, J.E. The Quick and the Dead: A Guide to Fast Phasing of Small Ribozyme and Riboswitch Crystal Structures. Methods Mol. Biol. 2016, 1490, 265–280.
- Batey, R.T.; Kieft, J.S. Soaking Hexammine Cations into RNA Crystals to Obtain Derivatives for Phasing Diffraction Data. Methods Mol. Biol. 2016, 1320, 219–232.
- Keel, A.Y.; Rambo, R.P.; Batey, R.T.; Kieft, J.S. A General Strategy to Solve the Phase Problem in RNA Crystallography. Structure 2007, 15, 761–772.
- Jou, R.; Cowan, J.A. Ribonuclease H Activation by Inert Transition-Metal Complexes. Mechanistic Probes for Metallocofactors: Insights on the Metallobiochemistry of Divalent Magnesium Ion. J. Am. Chem. Soc. 1991, 113, 6685–6686.
- Cate, J.H.; Yusupov, M.M.; Yusupova, G.Z.; Earnest, T.N.; Noller, H.F. X-Ray Crystal Structures of 70S Ribosome Functional Complexes. Science 1999, 285, 2095–2104.
- Cate, J.H.; Doudna, J.A. Metal-Binding Sites in the Major Groove of a Large Ribozyme Domain. Structure 1996, 4, 1221–1229.
- Kim, S.H.; Suddath, F.L.; Quigley, G.J.; McPherson, A.; Sussman, J.L.; Wang, A.H.; Seeman, N.C.; Rich, A. Three-Dimensional Tertiary Structure of Yeast Phenylalanine Transfer RNA. Science 1974, 185, 435–440.
- Robertus, J.D.; Ladner, J.E.; Finch, J.T.; Rhodes, D.; Brown, R.S.; Clark, B.F.; Klug, A. Structure of Yeast Phenylalanine tRNA at 3 A Resolution. Nature 1974, 250, 546–551.
- Golden, B.L. Heavy Atom Derivatives of RNA. In Methods in Enzymology; RNA—Ligand Interactions, Part A; Academic Press: Cambridge, MA, USA, 2000; Volume 317, pp. 124–132.
- Golden, B.L.; Gooding, A.R.; Podell, E.R.; Cech, T.R. X-Ray Crystallography of Large RNAs: Heavy-Atom Derivatives by RNA Engineering. RNA 1996, 2, 1295–1305.
- Wedekind, J.E.; McKay, D.B. Purification, Crystallization, and X-Ray Diffraction Analysis of Small Ribozymes. Meth. Enzymol. 2000, 317, 149–168.
- Masquida, B.; Westhof, E. On the Wobble GoU and Related Pairs. RNA 2000, 6, 9–15.
- Varani, G.; McClain, W.H. The G·U Wobble Base Pair. EMBO Rep. 2000, 1, 18–23.
- Colmenarejo, G.; Tinoco, I. Structure and Thermodynamics of Metal Binding in the P5 Helix of a Group I Intron Ribozyme. J. Mol. Biol. 1999, 290, 119–135.
- Stefan, L.R.; Zhang, R.; Levitan, A.G.; Hendrix, D.K.; Brenner, S.E.; Holbrook, S.R. MeRNA: A Database of Metal Ion Binding Sites in RNA Structures. Nucleic Acids Res. 2006, 34, D131–D134.
- Gilbert, S.D.; Rambo, R.P.; Van Tyne, D.; Batey, R.T. Structure of the SAM-II Riboswitch Bound to S-Adenosylmethionine. Nat. Struct. Mol. Biol. 2008, 15, 177–182.
- Sheng, J.; Huang, Z. Selenium Derivatization of Nucleic Acids for X-Ray Crystal-Structure and Function Studies. Chem. Biodivers. 2010, 7, 753–785.
- Jiang, J.; Sheng, J.; Carrasco, N.; Huang, Z. Selenium Derivatization of Nucleic Acids for Crystallography. Nucleic Acids Res. 2007, 35, 477–485.
- Höbartner, C.; Rieder, R.; Kreutz, C.; Puffer, B.; Lang, K.; Polonskaia, A.; Serganov, A.; Micura, R. Syntheses of RNAs with up to 100 Nucleotides Containing Site-Specific 2′-Methylseleno Labels for Use in X-Ray Crystallography. J. Am. Chem. Soc. 2005, 127, 12035–12045.
- Höbartner, C.; Micura, R. Chemical Synthesis of Selenium-Modified Oligoribonucleotides and Their Enzymatic Ligation Leading to an U6 SnRNA Stem-Loop Segment. J. Am. Chem. Soc. 2004, 126, 1141–1149.
- Dauter, M.; Dauter, Z. Many Ways to Derivatize Macromolecules and Their Crystals for Phasing. Methods Mol. Biol. 2017, 1607, 349–356.


