Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Patrizia Limonta | -- | 4611 | 2023-03-06 11:18:59 | | | |
| 2 | Rita Xu | -3 word(s) | 4608 | 2023-03-08 06:48:39 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Fontana, F.; Anselmi, M.; Limonta, P. Mitochondrial Metabolism and Dynamics in Prostate Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/41896 (accessed on 30 June 2026).
Fontana F, Anselmi M, Limonta P. Mitochondrial Metabolism and Dynamics in Prostate Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/41896. Accessed June 30, 2026.
Fontana, Fabrizio, Martina Anselmi, Patrizia Limonta. "Mitochondrial Metabolism and Dynamics in Prostate Cancer" Encyclopedia, https://encyclopedia.pub/entry/41896 (accessed June 30, 2026).
Fontana, F., Anselmi, M., & Limonta, P. (2023, March 06). Mitochondrial Metabolism and Dynamics in Prostate Cancer. In Encyclopedia. https://encyclopedia.pub/entry/41896
Fontana, Fabrizio, et al. "Mitochondrial Metabolism and Dynamics in Prostate Cancer." Encyclopedia. Web. 06 March, 2023.
Copy Citation
Prostate cancer (PCa) is the second leading cause of cancer deaths among men in Western countries. Mitochondria, the “powerhouse” of cells, undergo distinctive metabolic and structural dynamics in different types of cancer. PCa cells experience peculiar metabolic changes during their progression from normal epithelial cells to early-stage and, progressively, to late-stage cancer cells.
prostate cancer
mitochondrial metabolism
mitochondrial dynamics
1. Introduction
Mitochondria are organelles involved in different cellular processes, including cell proliferation and intrinsic apoptosis, redox and Ca2+ homeostasis as well as cell stemness. They are also known as the master producers of ATP, being deeply involved in cellular energy metabolism; in addition, being dynamic organelles, they often undergo structural changes, including biogenesis, fusion/fission and mitophagy. Specifically, it is now well recognized that mitochondria undergo complex functional and structural dynamics in cancer cells during the different phases of tumor growth and progression.
1.1. Mitochondrial Metabolism in Cancer Cells
It is now recognized that cancer cells, growing in a hypoxic and hyponutrient microenvironment, are forced to adapt their metabolism (“metabolic reprogramming”) to obtain the required amount of biomass and energy to sustain their uncontrolled proliferation and aggressive behavior.
According to the theory proposed by Otto Warburg in the 1920s (named the Warburg effect), cancer cells are characterized by high rates of glucose uptake and preferentially metabolize it through the glycolytic pathway, even in the presence of adequate amounts of oxygen (aerobic glycolysis) and functional mitochondria [1][2]. Although a small amount of ATP per mole of glucose is produced through glycolysis, it is believed that this metabolic process rapidly produces high levels of metabolites to sustain the biosynthesis of the molecules (i.e., amino acids, fatty acids and nucleotides) required for cancer cell growth and division [3][4][5][6][7][8]. Moreover, the high levels of lactate produced at the end of glycolysis by lactate dehydrogenase (LDH) are secreted by cancer cells to generate an acidic tumor microenvironment promoting their transition to the most aggressive (i.e., migratory, invasive) phenotype and affecting the immune microenvironment to induce an immune tolerant condition [9][10].
Despite the presence of an active glycolytic pathway, several recent data strongly support the coexistence of functional mitochondria in cancer cells, even in the metastatic phase [6][11][12][13][14][15][16][17][18][19][20]. Mitochondria, known as the “powerhouse of the cell”, are deeply involved in the cellular metabolic dynamics, being the major intracellular producers of ATP through the oxidative phosphorylation (OXPHOS) pathway; these organelles are also the “venue” where the tricarboxylic acid (TCA) cycle takes place to provide the most building blocks for the synthesis of biomolecules. A high level of fatty acid β-oxidation also occurs in mitochondria to sustain the production of citrate. Moreover, glutaminolysis is activated to convert glutamine into intermediates for the synthesis of amino acids and nucleotides, as well as into glutamate to fuel the TCA cycle. Together, these metabolic pathways are necessary for cell anabolism to trigger cell proliferation and metastasis in cancers [8][20][21][22][23][24][25][26][27][28][29][30].
A metabolic remodeling based on a shift towards the OXPHOS machinery has also been shown to be involved in the development of drug resistance in tumor cells [31][32][33][34] and to occur in the subpopulation of cancer stem cells (CSCs), known to play a key role in tumor relapse, to provide sufficient amounts of energy and metabolites for their self-renewal and evasion from cell death induced by anticancer drugs [35][36][37][38][39][40][41][42][43].
Cancer cell metabolic plasticity has also been reported to be regulated by neighboring cells in the tumor microenvironment through both mechanical and chemical factors [18][44]. For instance, it has been shown that cancer-associated fibroblasts (CAFs) secrete lactate that is taken up by tumor cells to trigger their metabolic switch towards the OXPHOS energy-producing pathway and reactive oxygen species (ROS) generation [45][46][47][48].
The key role played by mitochondrial metabolism in cancer is strongly supported by the observation that different compounds were shown to exert their anticancer activity by targeting the oxidative phosphorylation pathways [42][49][50][51][52][53][54][55][56].
1.2. Mitochondrial Dynamics in Cancer Cells
Mitochondria are also highly dynamic organelles undergoing changes in number and structure through different processes such as biogenesis, fusion, fission (fragmentation) and mitophagy (removal of impaired mitochondria). A balance between these processes is required for the maintenance of homeostasis in healthy cells; on the other hand, adaptations of mitochondrial dynamics have been widely reported in cells undergoing metabolic and stressful conditions (i.e., glucose starvation, hypoxia), such as cancer cells [8][57][58][59][60][61][62].
Mitochondrial biogenesis is the generation of new organelles from pre-existing ones. In cancer cells, an increase in mitochondrial mass is induced by a variety of stressful signals and has been found to correlate with cell growth, invasiveness, metastasis and drug resistance [63][64]. The master regulator of mitochondrial biogenesis is peroxisome proliferator-activated receptor gamma coactivator 1α (PGC1α) which cooperates with different transcription factors to increase the expression of the mitochondrial transcription factor (TFAM), the final effector of the increase of mitochondrial mass [65]. PGC1α, activated by phosphorylation by the energy sensor adenosine monophosphate-activated protein kinase (AMPK) and by deacetylation by silent information regulator 1 (SIRT1), also triggers the transcription of both nuclear and mitochondrial genes, leading to increased mitochondrial mass, OXPHOS activity and ATP production [66][67][68]. Mitochondrial biogenesis has been shown to mediate the ability of CSCs to overcome antitumor therapies [69][70][71].
An imbalance in the mitochondrial fusion/fission leads to peculiar changes of the morphological features of these organelles (interconnected vs. fragmented). GTPases belonging to the dynamin family play a pivotal role in mediating both of these processes [16][59][62][72]. Mitochondrial fusion is the physical merging of the outer membranes (OMM) and the inner membranes (IMM) of distinct mitochondria and depends on GTP hydrolysis. This process foresees the activity of three GTPases, mitofusin (MFN) 1 and 2 on the OMM and optic atrophy protein 1 (OPA1) on the IMM. MFN 1 and 2 interact to induce the formation of strict connections between adjacent mitochondria, leading to the fusion of the OMM. Then, OPA1 interacts with MFNs forming intermembrane protein complexes, thus coupling the fusion of OMMs with IMMs [73][74][75][76]. Mitochondrial fission is a multi-step process allowing the division of one mitochondrion, leading to the formation of new organelles. The key protein involved in this process is dynamin-related protein 1 (DRP1), a cytosolic GTPase. Endoplasmic reticulum (ER) membranes get in touch with mitochondria allowing a Ca2+ flux from the ER into the mitochondria, thus favoring actin polymerization and inner mitochondrial membrane constriction at this site. At the level of ER-mitochondria contact sites, different proteins such as mitochondrial dynamics 49 and 51 (MID49 and MID51), MFF and mitochondrial fission 1 (FIS1) proteins, identified as DRP1 receptors, are also located. DRP1 binds to these proteins to encircle and shrink the mitochondria, finally leading to their fission. Accumulating evidence demonstrates that an imbalance in these mitochondrial dynamics occurs in different types of cancer [59][77][78][79][80].
2. Mitochondrial Metabolism and Dynamics in PCa
Prostate cancer (PCa) represents the most common malignancy among men and the second leading cause of cancer deaths in developed countries [81]. Androgen-deprivation therapy (ADT), based on gonadotropin-releasing hormone (GnRH) agonists and antagonists, either alone or in combination with androgen receptor antagonists (enzalutamide, apalutamide, darolutamide), still remains the most common treatment for androgen-dependent PCa patients [82][83][84][85][86][87][88]. However, PCa often progresses towards the castration-resistant phase (CRPC), a condition characterized by the acquisition of a more aggressive and metastatic behavior even in the absence of circulating androgens [89][90]. The standard treatment of CRPC patients is presently based on chemotherapy (docetaxel) either alone or in combination with GnRH analogs, antiandrogens or inhibitors of androgen synthesis (abiraterone) [91][92][93][94][95][96][97][98][99][100][101]. Novel therapeutic strategies, such as immune check-point inhibitors or CAR-T approaches, are under investigation [102][103][104][105]. The elucidation of novel molecular hallmarks of tumor progression (i.e., mitochondrial metabolism and dynamics) will likely pave the way for the development of novel therapeutic approaches for PCa patients. Based on the above considerations, as well as on the progressively accumulating data in the literature, researchers aim to highlight and discuss the recent findings on the involvement of mitochondrial functional reprogramming and structural dynamics in PCa, specifically in its CRPC stage; researchers also address the impact of these mechanisms as molecular targets of novel and effective antitumor strategies for this aggressive disease.
2.1. Mitochondrial Metabolism
Metabolic reprogramming is a well-recognized hallmark of cancer, enabling cancer cells to acquire properties that support cell survival, proliferation and acquisition of aggressive (invasive, metastatic) features. However, peculiar molecular mechanisms of this metabolic rewiring have been reported to occur in different types of tumor cells, and this metabolic heterogeneity confers differences in their proliferative/metastatic potential. In prostate epithelial cells, distinctive changes of cell metabolism have been highlighted during the different phases of their conversion from healthy cells to early-stage and, progressively, to late-stage cancer cells [31][106][107][108][109].
2.1.1. Metabolic Rewiring
Healthy epithelial cells, located in the peripheral zone of the prostate, exhibit a peculiar metabolic programming aimed at producing and secreting citrate into the prostatic fluid, one of the most relevant functions of this gland [110]. In most mammalian cells, pyruvate, the end product of the glycolytic pathway, is transported into the mitochondria where it is decarboxylated to acetyl-CoA. Acetyl-CoA subsequently binds to oxaloacetic acid to form citrate that enters the TCA cycle. Citrate is then converted into its isomer isocitrate that is further oxidized into the TCA cycle for the progression to OXPHOS and ATP production; mitochondrial aconitase (m-aconitase) is the ROS-sensitive key enzyme responsible for the citrate-isocitrate conversion. In normal prostate epithelial cells, m-aconitase activity is inhibited, resulting in the impairment of citrate oxidation followed by its accumulation and secretion [111]. The inhibition of m-aconitase strictly correlates with the ability of these cells to accumulate zinc, due to their elevated expression of its transporter ZIP1; high intramitochondrial zinc levels increase ROS generation, leading to the inhibition of m-aconitase activity and resulting in a truncated TCA cycle [112]. As a consequence, the healthy “zinc-accumulating, citrate-producing” epithelial cells are characterized by an inefficient OXPHOS which is compensated by an increased glycolytic pathway to support citrate production [113]. High zinc levels were also found to be associated with a mitochondrial apoptotic phenotype mediated by the release of cytochrome c and caspase activation [114].
On other hand, it is now well established that prostate epithelial cells undergo a peculiar metabolic rewiring during the early phases of cancer development. Specifically, elevated levels of the TCA cycle enzymes and intermediate metabolites could be detected in prostate cancer tissues in comparison to adjacent normal tissues [115][116][117]. Intracellular zinc levels were found to be significantly reduced in PCa cells, thus leading to the reactivation of m-aconitase, citrate oxidation, TCA cycle pathways and oxidative phosphorylation [118]. This reduction was shown to be related to a decreased expression of zinc transporters, such as ZIP1 and 3 [119], mediated by the hypermethylation of their gene promoters [120]. The low levels of zinc also allow cancer cells to avoid apoptosis; actually, it has been reported that zinc treatments trigger cell death and promote chemosensitivity in PCa cells [121]. In line with these observations, very low zinc levels were observed in PCa tissues [122]. Taken together, these data support that the transformation of prostate epithelial cells into their tumoral phenotype is associated with an efficient reactivation of the TCA cycle/OXPHOS metabolic pathway to meet their increased energy and metabolite demand [123].
Interestingly, prostate epithelial cells seem to possess a markedly metabolic plasticity by changing their mitochondrial metabolic features again when progressing from the early-stage towards the late-stage (i.e., metastasis) of cancer, even in the presence of low intracellular zinc levels. Specifically, the Warburg effect (i.e., increased glycolytic activity) has been proposed as the prominent metabolic feature of metastatic prostate tumors [123][124]. Mechanistically, the PI3K-AKT-mammalian target of the rapamycin (mTOR) pathway, a key driver of tumor progression, was shown to play a causal role in prostate tumorigenesis through the up-regulation of pyruvate kinase isoenzyme type M2 (PKM2), the rate-limiting enzyme catalyzing the final reaction of the glycolytic pathway [125][126]. Mutations of the tumor suppressor p53, frequently occurring in advanced prostate cancers, were reported to trigger the Warburg effect. Moreover, deletions of the tumor suppressor PTEN, often observed in aggressive prostate tumors, were demonstrated to correlate with an increased expression of hexokinase 2 (HK2), the initial enzyme of glycolysis, catalyzing the phosphorylation of glucose by ATP to glucose-6-P through the AKT-mTOR pathway [123][127]. Taken together, these observations support that PTEN and p53 tumor suppressors, together with the PI3K-AKT-mTOR pathway, are essential drivers of the Warburg effect to maintain a sufficient energy and metabolites supply for PCa growth and progression [128]. In addition, it has been demonstrated that, in prostatic carcinoma cell lines, the hypoxic conditions of the tumor microenvironment trigger the expression of SUMO1/sentrin-specific peptidase 1 (SENP1) that in turn interacts with HIF1α to promote the Warburg effect and sustain cell proliferation [129]. In line with these data, Sun and coworkers recently reported that HK2 and HIF1α are highly expressed in PCa tissues and their expression correlates with tumor growth and metastasis [130].
There is also consistent evidence that an association exists between obesity and the risk of PCa growth [131]. A deleterious bidirectional cross-talk between PCa cells and adipocytes in their microenvironment has been demonstrated [132]. Specifically, it has been reported that PCa cells educate neighboring adipocytes towards a lipolytic phenotype, resulting in free glycerol production and secretion; adipocyte-derived glycerol is then uptaken by PCa cells to enter and fuel the glycolytic pathway [133]. Moreover, adipocyte conditioning of PCa cells leads to an increased expression of glycolytic genes, resulting in lactate production and OXPHOS inhibition [133][134]. In line with these observations, researchers recently reported that adipocyte-released extracellular vesicles significantly decrease the sensitivity of PCa cells to the chemotherapeutic drug docetaxel and this effect is associated with an AKT/HIF-1α axis-related Warburg effect, which is characterized by enhanced glucose consumption, lactate release and ATP production [135].
To confirm that PCa cells undergo dynamic metabolic changes at each stage of tumor development, Vayalil and Landar introduced the “mitochondrial oncobioenergetic index (MOBI)” (i.e., the mathematical representation of the oncobioenergetic features of a tumor cell). In PCa cells with progressive malignant behaviors, they demonstrated that MOBI values (representative of OXPHOS activity) are high in premalignant prostate cells and significantly decrease with increasing malignancy [107].
Taken together, these observations support that PCa cells reprogram their metabolism towards the aerobic glycolysis (the Warburg effect) in the context of tumor progression. However, contrasting results, demonstrating that the high levels of OXPHOS activity observed in primary tumors still persists during the progression of the pathology towards its metastatic stage, have also been reported in the literature [136][137][138][139].
Galbraith and coworkers recently demonstrated an association between peroxisome proliferator-activated receptor gamma (PPARG) expression and metastatic features in PCa. By means of in vitro and in vivo studies, these authors could show that, in PCa cells, PPARG overexpression induces AKT3 expression leading to increased mitochondrial biogenesis and ATP production, finally fueling tumor cell epithelial-to-mesenchymal transition (EMT) and metastatic behavior [140]. Pyruvate dehydrogenase complex (PDC) is the multi-protein complex that catalyzes the conversion of pyruvate to acetyl-CoA, thus fostering mitochondrial activity. It was reported that, in PCa cells, knockout of the major subunit of PDC (PDHA1) is accompanied by lower levels of the TCA cycle activity, resulting in impaired OXPHOS activity and growth of these cells when xenografted in nude mice [137]. Pyruvate kinase isozyme 2 (PKM2) has been shown to be highly expressed in many types of cancer cells, including PCa cells. Interestingly, in these cells, PKM2 has been reported to be also deeply involved in glucose metabolism (OXPHOS activity) and to mediate proliferation, metastatic behavior and acquisition of stem cell properties [141][142][143]. Mitochondrial pyruvate carrier (MPC) is the hetero-dimeric complex (formed by MPC1 and MPC2) responsible for the import of pyruvate into the mitochondria where it is converted to acetyl-CoA and then further enters the TCA cycle to fuel the OXPHOS machinery. MPC2 expression was found to correlate with tumor aggressiveness in PCa specimens [144][145]. Transcriptional enhanced associate domain 4 (TEAD4) is a transcription factor previously shown to be involved in the regulation of the expression of mitochondrial genes involved in the OXPHOS pathways [146]. TEAD4 is expressed in PCa cells, and its expression has been reported to be critical in increasing OXPHOS activity. In a recent paper, Chen and coworkers reported that TEAD4 expression is epigenetically regulated by the semi-essential amino acid arginine to modulate OXPHOS functions in hormone-refractory PCa cells [147].
Interestingly, elevated OXPHOS and mitochondrial mass have been observed in the aggressive stem cell subpopulation of different tumors, including PCa [148]. Sotgia and coworkers proposed the development of a “mitochondrial based oncology platform” for specifically targeting CSC metabolism [149][150]. In line with this observation, metformin has been proposed as an effective anticancer agent based on its ability to specifically target OXPHOS and ATP production in prostate CSCs [151]. On the other hand, impaired mitochondrial OXPHOS and upregulated glycolysis were observed in these cells [152]. Thus, the presence of exacerbated OXPHOS in PCa stem cells still remains a controversial issue.
Given the crucial role of the tumor-stroma cross-talk in shaping cancer cell metabolism, Ippolito et al. investigated how CAFs might regulate mitochondrial dynamics in PCa cells. They found that tumor-associated CAFs significantly enhance mitochondrial respiration, mediated by a lactate shuttle, and favor mitochondria transfer in PCa cells, thus promoting their malignant behavior [46]. In line with these results, Grupp et al. demonstrated that a high mitochondria content in PCa specimens correlates positively with PCa progression and represents an effective predictor of a poor clinical prognosis and outcome [106]. Last but not least, a switch from glycolysis to OXPHOS activity has been observed in PCa cells undergoing the development of resistance to standard therapies (i.e., enzalutamide, docetaxel) [153].
Based on these observations, it is now accepted that targeting both glycolysis and mitochondrial OXPHOS pathway might represent an effective therapeutic strategy for advanced, metastatic and drug-resistant PCa [154].
A schematic overview of the metabolic rewiring occurring in prostate epithelial cells during the different stages of cancer progression is given in Figure 1.
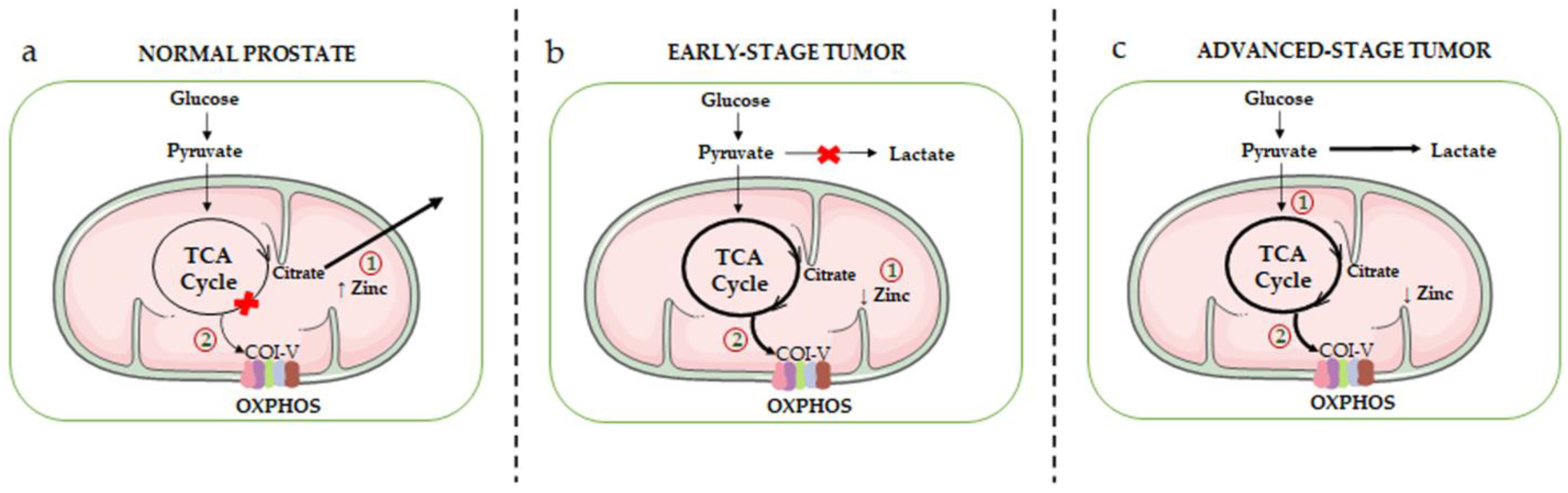
Figure 1. Schematic overview of the mitochondrial metabolic rewiring occurring in prostate epithelial cells during the different stages of cancer progression. (a) Healthy prostate epithelial cells accumulate high levels of zinc (due to the overexpression of its transporter ZIP1), resulting in the inhibition of mitochondrial m-aconitase, the key enzyme responsible for the citrate-isocitrate conversion in the TCA cycle (1). This inhibition ultimately leads to the truncation of the TCA cycle and to citrate accumulation and secretion. As a result, normal prostate epithelial cells are characterized by an inefficient OXPHOS machinery (complexes I-V, COI-V) (2). (b) In the early stage of tumor progression, intracellular zinc levels are significantly reduced (due to a decreased expression of its transporter) (1); this leads to the reactivation of m-aconitase, restoring the citrate-isocitrate conversion, and consequently of the TCA cycle and OXPHOS metabolic pathways (2). (c) In the advanced stage of tumor progression, PCa cells exhibit the Warburg effect, an active aerobic glycolysis accompanied by high levels of lactate production (1). However, it is now well established that a high activity of the TCA cycle/OXPHOS pathways still persists in these cells, and it is even exacerbated in PCa stem cells as well as in drug-resistant PCa cells (2).
2.1.2. The AR-Mitochondria Axis
ADT still represents the therapy of choice for early-stage, androgen-dependent PCa. However, most patients progress towards the aggressive CRPC stage characterized by a high rate of cell proliferation, invasiveness and metastatic behavior; interestingly, in most CRPC patients (about 80%) a reactivation of the androgen receptor (AR) has been observed. The persistent activity of AR in this stage has been shown to involve different mechanisms, including AR gene amplification, AR mutations, AR gene alternative splicing, generating different receptor splice variants and intratumoral synthesis of androgens. Since PCa progression is also associated with a peculiar metabolic reprogramming, as discussed above, it has been postulated that AR might be a master regulator of PCa cell metabolism. In line with this hypothesis, genomic, transcriptomic and metabolomic functional studies pointed out that AR regulates different metabolic pathways, including glucose uptake (through the induction of different glucose transporters), glycolysis, TCA cycle, mitochondrial biogenesis and respiration, de novo lipid synthesis and fatty acid β-oxidation [117][144][155][156][157][158].
Different intracellular signaling pathways were reported to be involved in this AR-driven metabolic reprogramming. Importantly, AR activation was demonstrated to induce mTOR translocation into the nucleus where it binds to the promoter regions of metabolic genes (such as HK2), thereby regulating their expression; accordingly, inhibition of the mTOR pathway resulted in impaired glycolytic activities and reduced proliferation in PCa cells [157]. In AR-expressing PCa cells, it was shown that androgens promote the activity of the AMPK/PGC1α signaling cascade, leading to increased glycolytic rates, mitochondrial biogenesis, OXPHOS, intracellular ATP levels and cell growth [156].
An additional interesting mediator of the AR metabolic activity is MPC (specifically the MPC2 isoform), reported to be highly expressed in AR-positive PCa cells (both hormone-dependent and CRPC cells) but almost absent in AR-negative PCa cells and to play a pivotal role in supporting a functional TCA cycle. Bader and coworkers demonstrated that MPC is transcriptionally upregulated by AR in PCa cells, and its inhibition impairs O2 consumption, TCA cycle metabolite levels and oxidative phosphorylation, thus halting cell proliferation. Moreover, these authors could show that targeting MPC with the MPC inhibitors UK5099 and MSDC0160 (this inhibitor is orally administered and clinically viable) results in the suppression of the growth of AR-expressing, but not AR-negative, PCa cells in in vitro and in vivo studies [117][144][159]. In line with these results, it has been reported that, in androgen-sensitive and CRPC cells, activation of the AR signaling upregulates the expression of DRP1 (the mediator of the mitochondrial fission) to induce the formation of the VDAC/MPC2 complex and thereby the pyruvate transport into the mitochondria and sustains mitochondrial metabolic pathways, such as OXPHOS.
2.1.3. mtDNA Mutations
So far, most studies addressing the relevance of genetic mutations in PCa development have been focused on the nuclear genome. Mitochondria, maternally inherited organelles, are deeply involved in the process of tumorigenesis by orchestrating metabolic and energy production pathways, ROS signaling and apoptosis [8][160][161]. Thus, dissecting the mitochondrial genome is currently considered an essential step to obtain a complete view of the genetic alteration profile in PCas.
The majority of the proteins of the four ETC complexes (COI-IV) involved in OXPHOS are encoded by nuclear DNA; however, 13 proteins in these complexes are encoded by mtDNA, the small circular DNA molecule found inside mitochondria. The mtDNA is characterized by a high mutation rate, mainly linked to high levels of ETC-derived ROS and a low efficient DNA repair system in these organelles [162]. Mutations of mtDNA have been found in different types of human cancers, including PCa [136][163][164][165][166][167], although their functional role still needs to be fully elucidated. Gomez-Zaera and coworkers analyzed the presence of mtDNA sequence variants in human PCa tissues; they reported that the most frequent variants were present in the following genes: mt-RNR2, encoding the large 16S mitochondrial ribosomal RNA (rRNA) subunit; mt-D-loop (displacement loop, control sites for the expression of the mitochondrial genome); and mt-ND4, encoding the protein NADH dehydrogenase 4, part of the COI of the ETC pathway [168].
The analysis of somatic mutations in tumor tissues from PCa patients pointed out their presence in genes coding for rRNA (mt-RNR1 and mt-RNR2), transfer RNA (tRNA) and the protein-coding gene mitochondrially encoded ATP synthase membrane subunit 6 (mt-ATP6), that encodes the ATP synthase Fo subunit 6 (or subunit/chain A). Moreover, somatic mutations in the entire mitochondrial genome were found to be associated with high PSA levels in PCa patients [169]. By using a yeast model organism, it was shown that the mutation mt-ATP6-P136S specifically found in PCa tissues positively correlates with tumor progression and may be involved in cancer cell escape from apoptosis [170]. Hopkins et al. reported that mutations in mitochondrial rRNA, tRNA as well as in the protein-coding genes mt-ATP6, mt-ND1, mt-ND2 and mitochondrially encoded cytochrome c oxidase I (mt-CO1), component of the complex IV, cytochrome c oxidase, the last enzyme in the mitochondrial electron transport chain which drives OXPHOS, are frequent in PCa tissues and are drivers of PCa aggressive behavior [167]. In line with these observations, mutations in genes encoding for proteins of the mitochondrial complex I (mt-NDs) were reported to be frequent in high grade PCa tissues and to be associated with a reduced activity of the NADH dehydrogenase pathway and an increased, compensatory, activity of the succinate-using FADH2 pathway [171]. Interestingly, Sun and coworkers demonstrated that the presence of a mutant mt-CO1 gene results in the resistance of PCa cells to the pro-death activity of simvastatin [172].
2.2. Mitochondrial Dynamics
It is now well established that alterations of the mitochondrial structural dynamics (biogenesis, fusion, fission and mitophagy) are deeply involved in the different steps of cancer growth, progression and development of drug resistance [16][60][62][173][174]. However, current data on the role of the mitochondrial structural alterations in PCa are still scanty.
PGC1α is the well-recognized master regulator of mitochondrial biogenesis [175]; it is also involved in the control of the mitochondrial fusion/fission balance by promoting fusion, through the activation of MFN1 and 2, and impairing DRP1 expression, through the binding to its promoter region [176]. The expression of this gene, together with the mitochondrial number, was found to be upregulated in tumors, including PCa, of African American cancer patients known to be exposed to a higher risk of cancer and mortality compared to European American patients [177]. PGC1α has been observed to be highly expressed in PCa cells harboring either deletion or mutation of the classic tumor suppressor protein p53, and its expression positively correlates with cancer cell metastatic behavior [178]. It has been demonstrated that, in CRPC PC3 cells, overexpression of p53 decreases the expression and activity (i.e., nuclear localization) of PGC-1α, thereby leading to a reduced mitochondrial mass and a significant change in the expression levels of genes and proteins involved in the fusion/fission balance [179]. More recently, Galbraith and coworkers reported that, in PCa cells, the activity of PGC1α is also regulated by the PPARG/AKT3 axis. Specifically, these authors found that, in CRPC cells, overexpression of the transcription factor PPARG induces the expression of the AKT3 kinase that, in turn, triggers the nuclear localization of PGC1α, thereby driving mitochondrial biogenesis and ATP production which may fuel the metastatic behavior of tumor cells [140].
Alterations of the fusion/fission balance have also shown to be deeply involved in tumorigenesis, although the data so far available on this issue in PCa cells are still limited. Generally, it is accepted that mitochondria fission, the division of mitochondria in smaller organelles, is a typical feature of cells undergoing apoptosis; moreover, this process foresees the translocation of the cytoplasmic DRP1 protein to the mitochondria where it interacts with its receptor FIS1. In PCa cells, it has been reported that the overload of Ca2+ in mitochondria triggers the interaction of DRP1 with FIS1, thereby leading to mitochondrial fragmentation and enhanced cell response to pro-apoptotic agents [180]. In line with these observations, researchers observed that, in CRPC cells, mitochondrial Ca2+ and ROS overload triggers mitochondrial fission and mitophagy to mediate the pro-death (apoptotic, paraptotic) activities of natural anticancer compounds [58]. Moreover, enhanced mitochondrial fusion, together with mutations of the complex I mtDNA, were found to be associated with PCa progression, as evaluated in cancer cell lines as well as in mice and human tissue samples [181].
On the other hand, mitochondrial fission has been recently reported to play a key role in the maintenance of stemness features in prostate CSCs. Specifically, Civenni and coworkers focused their attention on bromodomain and extra-terminal domain (BET) proteins, such as bromodomain containing 4 (BRD4), well known as epigenetic modifiers of gene transcription. These authors showed that the DRP1 receptor, and fission factor, MFF is upregulated in hormone-refractory human prostate tumors as well as in prostate CSCs. Moreover, they could show that BRD4 acts as a key driver of MFF transcription and, therefore, of mitochondrial fission, which is an essential biological event for the survival and self-renewal of CSCs; accordingly, they observed that the inhibition of the BRD4 activity and of the subsequent MFF transcription results in the accumulation of dysfunctional mitochondria and, consequently, in the acquisition of the senescent phenotype in these cells. Thus, mitochondrial fission is a crucial process for the maintenance of the self-renewal and tumorigenic potential of the CSC subpopulation in prostate tumors.
References
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530.
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314.
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337.
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874.
- Granchi, C.; Fancelli, D.; Minutolo, F. An update on therapeutic opportunities offered by cancer glycolytic metabolism. Bioorg. Med. Chem. Lett. 2014, 24, 4915–4925.
- Lu, J. The Warburg metabolism fuels tumor metastasis. Cancer Metastasis Rev. 2019, 38, 157–164.
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2020, 599, 1745–1757.
- Fontana, F.; Limonta, P. The multifaceted roles of mitochondria at the crossroads of cell life and death in cancer. Free. Radic. Biol. Med. 2021, 176, 203–221.
- Niu, D.; Wu, Y.; Lei, Z.; Zhang, M.; Xie, Z.; Tang, S. Lactic acid, a driver of tumor-stroma interactions. Int. Immunopharmacol. 2022, 106, 108597.
- Wang, X.; Liu, H.; Ni, Y.; Shen, P.; Han, X. Lactate shuttle: From substance exchange to regulatory mechanism. Hum. Cell 2022, 35, 1–14.
- Morandi, A.; Taddei, M.L.; Chiarugi, P.; Giannoni, E. Targeting the Metabolic Reprogramming That Controls Epithelial-to-Mesenchymal Transition in Aggressive Tumors. Front. Oncol. 2017, 7, 40.
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280.
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918.
- Cassim, S.; Vučetić, M.; Ždralević, M.; Pouyssegur, J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers 2020, 12, 1119.
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129.
- Grasso, D.; Zampieri, L.X.; Capelôa, T.; Van De Velde, J.A.; Sonveaux, P. Mitochondria in cancer. Cell Stress 2020, 4, 114–146.
- Roth, K.G.; Mambetsariev, I.; Kulkarni, P.; Salgia, R. The Mitochondrion as an Emerging Therapeutic Target in Cancer. Trends Mol. Med. 2020, 26, 119–134.
- Mosier, J.A.; Schwager, S.C.; Boyajian, D.A.; Reinhart-King, C.A. Cancer cell metabolic plasticity in migration and metastasis. Clin. Exp. Metastasis 2021, 38, 343–359.
- Tan, Y.Q.; Zhang, X.; Zhang, S.; Zhu, T.; Garg, M.; Lobie, P.E.; Pandey, V. Mitochondria: The metabolic switch of cellular oncogenic transformation. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2021, 1876, 188534.
- Zhang, X.; Su, Q.; Zhou, J.; Yang, Z.; Liu, Z.; Ji, L.; Gao, H.; Jiang, G. To betray or to fight? The dual identity of the mitochondria in cancer. Futur. Oncol. 2021, 17, 723–743.
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308.
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111.
- Scalise, M.; Pochini, L.; Galluccio, M.; Console, L.; Indiveri, C. Glutamine Transport and Mitochondrial Metabolism in Cancer Cell Growth. Front. Oncol. 2017, 7, 306.
- Herst, P.M.; Grasso, C.; Berridge, M.V. Metabolic reprogramming of mitochondrial respiration in metastatic cancer. Cancer Metastasis Rev. 2018, 37, 643–653.
- Bacci, M.; Lorito, N.; Ippolito, L.; Ramazzotti, M.; Luti, S.; Romagnoli, S.; Parri, M.; Bianchini, F.; Cappellesso, F.; Virga, F.; et al. Reprogramming of Amino Acid Transporters to Support Aspartate and Glutamate Dependency Sustains Endocrine Resistance in Breast Cancer. Cell Rep. 2019, 28, 104–118 e108.
- Lee, A.; Lau, P.; Kwan, Y.; Kong, S. Mitochondrial Fuel Dependence on Glutamine Drives Chemo-Resistance in the Cancer Stem Cells of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 3315.
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473.
- Icard, P.; Coquerel, A.; Wu, Z.; Gligorov, J.; Fuks, D.; Fournel, L.; Lincet, H.; Simula, L. Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update. Int. J. Mol. Sci. 2021, 22, 6587.
- Mortezaee, K. Redox tolerance and metabolic reprogramming in solid tumors. Cell Biol. Int. 2021, 45, 273–286.
- Oliveira, G.L.; Coelho, A.R.; Marques, R.; Oliveira, P.J. Cancer cell metabolism: Rewiring the mitochondrial hub. Biochim. et Biophys. Acta (BBA)—Mol. Basis Dis. 2020, 1867, 166016.
- Ippolito, L.; Marini, A.; Cavallini, L.; Morandi, A.; Pietrovito, L.; Pintus, G.; Giannoni, E.; Schrader, T.; Puhr, M.; Chiarugi, P.; et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget 2016, 7, 61890–61904.
- Pan, Y.; Cao, M.; Liu, J.; Yang, Q.; Miao, X.; Go, V.L.W.; Lee, P.W.N.; Xiao, G.G. Metabolic Regulation in Mitochondria and Drug Resistance. Adv. Exp. Med. Biol. 2017, 1038, 149–171.
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2019, 20, 3384.
- Salunkhe, S.; Mishra, S.V.; Ghorai, A.; Hole, A.; Chandrani, P.; Dutt, A.; Chilakapati, M.; Dutt, S. Metabolic rewiring in drug resistant cells exhibit higher OXPHOS and fatty acids as preferred major source to cellular energetics. Biochim. Biophys. Acta (BBA)—Bioenerg. 2020, 1861, 148300.
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634.
- Fiorillo, M.; Sotgia, F.; Lisanti, M.P. “Energetic” Cancer Stem Cells (e-CSCs): A New Hyper-Metabolic and Proliferative Tumor Cell Phenotype, Driven by Mitochondrial Energy. Front. Oncol. 2018, 8, 677.
- Bonekamp, N.A.; Peter, B.; Hillen, H.S.; Felser, A.; Bergbrede, T.; Choidas, A.; Horn, M.; Unger, A.; Di Lucrezia, R.; Atanassov, I.; et al. Small-molecule inhibitors of human mitochondrial DNA transcription. Nature 2020, 588, 712–716.
- Marzagalli, M.; Raimondi, M.; Fontana, F.; Marelli, M.M.; Moretti, R.M.; Limonta, P. Cellular and molecular biology of cancer stem cells in melanoma: Possible therapeutic implications. Semin. Cancer Biol. 2019, 59, 221–235.
- Sotgia, F.; Fiorillo, M.; Lisanti, M.P. Hallmarks of the cancer cell of origin: Comparisons with “energetic” cancer stem cells (e-CSCs). Aging 2019, 11, 1065–1068.
- García-Heredia, J.M.; Carnero, A. Role of Mitochondria in Cancer Stem Cell Resistance. Cells 2020, 9, 1693.
- Liu, G.; Luo, Q.; Li, H.; Liu, Q.; Ju, Y.; Song, G. Increased Oxidative Phosphorylation is Required for Stemness Maintenance in Liver Cancer Stem Cells from Hepatocellular Carcinoma Cell Line HCCLM3 Cells. Int. J. Mol. Sci. 2020, 21, 5276.
- Kaur, J.; Bhattacharyya, S. Cancer Stem Cells: Metabolic Characterization for Targeted Cancer Therapy. Front. Oncol. 2021, 11, 756888.
- Marzagalli, M.; Fontana, F.; Raimondi, M.; Limonta, P. Cancer Stem Cells—Key Players in Tumor Relapse. Cancers 2021, 13, 376.
- Zanotelli, M.R.; Goldblatt, Z.E.; Miller, J.P.; Bordeleau, F.; Li, J.; VanderBurgh, J.A.; Lampi, M.C.; King, M.R.; Reinhart-King, C.A. Regulation of ATP utilization during metastatic cell migration by collagen architecture. Mol. Biol. Cell 2018, 29, 1–9.
- Chiarugi, P.; Cirri, P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2016, 380, 272–280.
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355.
- Bacci, M.; Ippolito, L.; Magnelli, L.; Giannoni, E.; Chiarugi, P. Stromal-induced mitochondrial re-education: Impact on epithelial-to-mesenchymal transition and cancer aggressiveness. Semin. Cell Dev. Biol. 2020, 98, 71–79.
- Li, Z.; Sun, C.; Qin, Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics 2021, 11, 8322–8336.
- Modica-Napolitano, J.S.; Weissig, V. Treatment Strategies that Enhance the Efficacy and Selectivity of Mitochondria-Targeted Anticancer Agents. Int. J. Mol. Sci. 2015, 16, 17394–17421.
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490.
- Fontana, F.; Raimondi, M.; Marzagalli, M.; Di Domizio, A.; Limonta, P. Natural Compounds in Prostate Cancer Prevention and Treatment: Mechanisms of Action and Molecular Targets. Cells 2020, 9, 460.
- Lee, J.-S.; Lee, H.; Jang, H.; Woo, S.M.; Park, J.B.; Lee, S.-H.; Kang, J.H.; Kim, H.Y.; Song, J.; Kim, S.-Y. Targeting Oxidative Phosphorylation Reverses Drug Resistance in Cancer Cells by Blocking Autophagy Recycling. Cells 2020, 9, 2013.
- Iessi, E.; Vona, R.; Cittadini, C.; Matarrese, P. Targeting the Interplay between Cancer Metabolic Reprogramming and Cell Death Pathways as a Viable Therapeutic Path. Biomedicines 2021, 9, 1942.
- Raimondi, M.; Fontana, F.; Marzagalli, M.; Audano, M.; Beretta, G.; Procacci, P.; Sartori, P.; Mitro, N.; Limonta, P. Ca(2+) overload- and ROS-associated mitochondrial dysfunction contributes to delta-tocotrienol-mediated paraptosis in melanoma cells. Apoptosis 2021, 26, 277–292.
- Wu, Z.; Ho, W.S.; Lu, R. Targeting Mitochondrial Oxidative Phosphorylation in Glioblastoma Therapy. NeuroMol. Med. 2022, 24, 18–22.
- Yu, H.-J.; Xiao, G.-L.; Zhao, Y.-Y.; Wang, X.-X.; Lan, R. Targeting Mitochondrial Metabolism and RNA Polymerase POLRMT to Overcome Multidrug Resistance in Cancer. Front. Chem. 2021, 9, 775226.
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195.
- Fontana, F.; Raimondi, M.; Marzagalli, M.; Audano, M.; Beretta, G.; Procacci, P.; Sartori, P.; Mitro, N.; Limonta, P. Mitochondrial functional and structural impairment is involved in the antitumor activity of delta-tocotrienol in prostate cancer cells. Free. Radic. Biol. Med. 2020, 160, 376–390.
- Ma, Y.; Wang, L.; Jia, R. The role of mitochondrial dynamics in human cancers. Am. J. Cancer Res. 2020, 10, 1278–1293.
- Lee, Y.G.; Park, D.H.; Chae, Y.C. Role of Mitochondrial Stress Response in Cancer Progression. Cells 2022, 11, 771.
- Liu, H.; Zang, C.; Yuan, F.; Ju, C.; Shang, M.; Ning, J.; Yang, Y.; Ma, J.; Li, G.; Bao, X.; et al. The role of FUNDC1 in mitophagy, mitochondrial dynamics and human diseases. Biochem. Pharmacol. 2022, 197, 114891.
- Wang, S.; Tan, J.; Miao, Y.; Zhang, Q. Mitochondrial Dynamics, Mitophagy, and Mitochondria–Endoplasmic Reticulum Contact Sites Crosstalk Under Hypoxia. Front. Cell Dev. Biol. 2022, 10, 848214.
- Grasso, D.; Medeiros, H.; Zampieri, L.X.; Bol, V.; Danhier, P.; Van Gisbergen, M.W.; Bouzin, C.; Brusa, D.; Grégoire, V.; Smeets, H.; et al. Fitter Mitochondria Are Associated with Radioresistance in Human Head and Neck SQD9 Cancer Cells. Front. Pharmacol. 2020, 11, 263.
- Popov, L. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899.
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.-G. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944.
- Canto, C.; Auwerx, J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105.
- Bost, F.; Kaminski, L. The metabolic modulator PGC-1α in cancer. Am. J. Cancer Res. 2019, 9, 198–211.
- Zhang, Q.; Chen, W.; Xie, C.; Dai, X.; Ma, J.; Lu, J. The Role of PGC-1α in Digestive System Malignant Tumours. Anti-Cancer Agents Med. Chem. 2020, 20, 276–285.
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015, 6, 30472–30486.
- Raggi, C.; Taddei, M.L.; Sacco, E.; Navari, N.; Correnti, M.; Piombanti, B.; Pastore, M.; Campani, C.; Pranzini, E.; Iorio, J.; et al. Mitochondrial oxidative metabolism contributes to a cancer stem cell phenotype in cholangiocarcinoma. J. Hepatol. 2021, 74, 1373–1385.
- Praharaj, P.P.; Panigrahi, D.P.; Bhol, C.S.; Patra, S.; Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Singh, A.; Patil, S.; Bhutia, S.K. Mitochondrial rewiring through mitophagy and mitochondrial biogenesis in cancer stem cells: A potential target for anti-CSC cancer therapy. Cancer Lett. 2021, 498, 217–228.
- Brillo, V.; Chieregato, L.; Leanza, L.; Muccioli, S.; Costa, R. Mitochondrial Dynamics, ROS, and Cell Signaling: A Blended Overview. Life 2021, 11, 332.
- Chen, H.C.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200.
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural Basis of Mitochondrial Tethering by Mitofusin Complexes. Science 2004, 305, 858–862.
- Martens, S.; McMahon, H.T. Mechanisms of membrane fusion: Disparate players and common principles. Nat. Rev. Mol. Cell Biol. 2008, 9, 543–556.
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863.
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362.
- Lee, H.; Yoon, Y. Mitochondrial fission and fusion. Biochem. Soc. Trans. 2016, 44, 1725–1735.
- Steffen, J.; Koehler, C.M. ER–mitochondria contacts: Actin dynamics at the ER control mitochondrial fission via calcium release. J. Cell Biol. 2017, 217, 15–17.
- Banerjee, R.; Mukherjee, A.; Nagotu, S. Mitochondrial dynamics and its impact on human health and diseases: Inside the DRP1 blackbox. J. Mol. Med. 2021, 100, 1–21.
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2022, 72, 7–33.
- Labrie, F.; Candas, B.; Gomez, J.-L.; Cusan, L. Can combined androgen blockade provide long-term control or possible cure of localized prostate cancer? Urology 2002, 60, 115–119.
- James, N.D.; De Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.S.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med. 2017, 377, 338–351.
- Litwin, M.S.; Tan, H.-J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542.
- Onozawa, M.; Akaza, H.; Hinotsu, S.; Oya, M.; Ogawa, O.; Kitamura, T.; Suzuki, K.; Naito, S.; Namiki, M.; Nishimura, K.; et al. Combined androgen blockade achieved better oncological outcome in androgen deprivation therapy for prostate cancer: Analysis of community-based multi-institutional database across Japan using propensity score matching. Cancer Med. 2018, 7, 4893–4902.
- Armstrong, A.J.; Szmulewitz, R.Z.; Petrylak, D.P.; Holzbeierlein, J.; Villers, A.; Azad, A.; Alcaraz, A.; Alekseev, B.; Iguchi, T.; Shore, N.D.; et al. ARCHES: A Randomized, Phase III Study of Androgen Deprivation Therapy With Enzalutamide or Placebo in Men With Metastatic Hormone-Sensitive Prostate Cancer. J. Clin. Oncol. 2019, 37, 2974–2986.
- Chi, K.N.; Agarwal, N.; Bjartell, A.; Chung, B.H.; Pereira de Santana Gomes, A.J.; Given, R.; Juarez Soto, A.; Merseburger, A.S.; Ozguroglu, M.; Uemura, H.; et al. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2019, 381, 13–24.
- Kim, T.; Lee, Y.; Koo, K. Current Status and Future Perspectives of Androgen Receptor Inhibition Therapy for Prostate Cancer: A Comprehensive Review. Biomolecules 2021, 11, 492.
- Perner, S.; Cronauer, M.V.; Schrader, A.J.; Klocker, H.; Culig, Z.; Baniahmad, A. Adaptive responses of androgen receptor signaling in castration-resistant prostate cancer. Oncotarget 2015, 6, 35542–35555.
- Galletti, G.; Leach, B.I.; Lam, L.; Tagawa, S.T. Mechanisms of resistance to systemic therapy in metastatic castration-resistant prostate cancer. Cancer Treat. Rev. 2017, 57, 16–27.
- Marelli, M.M.; Moretti, R.M.; Januszkiewicz-Caulier, J.; Motta, M.; Limonta, P. Gonadotropin-Releasing Hormone (GnRH) Receptors in Tumors: A New Rationale for the Therapeutical Application of GnRH Analogs in Cancer Patients? Curr. Cancer Drug Targets 2006, 6, 257–269.
- Limonta, P.; Marelli, M.M.; Mai, S.; Motta, M.; Martini, L.; Moretti, R.M. GnRH Receptors in Cancer: From Cell Biology to Novel Targeted Therapeutic Strategies. Endocr. Rev. 2012, 33, 784–811.
- Limonta, P.; Manea, M. Gonadotropin-releasing hormone receptors as molecular therapeutic targets in prostate cancer: Current options and emerging strategies. Cancer Treat. Rev. 2013, 39, 647–663.
- Manea, M.; Marelli, M.M.; Moretti, R.; Maggi, R.; Marzagalli, M.; Limonta, P. Targeting Hormonal Signaling Pathways in Castration Resistant Prostate Cancer. Recent Patents Anti-Cancer Drug Discov. 2014, 9, 267–285.
- Jang, H.S.; Koo, K.C.; Cho, K.S.; Chung, B.H. Survival Outcomes of Concurrent Treatment with Docetaxel and Androgen Deprivation Therapy in Metastatic Castration-Resistant Prostate Cancer. Yonsei Med. J. 2016, 57, 1070–1078.
- Komura, K.; Sweeney, C.J.; Inamoto, T.; Ibuki, N.; Azuma, H.; Kantoff, P.W. Current treatment strategies for advanced prostate cancer. Int. J. Urol. 2018, 25, 220–231.
- Saad, F.; Shore, N.; Zhang, T.; Sharma, S.; Cho, H.K.; Jacobs, I.A. Emerging therapeutic targets for patients with advanced prostate cancer. Cancer Treat. Rev. 2019, 76, 1–9.
- Fontana, F.; Marzagalli, M.; Marelli, M.M.; Raimondi, M.; Moretti, R.; Limonta, P. Gonadotropin-Releasing Hormone Receptors in Prostate Cancer: Molecular Aspects and Biological Functions. Int. J. Mol. Sci. 2020, 21, 9511.
- Fontana, F.; Limonta, P. Dissecting the Hormonal Signaling Landscape in Castration-Resistant Prostate Cancer. Cells 2021, 10, 1133.
- Hou, Z.; Huang, S.; Li, Z. Androgens in prostate cancer: A tale that never ends. Cancer Lett. 2021, 516, 1–12.
- Fontana, F.; Anselmi, M.; Limonta, P. Molecular mechanisms and genetic alterations in prostate cancer: From diagnosis to targeted therapy. Cancer Lett. 2022, 534, 215619.
- Grigor, E.J.; Fergusson, D.; Kekre, N.; Montroy, J.; Atkins, H.; Seftel, M.D.; Daugaard, M.; Presseau, J.; Thavorn, K.; Hutton, B.; et al. Risks and Benefits of Chimeric Antigen Receptor T-Cell (CAR-T) Therapy in Cancer: A Systematic Review and Meta-Analysis. Transfus. Med. Rev. 2019, 33, 98–110.
- Markowski, M.C.; Shenderov, E.; Eisenberger, M.A.; Kachhap, S.; Pardoll, D.M.; Denmeade, S.R.; Antonarakis, E.S. Extreme responses to immune checkpoint blockade following bipolar androgen therapy and enzalutamide in patients with metastatic castration resistant prostate cancer. Prostate 2020, 80, 407–411.
- Sun, B.L. Immunotherapy in treatment of metastatic prostate cancer: An approach to circumvent immunosuppressive tumor microenvironment. Prostate 2021, 81, 1125–1134.
- Siewe, N.; Friedman, A. Combination therapy for mCRPC with immune checkpoint inhibitors, ADT and vaccine: A mathematical model. PLoS ONE 2022, 17, e0262453.
- Grupp, K.; Jedrzejewska, K.; Tsourlakis, M.C.; Koop, C.; Wilczak, W.; Adam, M.; Quaas, A.; Sauter, G.; Simon, R.; Izbicki, J.R.; et al. High mitochondria content is associated with prostate cancer disease progression. Mol. Cancer 2013, 12, 145.
- Vayalil, P.K.; Landar, A. Mitochondrial oncobioenergetic index: A potential biomarker to predict progression from indolent to aggressive prostate cancer. Oncotarget 2015, 6, 43065–43080.
- Kelly, R.S.; Sinnott, J.A.; Rider, J.R.; Ebot, E.M.; Gerke, T.; Bowden, M.; Pettersson, A.; Loda, M.; Sesso, H.D.; Kantoff, P.W.; et al. The role of tumor metabolism as a driver of prostate cancer progression and lethal disease: Results from a nested case-control study. Cancer Metab. 2016, 4, 22.
- Eidelman, E.; Twum-Ampofo, J.; Ansari, J.; Siddiqui, M.M. The Metabolic Phenotype of Prostate Cancer. Front. Oncol. 2017, 7, 131.
- Costello, L.C.; Feng, P.; Milon, B.; Tan, M.; Franklin, R.B. Role of zinc in the pathogenesis and treatment of prostate cancer: Critical issues to resolve. Prostate Cancer Prostatic Dis. 2004, 7, 111–117.
- Costello, L.C.; Franklin, R.B. The clinical relevance of the metabolism of prostate cancer; zinc and tumor suppression: Connecting the dots. Mol. Cancer 2006, 5, 17.
- Costello, L.C.; Franklin, R.B.; Zou, J.; Feng, P.; Bok, R.; Swanson, M.G.; Kurhanewicz, J. Human prostate cancer ZIP1/zinc/citrate genetic/metabolic relationship in the TRAMP prostate cancer animal model. Cancer Biol. Ther. 2011, 12, 1078–1084.
- Costello, L.C.; Franklin, R.B. A comprehensive review of the role of zinc in normal prostate function and metabolism; and its implications in prostate cancer. Arch. Biochem. Biophys. 2016, 611, 100–112.
- Hacioglu, C.; Kacar, S.; Kar, F.; Kanbak, G.; Sahinturk, V. Concentration-Dependent Effects of Zinc Sulfate on DU-145 Human Prostate Cancer Cell Line: Oxidative, Apoptotic, Inflammatory, and Morphological Analyzes. Biol. Trace Element Res. 2020, 195, 436–444.
- Latonen, L.; Afyounian, E.; Jylhä, A.; Nättinen, J.; Aapola, U.; Annala, M.; Kivinummi, K.K.; Tammela, T.T.L.; Beuerman, R.W.; Uusitalo, H.; et al. Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression. Nat. Commun. 2018, 9, 1176.
- Shao, Y.; Ye, G.; Ren, S.; Piao, H.-L.; Zhao, X.; Lu, X.; Wang, F.; Ma, W.; Li, J.; Yin, P.; et al. Metabolomics and transcriptomics profiles reveal the dysregulation of the tricarboxylic acid cycle and related mechanisms in prostate cancer. Int. J. Cancer 2018, 143, 396–407.
- Bader, D.A.; McGuire, S.E. Tumour metabolism and its unique properties in prostate adenocarcinoma. Nat. Rev. Urol. 2020, 17, 214–231.
- Kolenko, V.; Teper, E.; Kutikov, A.; Uzzo, R. Zinc and zinc transporters in prostate carcinogenesis. Nat. Rev. Urol. 2013, 10, 219–226.
- Franz, M.-C.; Anderle, P.; Bürzle, M.; Suzuki, Y.; Freeman, M.; Hediger, M.; Kovacs, G. Zinc transporters in prostate cancer. Mol. Asp. Med. 2013, 34, 735–741.
- Makhov, P.B.; Golovine, K.V.; Kutikov, A.; Canter, D.J.; Rybko, V.A.; Roshchin, D.A.; Matveev, V.B.; Uzzo, R.G.; Kolenko, V.M. Reversal of epigenetic silencing of AP-2alpha results in increased zinc uptake in DU-145 and LNCaP prostate cancer cells. Carcinogenesis 2011, 32, 1773–1781.
- Xue, Y.N.; Yu, B.B.; Liu, Y.N.; Guo, R.; Li, L.J.; Zhang, L.C.; Su, J.; Sun, L.K.; Li, Y. Zinc promotes prostate cancer cell chemosensitivity to paclitaxel by inhibiting epithelial-mesenchymal transition and inducing apoptosis. Prostate 2019, 79, 647–656.
- Sauer, A.K.; Vela, H.; Vela, G.; Stark, P.; Barrera-Juarez, E.; Grabrucker, A.M. Zinc Deficiency in Men Over 50 and Its Implications in Prostate Disorders. Front. Oncol. 2020, 10, 1293.
- Ahmad, F.; Cherukuri, M.K.; Choyke, P.L. Metabolic reprogramming in prostate cancer. Br. J. Cancer 2021, 125, 1185–1196.
- Bazylianska, V.; Kalpage, H.A.; Wan, J.; Vaishnav, A.; Mahapatra, G.; Turner, A.A.; Chowdhury, D.D.; Kim, K.; Morse, P.T.; Lee, I.; et al. Lysine 53 Acetylation of Cytochrome c in Prostate Cancer: Warburg Metabolism and Evasion of Apoptosis. Cells 2021, 10, 802.
- Fang, M.; Shen, Z.; Huang, S.; Zhao, L.; Chen, S.; Mak, T.W.; Wang, X. The ER UDPase ENTPD5 Promotes Protein N-Glycosylation, the Warburg Effect, and Proliferation in the PTEN Pathway. Cell 2010, 143, 711–724.
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134.
- Wang, L.; Xiong, H.; Wu, F.; Zhang, Y.; Wang, J.; Zhao, L.; Guo, X.; Chang, L.-J.; Zhang, Y.; You, M.J.; et al. Hexokinase 2-Mediated Warburg Effect Is Required for PTEN- and p53-Deficiency-Driven Prostate Cancer Growth. Cell Rep. 2014, 8, 1461–1474.
- Singh, R.; Mills, I.G. The Interplay Between Prostate Cancer Genomics, Metabolism, and the Epigenome: Perspectives and Future Prospects. Front. Oncol. 2021, 11, 704353.
- Wang, C.; Tao, W.; Ni, S.; Chen, Q. SENP1 Interacts with HIF1α to Regulate Glycolysis of Prostatic Carcinoma Cells. Int. J. Biol. Sci. 2019, 15, 395–403.
- Sun, X.; Huang, Q.; Peng, F.; Wang, J.; Zhao, W.; Guo, G. Expression and Clinical Significance of HKII and HIF-1α in Grade Groups of Prostate Cancer. Front. Genet. 2021, 12, 680928.
- Nassar, Z.D.; Aref, A.T.; Miladinovic, D.; Mah, C.Y.; Raj, G.V.; Hoy, A.J.; Butler, L.M. Peri-prostatic adipose tissue: The metabolic microenvironment of prostate cancer. BJU Int. 2018, 121 (Suppl. S3), 9–21.
- Himbert, C.; Delphan, M.; Scherer, D.; Bowers, L.W.; Hursting, S.; Ulrich, C.M. Signals from the Adipose Microenvironment and the Obesity–Cancer Link—A Systematic Review. Cancer Prev. Res. 2017, 10, 494–506.
- Diedrich, J.D.; Rajagurubandara, E.; Herroon, M.K.; Mahapatra, G.; Hüttemann, M.; Podgorski, I. Bone marrow adipocytes promote the Warburg phenotype in metastatic prostate tumorsviaHIF-1α activation. Oncotarget 2016, 7, 64854–64877.
- Cutruzzolà, F.; Giardina, G.; Marani, M.; Macone, A.; Paiardini, A.; Rinaldo, S.; Paone, A. Glucose Metabolism in the Progression of Prostate Cancer. Front. Physiol. 2017, 8, 97.
- Fontana, F.; Anselmi, M.; Carollo, E.; Sartori, P.; Procacci, P.; Carter, D.; Limonta, P. Adipocyte-Derived Extracellular Vesicles Promote Prostate Cancer Cell Aggressiveness by Enabling Multiple Phenotypic and Metabolic Changes. Cells 2022, 11, 2388.
- Dakubo, G.D.; Parr, R.L.; Costello, L.C.; Franklin, R.B.; Thayer, R.E. Altered metabolism and mitochondrial genome in prostate cancer. J. Clin. Pathol. 2006, 59, 10–16.
- Chen, J.; Guccini, I.; Di Mitri, D.; Brina, D.; Revandkar, A.; Sarti, M.; Pasquini, E.; Alajati, A.; Pinton, S.; Losa, M.; et al. Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat. Genet. 2018, 50, 219–228.
- Oberhuber, M.; Pecoraro, M.; Rusz, M.; Oberhuber, G.; Wieselberg, M.; Haslinger, P.; Gurnhofer, E.; Schlederer, M.; Limberger, T.; Lagger, S.; et al. STAT 3 -dependent analysis reveals PDK 4 as independent predictor of recurrence in prostate cancer. Mol. Syst. Biol. 2020, 16, e9247.
- Chen, C.-L.; Lin, C.-Y.; Kung, H.-J. Targeting Mitochondrial OXPHOS and Their Regulatory Signals in Prostate Cancers. Int. J. Mol. Sci. 2021, 22, 13435.
- Galbraith, L.C.A.; Mui, E.; Nixon, C.; Hedley, A.; Strachan, D.; MacKay, G.; Sumpton, D.; Sansom, O.J.; Leung, H.Y.; Ahmad, I. PPAR-gamma induced AKT3 expression increases levels of mitochondrial biogenesis driving prostate cancer. Oncogene 2021, 40, 2355–2366.
- Giannoni, E.; Taddei, M.L.; Morandi, A.; Comito, G.; Calvani, M.; Bianchini, F.; Richichi, B.; Raugei, G.; Wong, N.; Tang, D.; et al. Targeting stromal-induced pyruvate kinase M2 nuclear translocation impairs OXPHOS and prostate cancer metastatic spread. Oncotarget 2015, 6, 24061–24074.
- Dayton, T.L.; Jacks, T.; Vander Heiden, M.G. PKM 2, cancer metabolism, and the road ahead. EMBO Rep. 2016, 17, 1721–1730.
- Hsu, M.-C.; Hung, W.-C. Pyruvate kinase M2 fuels multiple aspects of cancer cells: From cellular metabolism, transcriptional regulation to extracellular signaling. Mol. Cancer 2018, 17, 35.
- Bader, D.A.; Hartig, S.M.; Putluri, V.; Foley, C.; Hamilton, M.P.; Smith, E.A.; Saha, P.K.; Panigrahi, A.; Walker, C.; Zong, L.; et al. Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nat. Metab. 2018, 1, 70–85.
- Lee, Y.G.; Nam, Y.; Shin, K.J.; Yoon, S.; Park, W.S.; Joung, J.Y.; Seo, J.K.; Jang, J.; Lee, S.; Nam, D.; et al. Androgen-induced expression of DRP1 regulates mitochondrial metabolic reprogramming in prostate cancer. Cancer Lett. 2020, 471, 72–87.
- Kumar, R.P.; Ray, S.; Home, P.; Saha, B.; Bhattacharya, B.; Wilkins, H.M.; Chavan, H.; Ganguly, A.; Milano-Foster, J.; Paul, A.; et al. Regulation of energy metabolism during early mammalian development: TEAD4 controls mitochondrial transcription. Development 2018, 145, dev162644.
- Chen, C.-L.; Hsu, S.-C.; Chung, T.-Y.; Chu, C.-Y.; Wang, H.-J.; Hsiao, P.-W.; Yeh, S.-D.; Ann, D.K.; Yen, Y.; Kung, H.-J. Arginine is an epigenetic regulator targeting TEAD4 to modulate OXPHOS in prostate cancer cells. Nat. Commun. 2021, 12, 2398.
- Skvortsov, S.; Skvortsova, I.-I.; Tang, D.G.; Dubrovska, A. Concise Review: Prostate Cancer Stem Cells: Current Understanding. Stem Cells 2018, 36, 1457–1474.
- Sotgia, F.; Martinez-Outschoorn, U.E.; Lisanti, M.P. Cancer Metabolism: New Validated Targets for Drug Discovery. Oncotarget 2013, 4, 1309–1316.
- Sotgia, F.; Ozsvari, B.; Fiorillo, M.; De Francesco, E.M.; Bonuccelli, G.; Lisanti, M.P. A mitochondrial based oncology platform for targeting cancer stem cells (CSCs): MITO-ONC-RX. Cell Cycle 2018, 17, 2091–2100.
- Mayer, M.J.; Klotz, L.H.; Venkateswaran, V. Metformin and prostate cancer stem cells: A novel therapeutic target. Prostate Cancer Prostatic Dis. 2015, 18, 303–309.
- Zhong, Y.; Li, X.; Yu, D.; Li, X.; Li, Y.; Long, Y.; Yuan, Y.; Ji, Z.; Zhang, M.; Wen, J.-G.; et al. Application of mitochondrial pyruvate carrier blocker UK5099 creates metabolic reprogram and greater stem-like properties in LnCap prostate cancer cells in vitro. Oncotarget 2015, 6, 37758–37769.
- Basu, H.S.; Wilganowski, N.; Robertson, S.; Reuben, J.M.; Cohen, E.N.; Zurita, A.; Ramachandran, S.; Xiao, L.; Titus, M.; Wilding, G. Prostate cancer cells survive anti-androgen and mitochondrial metabolic inhibitors by modulating glycolysis and mitochondrial metabolic activities. Prostate 2021, 81, 799–811.
- Mamouni, K.; Kallifatidis, G.; Lokeshwar, B. Targeting Mitochondrial Metabolism in Prostate Cancer with Triterpenoids. Int. J. Mol. Sci. 2021, 22, 2466.
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733.
- Tennakoon, J.B.; Shi, Y.; Han, J.J.; Tsouko, E.; White, M.A.; Burns, A.R.; Zhang, A.; Xia, X.; Ilkayeva, O.R.; Xin, L.; et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1α-mediated metabolic switch. Oncogene 2013, 33, 5251–5261.
- Audet-Walsh, É.; Vernier, M.; Yee, T.; E Laflamme, C.; Li, S.; Chen, Y.; Giguère, V. SREBF1 Activity Is Regulated by an AR/mTOR Nuclear Axis in Prostate Cancer. Mol. Cancer Res. 2018, 16, 1396–1405.
- Gonthier, K.; Poluri, R.T.K.; Audet-Walsh, E. Functional genomic studies reveal the androgen receptor as a master regulator of cellular energy metabolism in prostate cancer. J. Steroid Biochem. Mol. Biol. 2019, 191, 105367.
- Stone, L. Mitochondrial metabolism: A target in AR-driven disease. Nat. Rev. Urol. 2019, 16, 1.
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698.
- Valcarcel-Jimenez, L.; Gaude, E.; Torrano, V.; Frezza, C.; Carracedo, A. Mitochondrial Metabolism: Yin and Yang for Tumor Progression. Trends Endocrinol. Metab. 2017, 28, 748–757.
- Quinlan, C.L.; Treberg, J.R.; Perevoshchikova, I.V.; Orr, A.L.; Brand, M.D. Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free. Radic. Biol. Med. 2012, 53, 1807–1817.
- Chen, J.Z.; Gokden, N.; Greene, G.F.; Mukunyadzi, P.; Kadlubar, F.F. Extensive somatic mitochondrial mutations in primary prostate cancer using laser capture microdissection. Cancer Res. 2002, 62, 6470–6474.
- Chen, J.Z.; Kadlubar, F.F.; Chen, J.Z. Mitochondrial Mutagenesis and Oxidative Stress in Human Prostate Cancer. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2004, 22, 1–12.
- Lindberg, J.; Mills, I.G.; Klevebring, D.; Liu, W.; Neiman, M.; Xu, J.; Wikström, P.; Wiklund, P.; Wiklund, F.; Egevad, L.; et al. The Mitochondrial and Autosomal Mutation Landscapes of Prostate Cancer. Eur. Urol. 2013, 63, 702–708.
- McCrow, J.P.; Petersen, D.C.; Louw, M.; Chan, E.K.F.; Harmeyer, K.; Vecchiarelli, S.; Lyons, R.J.; Bornman, M.S.R.; Hayes, V.M. Spectrum of mitochondrial genomic variation and associated clinical presentation of prostate cancer in South African men. Prostate 2016, 76, 349–358.
- Hopkins, J.F.; Sabelnykova, V.Y.; Weischenfeldt, J.; Simon, R.; Aguiar, J.A.; Alkallas, R.; Heisler, L.E.; Zhang, J.; Watson, J.D.; Chua, M.L.K.; et al. Mitochondrial mutations drive prostate cancer aggression. Nat. Commun. 2017, 8, 656.
- Gómez-Zaera, M.; Abril, J.; González, L.; Aguiló, F.; Condom, E.; Nadal, M.; Nunes, V. Identification of somatic and germline mitochondrial DNA sequence variants in prostate cancer patients. Mutat. Res. 2006, 595, 42–51.
- Kloss-Brandstätter, A.; Schäfer, G.; Erhart, G.; Hüttenhofer, A.; Coassin, S.; Seifarth, C.; Summerer, M.; Bektic, J.; Klocker, H.; Kronenberg, F. Somatic Mutations throughout the Entire Mitochondrial Genome Are Associated with Elevated PSA Levels in Prostate Cancer Patients. Am. J. Hum. Genet. 2010, 87, 802–812.
- Niedzwiecka, K.; Tisi, R.; Penna, S.; Lichocka, M.; Plochocka, D.; Kucharczyk, R. Two mutations in mitochondrial ATP6 gene of ATP synthase, related to human cancer, affect ROS, calcium homeostasis and mitochondrial permeability transition in yeast. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2018, 1865, 117–131.
- Schöpf, B.; Weissensteiner, H.; Schäfer, G.; Fazzini, F.; Charoentong, P.; Naschberger, A.; Rupp, B.; Fendt, L.; Bukur, V.; Giese, I.; et al. OXPHOS remodeling in high-grade prostate cancer involves mtDNA mutations and increased succinate oxidation. Nat. Commun. 2020, 11, 1487.
- Sun, Q.; Arnold, R.S.; Sun, C.Q.; Petros, J.A. A mitochondrial DNA mutation influences the apoptotic effect of statins on prostate cancer. Prostate 2015, 75, 1916–1925.
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824.
- Audano, M.; Pedretti, S.; Ligorio, S.; Crestani, M.; Caruso, D.; De Fabiani, E.; Mitro, N. “The Loss of Golden Touch”: Mitochondria-Organelle Interactions, Metabolism, and Cancer. Cells 2020, 9, 2519.
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124.
- Dabrowska, A.; Venero, J.L.; Iwasawa, R.; Hankir, M.-K.; Rahman, S.; Boobis, A.; Hajji, N. PGC-1α controls mitochondrial biogenesis and dynamics in lead-induced neurotoxicity. Aging 2015, 7, 629–647.
- Piyarathna, D.W.B.; Balasubramanian, A.; Arnold, J.M.; Lloyd, S.M.; Karanam, B.; Castro, P.; Ittmann, M.M.; Putluri, N.; Navone, N.; Jones, J.A.; et al. ERR1- and PGC1α-associated mitochondrial alterations correlate with pan-cancer disparity in African Americans. J. Clin. Investig. 2019, 129, 2351–2356.
- Shiota, M.; Yokomizo, A.; Tada, Y.; Inokuchi, J.; Tatsugami, K.; Kuroiwa, K.; Uchiumi, T.; Fujimoto, N.; Seki, N.; Naito, S. Peroxisome Proliferator-Activated Receptor γ Coactivator-1α Interacts with the Androgen Receptor (AR) and Promotes Prostate Cancer Cell Growth by Activating the AR. Mol. Endocrinol. 2010, 24, 114–127.
- Li, J.; Li, Y.; Chen, L.; Yu, B.; Xue, Y.; Guo, R.; Su, J.; Liu, Y.; Sun, L. p53/PGC-1α-mediated mitochondrial dysfunction promotes PC3 prostate cancer cell apoptosis. Mol. Med. Rep. 2020, 22, 155–164.
- Kaddour-Djebbar, I.; Choudhary, V.; Brooks, C.; Ghazaly, T.; Lakshmikanthan, V.; Dong, Z.; Kumar, M.V. Specific mitochondrial calcium overload induces mitochondrial fission in prostate cancer cells. Int. J. Oncol. 2010, 36, 1437–1444.
- Philley, J.V.; Kannan, A.; Qin, W.; Sauter, E.R.; Ikebe, M.; Hertweck, K.L.; Troyer, D.A.; Semmes, O.J.; Dasgupta, S. Complex-I Alteration and Enhanced Mitochondrial Fusion Are Associated With Prostate Cancer Progression. J. Cell. Physiol. 2016, 231, 1364–1374.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
696
Revisions:
2 times
(View History)
Update Date:
08 Mar 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No