Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ahmad Machmouchi | -- | 1957 | 2023-03-03 10:00:00 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Machmouchi, A.; Chehade, L.; Temraz, S.; Shamseddine, A. EGFR Resistance in Metastatic Colorectal Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/41845 (accessed on 05 July 2026).
Machmouchi A, Chehade L, Temraz S, Shamseddine A. EGFR Resistance in Metastatic Colorectal Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/41845. Accessed July 05, 2026.
Machmouchi, Ahmad, Laudy Chehade, Sally Temraz, Ali Shamseddine. "EGFR Resistance in Metastatic Colorectal Cancer" Encyclopedia, https://encyclopedia.pub/entry/41845 (accessed July 05, 2026).
Machmouchi, A., Chehade, L., Temraz, S., & Shamseddine, A. (2023, March 03). EGFR Resistance in Metastatic Colorectal Cancer. In Encyclopedia. https://encyclopedia.pub/entry/41845
Machmouchi, Ahmad, et al. "EGFR Resistance in Metastatic Colorectal Cancer." Encyclopedia. Web. 03 March, 2023.
Copy Citation
Overcoming EGFR Resistance in Metastatic Colorectal Cancer (mCRC) Using Vitamin C.
vitamin C
ascorbic acid
colorectal cancer
EGFR Resistance
1. Introduction
Many efforts have been made with the aim of finding the optimal treatment plan to improve the prognosis of CRC. Specifically, in RAS, BRAF wild and MSS tumors, cytotoxic/cytostatic chemotherapy (5-FU), Vascular Endothelial Growth Factor Inhibitor (VEGF), and Multi-Kinase targeted agents, in addition to targeted monoclonal antibodies (against Epidermal Growth Factor Receptor (EGFR), have been widely used as the leading treatment modality against metastatic CRC (mCRC) [2,3]. Indeed, EGFR targeted therapy was found to increase overall survival by 10–20% in colorectal cancer [4]. However, resistance to this therapy was inevitable with the emergence of KRAS and BRAF mutations, driven by intrinsic and extrinsic mechanisms affecting both cellular pathways and tumor microenvironment, respectively.
2. Mechanisms of EGFR Resistance in mCRC
Numerous therapeutic strategies have been conducted and investigated to overcome the resistance to anti-EGFR mAbs.
Intrinsic mechanisms include the activation of RAS/RAF/MEK/ERK and PI3K/AKT/mTOR cascades through genomic alterations and protein phosphorylation (as shown in Figure 1). Furthermore, compensatory feedback loop signaling of EGFR is stimulated by ERBB2/MET amplification and abnormal IGF-1R activation. In addition, epithelial-to-mesenchymal transition, glycolysis, lipid synthesis, fatty acid oxidation, and vitamin deficiency are also contributors to resistance [11].
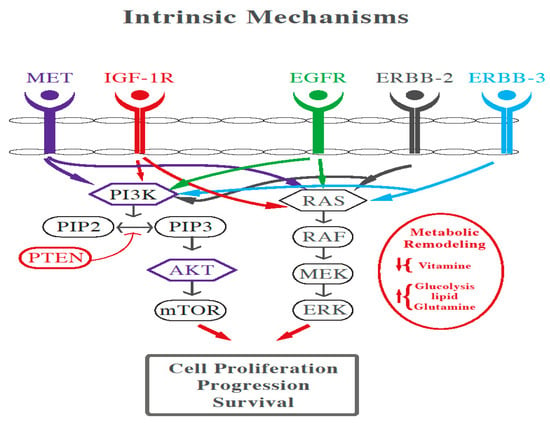
Figure 1. Cross talk between MAPK, PI3K, and Wnt pathway in CRC.
On the other hand, the tumor microenvironment also plays a role in conferring extrinsic resistance to anti-EGFR therapy. This can include dysfunction of natural killer (NK) cells and macrophages that decrease the anti-EGFR antibody-dependent cellular cytotoxicity, and decreased density of effector T-cells and increased PD-L1 expression which assist cancer survival. Other factors leading to drug resistance are cancer-associated fibroblasts secreting mitogenic growth factors that activate RAS or MET pathways, in addition to abnormal angiogenesis [11].
Another aspect leading to acquired resistance in mCRC is caused by the emergence of a heterogeneous resistant population of surviving clones (i.e., persister cells, drug-sensitive RAS/BRAF wild type cells which were not eliminated by anti-EGFR targeted therapies) characterized by a drug tolerant state instigated by prolonged drug exposure, relying on different mechanisms, either genetic or non-genetic [12].
3. The Role of High Dose Vitamin C in Cancer
3.1. Vitamin C Bioavailability and Requirements
Oral vitamin C produces tissue and plasma concentrations that the body tightly controls [13,14]. Normally, total body content of vitamin C ranges from 300 mg (at near scurvy) to about 2 g [14,15]. Plasma concentration of vitamin C is tightly controlled, and generally does not surpass 100 μM. Roughly, at moderate intakes of 30–180 mg/day, 70–90% of vitamin C is absorbed [15]. However, at doses above 1 g/day, absorption decreases by less than 50% and absorbed ascorbic acid is excreted in the urine [14,16]. However, when vitamin C is administered by IV route, it bypasses the gastrointestinal regulation and attains a dose-dependent plasma concentration. Nonetheless, its half-life is less than 2 h [17].
3.2. High Dose Vitamin C in Cancer Clinical Trials
Several clinical trials were conducted to test the safety of high dose vitamin C as monotherapy or in combination with other chemotherapeutic agents, as well as to determine the maximal tolerable dose that could be used in phase II and III trials. For instance, Wang et al., administered vitamin C with FOLFOX or FOLFIRI with or without bevacizumab to patients with colorectal or gastric cancer. The study reported no drug limiting toxicity and showed decreased hematological and gastrointestinal toxicities compared to other trials employing the same chemotherapy regimens for these two cancers [28,29,30,31,32]. Monti et al., administered vitamin C to 14 stage IV patients with pancreatic cancer receiving gemcitabine and erlotinib. Eight patients had a decrease in the tumor size, seven had a stable disease and two had disease progression, although progression free survival and overall survival were comparable to those on gemcitabine/erlotinib alone [33]. In two other studies also on pancreatic cancer, vitamin C was found to decrease the rate of severe toxicity in patients receiving gemcitabine 500 mg/m, irinotecan 80 mg/m, leucovorin 300 mg, 5-fluorouracil (5-FU) 400 mg/m, and gemcitabine monotherapy [34,35]. Additional studies investigating the efficacy of vitamin C in advanced cancers did not show objective tumor response [26,27]. With respect to toxicity, mild side effects were recorded in some trials, mainly due to the osmotic load of the vitamin C infusion, and were reversible with adequate hydration [26,33], and one study reported kidney stone formation and hypokalemia as possibly related to vitamin C [36]. More severe side effects in these trials were related to the administration of chemotherapy. Furthermore, caution should be made in patients with G6PD deficiency, as high dose vitamin C can induce hemolysis, and patients should be screened before administration of IV vitamin C [37]. Results from several studies demonstrated that the optimal dose of IV vitamin C that could be adopted in phase II trials was 1.5 g/kg or 70 to 80 g/m2 [32].
3.3. Role of Vitamin C in KRAS and BRAF Mutated Colorectal Cancer
RAS mutations are present in around 40% of mCRC, while BRAF mutations account for 10% [38]. Both mutations have been used as predictors of resistance to EGFR targeting drugs. In fact, testing for these mutations on tissue specimen of mCRC patients before the initiation of anti-EGFR therapy has become mandatory, particularly since resistance may be present originally or even may develop during the treatment in initially wild type patients; a phenomenon known as acquired (or secondary) resistance [39]. In order to identify these mutations, tissue and liquid biopsy method can be used as an analytical technique to detect tumor-derived biomarkers in body fluids such as circulating tumor DNA (ctDNA) [40]. Indeed, the detection of ctDNA released by cancer cells provides valuable information in relation to the prognosis and prediction of therapeutic resistance or sensitivity. Undeniably, improving detection of KRAS/BRAF mutations at different time points, enhancing correlation between its levels and survival and monitoring its response to therapy.
Furthermore, KRAS and BRAF mutations correlate with GLUT1 overexpression by cancer cells and excessive dependence on aerobic glycolysis as an energy source [41]. Aerobic glycolysis, also known as the Warburg effect, is a hallmark of cancer, in which glucose is converted to lactate despite the availability of oxygen. This is because pyruvate, the end product of glycolysis, is diverted from the mitochondria as a result of transcriptional activation of pyruvate dehydrogenase kinase 1, which in turn inactivates pyruvate dehydrogenase. As a result, the conversion of pyruvate to acetyl-CoA is hindered, and pyruvate is diverted into the cytosol where it is converted to lactate [42]. Although the shift from oxidative phosphorylation to aerobic glycolysis may seem to generate less energy per mol of glucose, the latter process is around 10 to 100 times faster and thus generates more ATP per unit time compared to oxidative phosphorylation [43,44]. Even though aerobic glycolysis is not exclusive to cancer cells and occurs in normal rapidly growing cells [45,46], its activation is enhanced and sustained in cancer cells because of activation of oncogenes and loss of tumor suppressor genes [43]. In addition, aerobic glycolysis sustains the production of metabolic intermediates (carbon moieties) for the synthesis of cellular components of the growing tumor [15], producing reducing equivalents when these intermediates are shunted into the pentose phosphate pathway (PPP) and reducing the production of reactive oxygen species (ROS), which enhances cellular proliferation [14]. Finally, the accumulation of lactic acid renders the tumor microenvironment more acidic, which in turn drives genetic instability, favors tumor invasion, cell motility, epithelial-to-mesenchymal transition, metastasis, resistance to apoptosis, immune evasion, and enhances angiogenesis [13,14].
Therefore, targeting this rewired glucose metabolism can be an effective therapeutic option for KRAS and BRAF-mutant CRC. GLUT1 and GLUT3 transport the oxidized form of vitamin C, dehydroascorbate (DHA) into the cells, where it is reduced back to vitamin C at the expense of glutathione (GSH), thioredoxin, and NADPH. Yun et al., demonstrated that DHA transport was increased in KRAS and BRAF-mutant cells, and this was mediated by GLUT1 overexpression. The rapid uptake of DHA and its intra-cellular reduction to vitamin C depletes the reserves of glutathione, leading to ROS accumulation and GADPH inactivation (as shown in Figure 2). The end result is an energy crisis and apoptosis in KRAS and BRAF mutated cells, which is not observed in wild-type CRC cells [42]. Aguilera et al., also demonstrated the vitamin C induced disruption of the Warburg metabolism in KRAS mutant CRC cells. Intracellular vitamin C causes detachment of RAS from the plasma membrane, thereby blocking the phosphorylation of PKM2 (pyruvate kinase M2), leading to downregulation of GLUT-1 expression [43,44].
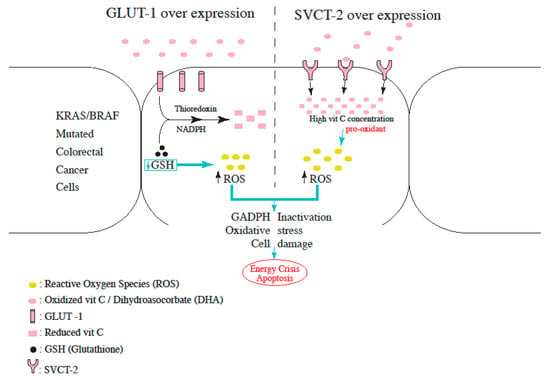
Figure 2. Proposed mechanism for increased ROS production in L-Ascorbic Acid-treated cells overexpressing either GLUT-1 or SVCT-2.
In a study conducted by Jung et al., L-ascorbic acid induced cell death when partnered with cetuximab. This was mainly demonstrated in human colon cancer cells with a mutant KRAS gene, influenced by sodium-dependent vitamin C transporter 2 (SVCT-2) with administration of daily doses of 10 g of L-ascorbic acid for 6 h. Specifically, the knockdown of endogenous SVCT-2 induced resistance to L-ascorbic acid treatment in SVCT-2-positive cells, whereas ectopic expression of SVCT-2 induced sensitivity to L-ascorbic acid treatment in human CRCs that do not express SVCT-2 [45]. In addition, differences in SVCT-2 expression revealed a clear correlation with sensitivities to cetuximab and L-ascorbic acid (as shown in Figure 2). Particularly, recent studies that showed flow of L-ascorbic acid into the cell via SVCT-2 but not SVCT-1 support these findings. Taken together, these outcomes suggest that SVCT-2 expression may enable bypassing resistance to cetuximab in human colon cancer patients with a mutant KRAS by L-ascorbic acid.
Another approach to overcoming secondary resistance to EGFR blockade is targeting cellular proliferation axes with a variety of drugs. At first, MAPK signaling pathway was assessed; particularly over the past 30 years, research has proven that it plays a crucial role in initiating a wide range of cellular responses (proliferation, migration, differentiation, and apoptosis) by converting extracellular stimuli [46]. However, since no significant changes were noted when evaluating the MAPK signaling pathway upon the addition of cetuximab, focus was shifted towards changes in the RAF-MEK-ERK pathway after L-ascorbic acid exposure. Remarkably, decreased phospho-MEK, phospho-ERK, phospho-BRAF, and phospho-CRAF were noted when both drugs were used in the treatment [45], all of which are known to be key molecules for EGFR resistance in mutant KRAS human CRC cells expressing SVCT-2.
CRAF has been known to bind to ASK-1, suppressing its pro-apoptotic activity. According to their study, Jung et al., revealed that activation of ASK-1 and p38 pathway was induced by L-ascorbic acid and cetuximab in SVCT-2 expressing cells [47]. These findings suggest that SVCT-2-dependent reactive oxygen species production induces the activation of the ASK-1 p38 pathway, modulating cellular apoptosis. Changes in these signaling molecules were observed only in the tissues from the mutant KRAS and SVCT-2-positive human colon cancer cell line SW620 but not in tissues from the mutant KRAS and SVCT-2-negative cell line HCT116.
While long-term and larger studies are still lacking, available data supports the notion that L-ascorbic acid overcomes resistance to cetuximab by initiating the ASK-1-mediated apoptosis pathway through the blockade of the MAPK signaling pathway.
Although not enough clinical evidence favors the use of high dose vitamin C in KRAS or BRAF mutated CRC, data from a phase III clinical trial revealed promising results. A total of 442 patients were assigned to receive either chemotherapy (control group) or chemotherapy plus high-dose IV vitamin C 1.5 g/kg/day on day 1–3 (experimental group) and were followed up for 24.5 months [48]. PFS, ORR, and OS were similar between the control and experimental group. At first, results revealed that chemotherapy alone yielded a superior PFS when compared to chemotherapy plus high dose vitamin C. However, the prespecified subgroup analysis of patients with RAS mutation showed improved PFS in the experimental group only in patients with mCRC and KRAS mutation (9.2 vs. 7.8 months, HR 0.67; 95% CI, 0.50–0.91; p = 0.01). Finally, treatment related adverse effect of grade 3 or higher occurred in 33.5% of the patients in the experimental group, as compared with 30.3% in the control group [48].
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
570
Revision:
1 time
(View History)
Update Date:
03 Mar 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No