Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pradeep Kumar | -- | 6047 | 2023-03-02 03:33:22 | | | |
| 2 | Rita Xu | -44 word(s) | 6003 | 2023-03-02 06:46:48 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kumar, A.; Singh, A.K.; Singh, H.; Vijayan, V.; Kumar, D.; Naik, J.; Thareja, S.; Yadav, J.P.; Pathak, P.; Grishina, M.; et al. Nitrogen Containing Heterocycles. Encyclopedia. Available online: https://encyclopedia.pub/entry/41799 (accessed on 06 August 2026).
Kumar A, Singh AK, Singh H, Vijayan V, Kumar D, Naik J, et al. Nitrogen Containing Heterocycles. Encyclopedia. Available at: https://encyclopedia.pub/entry/41799. Accessed August 06, 2026.
Kumar, Adarsh, Ankit Kumar Singh, Harshwardhan Singh, Veena Vijayan, Deepak Kumar, Jashwanth Naik, Suresh Thareja, Jagat Pal Yadav, Prateek Pathak, Maria Grishina, et al. "Nitrogen Containing Heterocycles" Encyclopedia, https://encyclopedia.pub/entry/41799 (accessed August 06, 2026).
Kumar, A., Singh, A.K., Singh, H., Vijayan, V., Kumar, D., Naik, J., Thareja, S., Yadav, J.P., Pathak, P., Grishina, M., Verma, A., Khalilullah, H., Jaremko, M., Emwas, A., & Kumar, P. (2023, March 02). Nitrogen Containing Heterocycles. In Encyclopedia. https://encyclopedia.pub/entry/41799
Kumar, Adarsh, et al. "Nitrogen Containing Heterocycles." Encyclopedia. Web. 02 March, 2023.
Copy Citation
Cancer is one of the major healthcare challenges across the globe. Several anticancer drugs are available on the market but they either lack specificity or have poor safety, severe side effects, and suffer from resistance. So, there is a dire need to develop safer and target-specific anticancer drugs. More than 85% of all physiologically active pharmaceuticals are heterocycles or contain at least one heteroatom. Nitrogen heterocycles constituting the most common heterocyclic framework.
heterocyclic
anti-cancer
nitrogen-containing heterocyclic
1. Introduction
Carcinoma is the abnormal growth of normal cells that typically grow beyond their original boundaries, invade surrounding areas, spread to other organs, and result in metastasis, which is one of the main causes of cancer-related death, the second most common cause of deaths across the globe [1]. Around 10.0 million cancer-related fatalities (9.9 million excluding squamous cell carcinoma) and 19.3 million new cases of cancer (18.1 million excluding squamous cells carcinoma) were estimated globally by 2020. Up to 25% of cancer cases are caused by cancer-causing illnesses such as hepatitis as well as human papillomavirus infections. The most common malignancies in both genders are breast, lung, stomach, colorectal, thyroid, liver, and ovarian. The most fatal cancers are lung (1.8 million), liver (830,000), stomach (769,000), breast cancer (627,000), and colorectal (935,000). The most commonly diagnosed cancers worldwide are lung (2.2 million), breast (2.09 million), colorectal (1.9 million), prostate (1.28 million), skin (1.04 million), and stomach (1.04 million). The prevalence of cancer is rising worldwide, burdening people and families emotionally and financially [2][3][4][5].
In wealthy nations, cancer has become one of the leading causes of mortality. A DNA mutation can cause cancer to develop. The etiology of the gene can be either adopted or hereditary. Both genetic and epigenetic variables are involved. The co-carcinogenic responses, hormonal impacts, and many other epigenetic variables are tumor promoters. Conversion of protooncogene to oncogene and the inactivation of tumor-suppressing genes are the two genetic factors responsible for cancer. Cancer cells can divide quickly or slowly, as in the cases of plasma tumor cells and Burkitt’s lymphoma, respectively. In the cancer cell growth, proliferation is the crucial element, which is very quick compared to the regular cells. When cancerous cells proliferate out of control, alterations occur in the proteins and growth factors, telomerase expression, tumor-associated angiogenesis, and intracellular signaling pathways that regulate apoptosis and cell cycles [6]. Sulfur mustards were utilized in battle throughout the First World War, producing marrow aplasia during that period and later being applied to the chemoprevention. In the last 30–50 years, several other types were developed, including folate analogues, pyrimidine inhibitors, and purine inhibitors. Depending on the type of cancer, different anti-cancer medicines were chosen for the treatment. Surgery, which is similar to plucking a seed from its shell, can eradicate benign tumors, typically referred as nonmalignant [7][8].
Heterocyclic are substances where the ring carbon atom has been replaced by one of the other three elements, oxygen, nitrogen, or sulphur, in the parent scaffold. The existence of the modified atoms as well as the size of the scaffold in the compound affects its physical and chemical properties. Modifications to a molecule’s heterocyclic ring structure can alter its anti-inflammatory, antibacterial, anti-tumor, antiviral, and antifungal properties. In nature, nitrogen-containing heterocyclic compounds are widely distributed and serve as the basis for many different substances, including alkaloids, vitamins, hormones, dyes, antibiotics, herbicides, and pharmaceuticals [9][10][11]. There are few examples of naturally occurring molecules with nitrogen atoms, including morphine, caffeine, nicotine, thiamine, and atropine, known as alkaloids. The number of nitrogen atoms found inside the ring, such as three, four, five, as well as six, is used to categorize these molecules. Pyrrole and azoles are five membered rings containing one nitrogen, while imidazole and pyrazole have two atoms of nitrogen. Pyridine, a six-membered ring with one nitrogen atom, and pyrimidine, a six-membered ring with two nitrogen atoms, are the best examples of nitrogen containing heterocycles [12].
For many years, nitrogen-containing heterocycles have attracted attention of scientists due to their structural variety and biological importance. The present study covers the most recent developments in nitrogen-containing heterocyclic compounds as potential cancer chemotherapeutics. With approximately 60% of unique small-molecules containing a nitrogen heterocyclic, a quick glance through FDA archives demonstrates the structural significance of nitrogen-based heterocycles in drug design. Due to the formation of hydrogen bonds between these heteroatoms and DNA, complexes containing heteroatoms are more stable. In reality, the strength of the binding between DNA and heterocyclic compounds corresponds with the anti-cancer impact [13][14][15]. Natural products, pharmaceuticals, organic materials, sensitizers, copolymers, dyestuff, dyes and corrosion inhibitors all contain nitrogen heterocycles in their skeleton [16][17][18][19][20].
In addition to their important structural role in herbal drugs, e.g., codeine, morphine, vinblastine, reserpine, procaine, papaverine, emetine, and cardiac glycosides, nitrogen-containing heterocyclic compounds are also widely present in synthetic drugs, e.g., azidothymidine, chlorpromazine, antipyrine, metronidazole, diazepam, captopril, isoniazid, chloroquinine, and barbituric acid [21]. Numerous active medicines and natural compounds contain heterocyclic scaffolds as their fundamental nuclei. According to statistics, ≥85% physiologically active molecules are heterocycles or contain one nitrogen atom in their intricate structures [22]. Researchers searched ‘‘Nitrogen containing heterocyclic compounds’’ on ChEMBL (https://www.ebi.ac.uk/chembl/, accessed on 22 November 2022), an open access biological database, and found 2,331,700 compounds on 467 targets. With the help of ChEMBL, researchers plotted a graph between the molecular weight of nitrogen containing heterocyclic compounds with log P values and color, which indicated the violation of rule of 5 (RO5). LogP values of the reported molecules were directly correlated with their molecular weight (Figure 1). As there are so many molecules violating RO5, to explore the correlation of RO5 with activity, researchers have plotted a heatmap of 929 nitrogen containing molecules violating RO5 against different biological targets, but certain molecules violating RO5 have biological activities (Figure 2). Drug development commonly employs Lipinski’s rule of five. This criterion makes it possible to determine whether a bioactive molecule would likely possess the physical and chemical traits necessary for oral bioavailability. According to the Lipinski rule, certain physicochemical features determine how a medicine will behave in terms of absorption, distribution, metabolism, and excretion. The PChEMBL average value indicates the activity count for particular compounds [23].
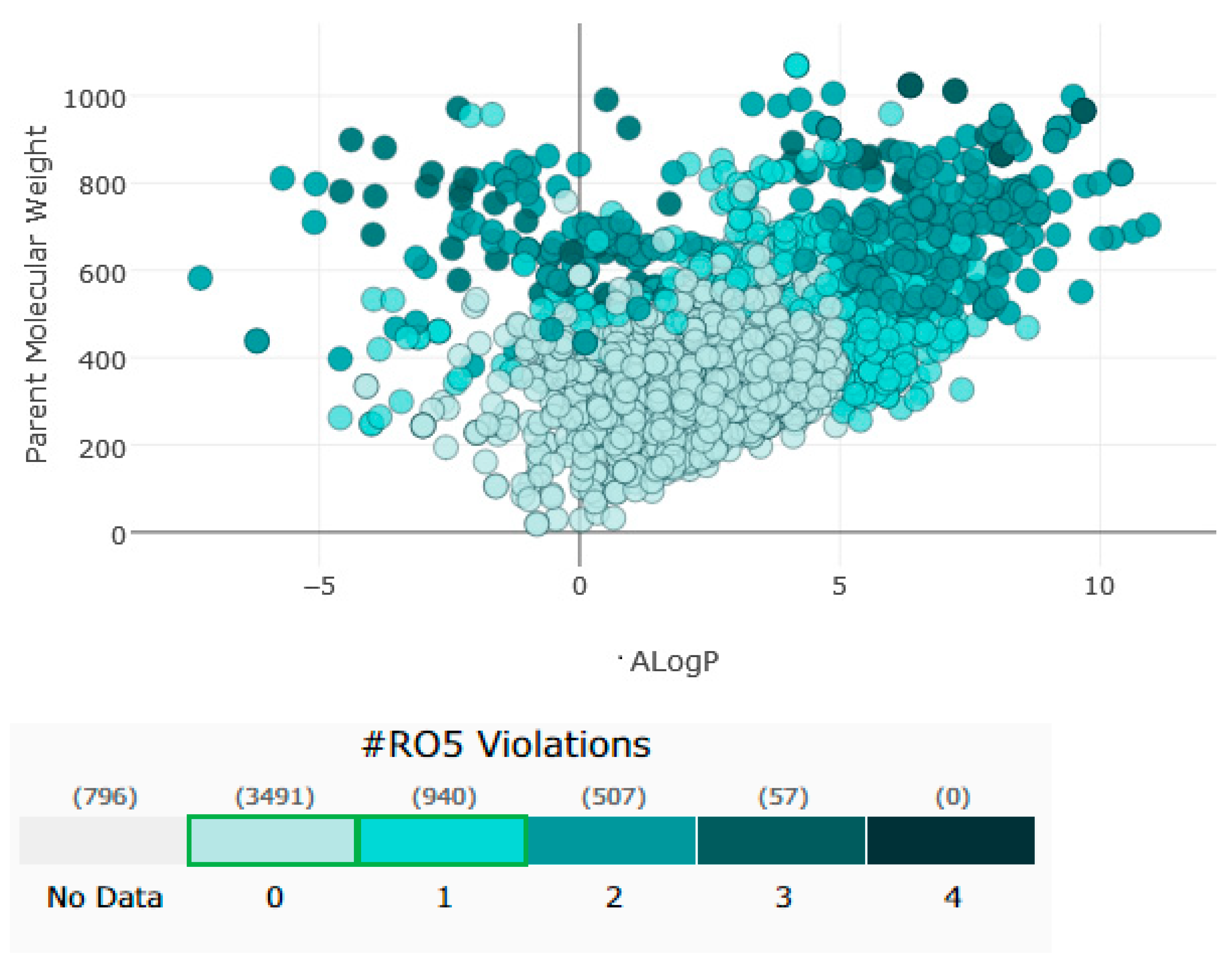
Figure 1. Correlation between molecular weight of nitrogen containing heterocyclic compounds with lop P.
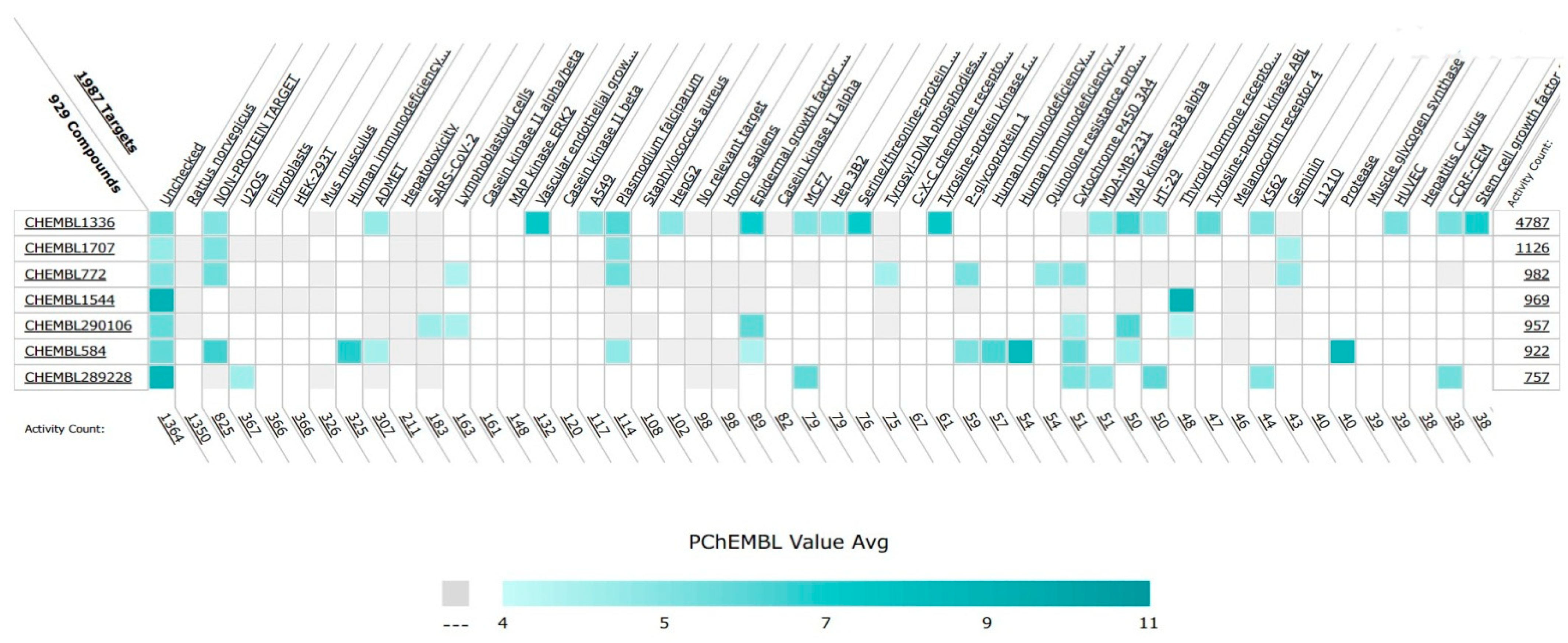
Figure 2. Correlation of nitrogen containing heterocyclic compounds violating RO5 with biological activities.
2. Current Advances in Nitrogen Containing Heterocycles as Anticancer Agents
2.1. Pyrimidine Derivatives as Anticancer Agents
Pyrimidines gained popularity in the history of organic chemistry as “m-Diazine” which is the product of uric acid catabolism. Brugnatelli discovered the first pyrimidine derivative, alloxan, in 1818 while oxidising uric acid with nitric acid. The heterocyclic six membered aromatic ring of pyrimidine contains nitrogen atoms in 1st and 3rd position. The melting and boiling temperatures of pyrimidine are 22.5 °C and 124 °C, respectively [24][25].
Fathalla et al. (2012) synthesized 10 pyrimidine derivatives and evaluated their antitumor activity against a liver cancer (HepG2) cell line by a comparison with the well-known anticancer drugs 5-Flurouracil and Doxorubicin. While comparing the synthesized compounds, growth inhibition effectiveness was shown on the tested tumor cell line at doses between 1 and 10 µg/mL. The most potent compound was found to be compound 1 with the IC50 value of 3.56 µg/mL, whereas doxorubicin and 5-flurouracil were having IC50 values 3.56 µg/mL and 5 µg/mL, respectively [26]. Ahmed et al. (2020) synthesized and evaluated the anti-tumor activity of 16 nitrogen heterocyclic compounds bearing a pyrimidine moiety. The newly developed pyrimidine derivatives were tested for in vitro anti-proliferative activity against human liver (HepG2), breast (MCF7), and normal fibroblast (WI–38) cell lines, and their efficacy was compared to Doxorubicin. Among all the tested compounds, compound 2 showed excellent anticancer activity with IC50 values of 7.36, 10.76, and 6.7 µM respectively, whereas IC50 values of doxorubicin were 4.5, 4.1, and 6.7 µM (WI-38) respectively [27]. Gupta et al. (2022) studied the anticancer activity of spiroisoquinoline-pyrimidine derivatives against the MCF-7 cancer cell line. Out of these, compound 3 having an ethoxy group from the acetylene molecule was found to be the most potent cytotoxic agent with an IC50 value of 98.8 µM as compared to the reference Doxorubicin. An MTT assay was carried out at the concentration of 50 µM and it showed 60% of cell viability with control doxorubicin having 100% cell viability [28]. Al-Issa (2013) synthesized fused pyrimidines and tested them in vitro anti-tumour activity against human cancer cell line HEPG2. Out of these compounds, the most potent anticancer activity was shown by the compound 4 with the IC50 value of 17.4 µg/mL as compared to the standard drug doxorubicin having an IC50 value 1.2 µg/mL [29]. Osmania et al. (2022) synthesized a new pyrimidine-triazole derivatives and carried out studies on its anticancer effect. A total of 10 novel Pyrimidine-Triazole derivatives were synthesized and they were evaluated against three cancer cell lines; A549, MCF-7, and NIH3T3. IC50 values were calculated at 24 h and 48 h (incubation time). Two compounds, namely compound 5a and 5b, were found to be potent anticancer agents. Compounds 5a and 5b have the IC50 values of 1.573 μM and 3.698 μM after 48 h on MCF-7 cell lines respectively. The selective index of these compounds at the end of incubation period was 28.59 and 5.51, respectively. The control was Cisplatin with the IC50 value of 49.23 µM (48 hr) and Doxorubicin at 0.958 µM (48 hr) against the MCF cell line, respectively. The aromatase enzyme inhibition effects of the compound 5a and 5b is also calculated having the IC50 values of 0.082 and 0.198 μM [30]. Qin et al. (2015) synthesized fifteen 2,4-diaminopyrimidines and evaluated their biological activity as selective Aurora A kinase inhibitors. Their cytotoxic activity was tested against five cell lines. The most potent compound was compound 6 with IC50 values of 3.6 µM (HCT-8), 0.5 µM (A-549), 0.9 µM (HeLa), and 2.4 µM (Hep-G2). VX680 was used as the standard, having IC50 of 44.6 µM (HCT-8), 19.4 µM (A-549), 27.3 µM (HeLa), and 63.4 µM (Hep-G2). Molecular docking studies revealed that compound 6 formed a major interaction with Aurora A, which showed 35-fold greater selectivity for Aurora A than for Aurora B. Additionally, compound 6 caused HeLa cells to arrest in the G2/M cell cycle. The IC50 values of compound 6 were 0.012 and 0.043 µM against Aurora A and Aurora B respectively whereas that of standard VX-680 were 0.261 and 0.453 µM respectively [31]. Filho et al. (2021) synthesized 6-ferrocene/heterocycle-2-aminopyrimidine and 5-ferrocene-1H-Pyrazole derivatives by microwave-assisted Atwal reaction and carried out docking, machine learning, and anti-proliferative activity studies of these agents. The compound 7 showed the potent anticancer activity. It was tested on 4 cancer cell lines like HCT116, PC3, HL60, and SNB19 with IC50 values of 56.99, 33.56, 70.26, and 85.11 µM respectively. The docking studies revealed that the compound 7 is the most active compound having the binding energy of −6.3 Kcal/mol with targeted protein (PDB:4HLW) [32].
El-Deen et al. (2022) designed and synthesized pyridothienopyrimidine derivatives and evaluated their anticancer and antimicrobial activity. The cytotoxic activity of newly synthesized compounds was evaluated against three cell lines, MCF-7, HepG-2, and WISH. The compounds that showed potent activity were examined as EGFR kinase inhibitors. The most potent anticancer agent was compound 8 with IC50 values of 1.17, 1.52, and 417.55 µM against the cell lines HepG-2, MCF-7, and WISH, respectively. It is compared with the control drug Doxorubicin with IC50 values of 2.85, 3.58, and 432.10 µM against the cell lines HepG-2, MCF-7, and WISH, respectively. In terms of in vitro enzymatic inhibitory activity against EGFR kinase, compound 8 showed IC50 of 7.27 nM as compared to control Erlotinib with IC50 value of 27.01 nM [33]. Al-Anazi et al. (2022) synthesized pyrazoline and pyrimidine derivatives as novel epidermal growth factor receptor (EGFR) inhibitors and tested their anticancer property. All the synthesized compounds showed good anticancer activity. Among them the compound 9 showed the most potent activity against MCF-7 cell line with the IC50 value of 5.5 µM and selective index 18.18 as compared to tamoxifen standard with IC50 value of 26.95 µM [34].
El-Metwally et al. (2021) synthesized thieno pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and evaluated their anti-cancer properties using MTT against the HepG2, HCT-116, and MCF-7 human cancer cell lines using sorafenib as a positive control. The most potent derivative reported was compound 10 with IC50 value 0.23 µM against the MCF-7 cell line which is similar to the control sorafenib with IC50 value 0.23 µM [35].
Madia et al. (2021) designed and synthesized pyrimidine-based analogues and evaluated their anticancer activity against HT-29, U-87 MG, MDA-MB231, CAL27, and FaDu cancer cell lines. Each of synthesized compounds showed excellent anticancer activity. Among them, compound 11 showed excellent activity at 24 and 48 h time intervals with EC50 values of 10.2, 22.0, 18.0, 9.7, and 26.2 and 5.4, 7.5, 7.9, 4.3, and 8.5 µM, respectively with control RDS 3442 (EC50 values of 51.8, 75.2, 34.8, 54.2, and 69.3 at 48 h respectively) [36].
All the reported pyrimidine derivatives (Figure 3 (1–11)) were acting as anticancer agents. The most potent compound among them was compound 10, having the lowest IC50 value of 0.23 µM against MCF-7 cell line. It has a hydrazine moiety joined by phenyl amino linkage and pyrimidine with benzothiophene nucleus which may be responsible for its excellent activity.
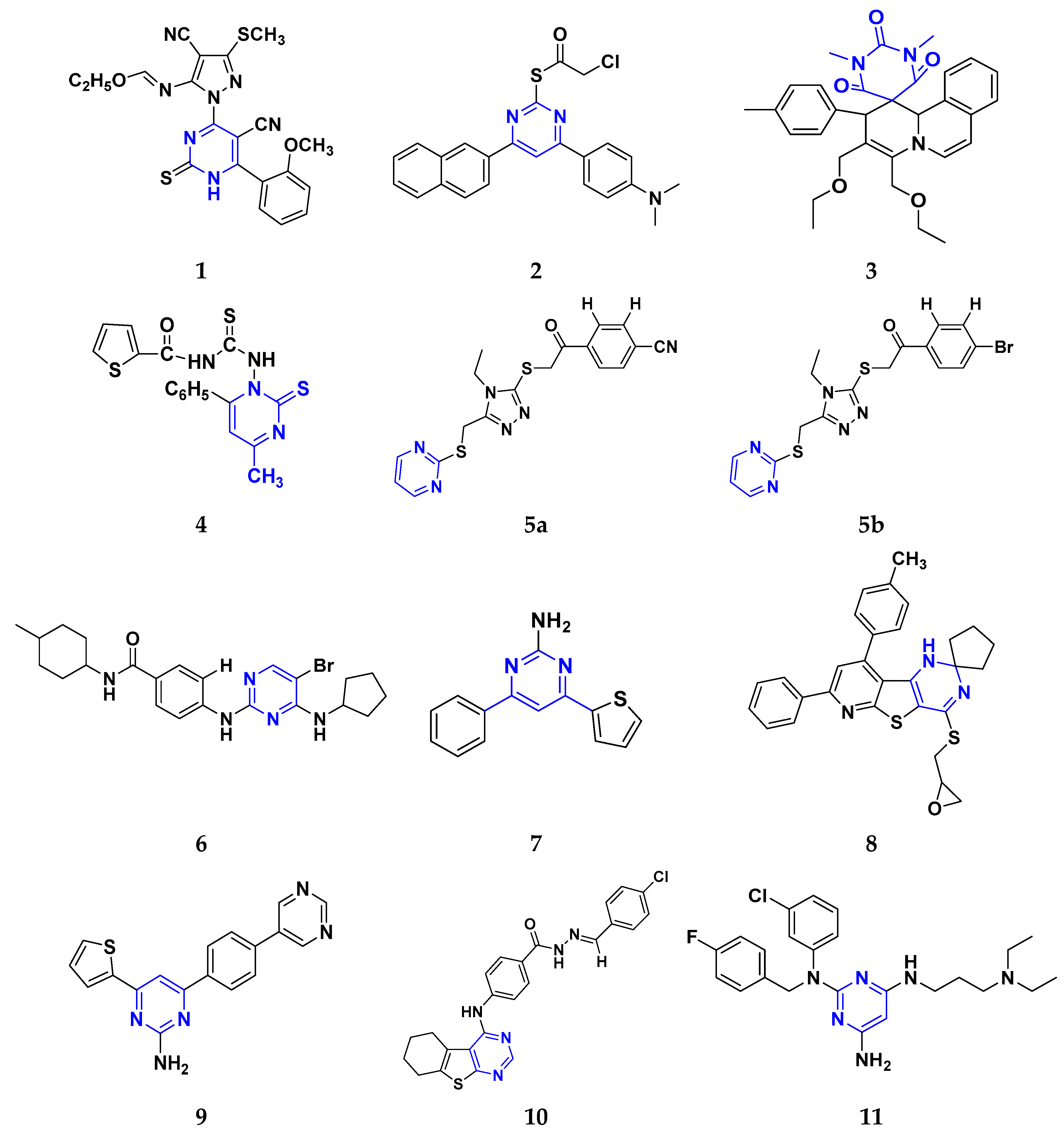
Figure 3. Pyrimidine derivatives (1–11) as anticancer agents.
2.2. Quinoline Derivatives as Anticancer Agents
Quinoline is a nitrogen containing heterocyclic aromatic compound, also known as 1-aza-napthalene or benzopyridine. Its molecular weight is 129.16 and molecular formula is C9H7N. The log P value is 2.04 while the pKb and pKa values are 4.85 and 9.5, respectively. Quinoline is a weak tertiary base and with acids it can produce salt and exhibits reactions akin to those of pyridine and benzene. Numerous naturally occurring chemicals (Cinchona Alkaloids) and pharmacologically active molecules with a wide range of biological activities include the quinoline nucleus in their structure. Diverse pharmacological activities (anticonvulsant, analgesic, cardiotonic, antibacterial, antifungal, anti-inflammatory, and anti-malarial) of quinoline have been reported [37].
Hamdy et al. (2019) conducted anticancer study on quinoline based heterocycles targeting Bcl-2. Out of these, Compound 12 showed excellent activity in MDA-MB-231, HeLa, KG1a, and Jurkat with the IC50 values of 0.54, 1.42,1.21, and >100 µM respectively. This compound showed the IC50 value of 0.15 µM against Bcl-2 compared with Gossypol (IC50 value of 0.60 µM). Compound 12 had sub-micromolar anti-proliferative activity in cancer cell lines that express Bcl-2, as well as a sub-micromolar IC50 value in an ELISA experiment using the Bcl2-Bim peptide [38]. Mathada et al. (2022) studied the anticancer effect of quinoline and its derivatives. Hence, 62 compounds were synthesized. Out of these, the most potent compound was found to be compound 13, which showed a potent anticancer effect against three cell lines, MDA-MB-231, HeLa, and SMMC-7721 with IC50 values of 0.12, 0.08, and 0.34 µM respectively. Etoposide was used as standard drug with IC50 values of 5.26, 2.98, and 3.48 µM respectively. Compound 13 had a unique capacity to induce apoptosis in HeLa cells, halt the cell cycle at the G0/G1 phase, elevate intracellular ROS (Reactive Oxygen Species) levels, and decrease mitochondrial membrane potential. Additionally, it had the ability to significantly reduce MEK1 kinase activity and disrupt the Ras/Raf/MEK/ERK transduction pathway [39].
Katariya et al. (2020) performed the studies on anticancer, antimicrobial activities of quinoline based hydrazone analogues. Nine compounds out of all the tested compounds showed significant anti-cancer activity at 10 µM. These compounds were then screened at 10-fold dilutions of five different concentrations (0.01, 0.1, 1, 1, and 100 µM) with GI50 values ranging from 0.33 to 4.87 µM and LC50 values ranging from 4.67 µM to >100 µM. The most active molecule, compound 13, had a mean graph midpoint (MG-MID) GI50 value of 1.58 µM, which was significantly lower than commercially available standards Bendamustine and chlorambucil (60 and 52 µM, respectively). The IC50 value of compound 14 was 132 µM against the NIH/3T3 cell line, which was significantly higher than the GI50 values of compound 14 against the NCI 60 cancer cell lines, which demonstrated that compound 14 was highly selective for cancer cells at concentrations that are lower than the effective concentration against healthy cell lines [40].
George et al. (2019) synthesized the new derivatives of quinoline, i.e., 4,5- dihydropyrazoles as EGFR inhibitors and evaluated their anti-proliferative activity. The newly synthesized compounds were tested against HeLa, MCF-7, and DLD1 cancer cell lines as well as normal fibroblast WI-38. Eight compounds showed potent activity towards the DLD1 cell line and it was safe to normal cell lines. The most active compound was compound 15, with IC50 values of 0.227, 0.136, 1.277, and >94.5 µM against the cell lines MCF-7, HeLa, DLD1, and WI-38, and it was compared to the control drug CHS 828 with the IC50 values of 0.018, 0.040, 2.315, and >134.71 µM respectively. Compound 15 showed the EGFR inhibitory activity with IC50 value of 31.8 nM as compared to the control Gefitinib 29.16 nM [41]. Koprulu et al. (2021) had conducted anticancer activity and molecular docking studies of quinoline derivatives. The cytotoxic activity of synthesized piperazine substituted quinoline derivatives were tested against three cell lines, namely rat glioblastoma (C6), human cervical cancer (HeLa), and human adenocarcinoma (HT29). The docking studies revealed that compound 16 was the most potent compound for metastatic cancer treatment due to its binding affinity to PLCγ1. The IC50 values of compound 16 were 100, 144.8, and 117.6 µM against the cell lines, C6, HeLa, and HT29. Further, 5-Fluro Uracil was used as the standard drug with IC50 values of 163, 469.6, and 501.2 µM [42].
Ramya et al. (2018) synthesized and evaluated anticancer potential of curcumin inspired 2-chloro/phenoxy quinoline analogues. This research described the synthesis of twenty novel 2-chloro/phenoxyquinoline derivatives inspired by curcumin. The cytotoxic activity of the acquired novel chemical entities toward various tumor cell lines was examined in vitro. The most active compound 17 showed potential cytotoxicity against different cell lines HeLa, HGC, NCI-H460, DU-145, PC-3, and 4T1 with IC50 values of 5.41, 8.73, 3.96, 3.99, 3.12, and 1.81 µM respectively. The reference drug curcumin had the IC50 values of 17.11, 33.15, 18.65, and 18.65 µM against the cell lines NCI-H460, DU-145, PC-3, and 4T1, respectively. Additionally, study of ROS levels, DAPI staining, AO-EB labelling, and annexin binding assay showed that the promising compound 16 may cause apoptosis in PC-3 cells and G2/M cell cycle arrest [43].
Upadhyay et al. (2018) conducted the synthesis and screening of pyrano[3,2-c],quinolones derivatives. One pot multicomponent condensation was carried out between malononitrile, 2,4-dihydroxy-1-methylquinoline, and substituted aromatic aldehydes to create a number of pyrano[3,2-c]quinoline based structural analogues. The compounds were accessed for their cytotoxic and anti-inflammatory activity. Out of these, compound 18 was found to be the most potent anticancer agent and showed 81–53% anti-proliferative inhibition at 1 µM concentration against all cell lines, including ACHN, Panc-1, HCT-116, H-460, and Calu-1, with IC50 values of 0.1, 1.0, 0.3, 0.3, and 0.5 µM respectively. Flavopiridol, having the IC50 values of 71, 78, 71, 88, and 74 µM, and gemcitabine, with 73, 74, 73, 71, and 79 µM, respectively, were used as standard [44].
Hagras et al. (2021) designed, synthesized, and conducted docking studies and anti-proliferative evaluation of newly discovered quinolines. The most effective anti-tubulin polymerization agents are colchicine binding site inhibitors. In order to exhibit the same fundamental pharmacophoric characteristics as colchicine binding site inhibitors, novel quinoline compounds have been developed and synthesized. Using colchicine as a positive control, the synthesized compounds were evaluated in vitro against three human cancer cell lines; MCF-7, HepG-2, and HCT-116. The most potent compound 19 have the IC50 values of 1.89, 1.43, and 4.21 µM against the cell lines HePG2, HCT-116, and MCF-7 respectively. The control drug colchicine with IC50 values of 7.4, 9.3, and 10.4 µM, respectively, and its effect on cell cycle distribution were evaluated. The outcomes showed that compound 19 had the ability to stop the cell cycle at the G2/M phase. A double-staining test with annexin V and PI was performed to investigate the apoptotic effect of the synthesized compounds. Compound 19 caused HepG-2 cells to apoptosis thirteen times more frequently than control cells [45]. Mirzaei et al. (2019) synthesized novel quinoline chalcone hybrids and conducted studies on the structure–activity relationship and docking studies for their anticancer potential as active tubulin inhibitors. Four different human cancer cell lines, including A2780/RCIS (Cisplatin-resistant human ovarian carcinoma), A2780 (human ovarian carcinoma), normal Huvec cells, MCF-7 (human breast cancer cells), and MCF-7/MX (Mitoxantrone-resistant human breast cancer cells), were used to test the cytotoxic activity of synthesized substances. The most potent compound was compound 20 with IC50 values of 2.32, 2.615, 4.96, 2.32, and 4.44 µM against the cell lines A2780, A2780/RCIS, MCF-7, MCF-7/MX, HUVEC respectively. It is compared with the standard drug CA-4 with IC50 values of 0.24, 0.22, 0.43, and 1.49 µM against the cell lines A2780, A2780/RCIS, MCF-7, and MCF-7/MX, respectively [46]. Chate et al. (2018) investigated novel spiro-pyrimido[5,4-b]quinoline-10,50-pyrrolo[2,3-d]pyrimidine] derivatives as promising anticancer agents. By using the MTT assay, the newly synthesized compounds’ anticancer effects were assessed in vitro against the A431, PC-3, MCF-7, and MCF-10A cancer cell lines. Out of these, the most potent was compound 21, having an IC50 value of 36.25 µM for MCF-10A (normal breast epithelial cell lines), indicating five times more selectivity for MCF-7 cancer cells with the IC50 value of 7.82 µM. Sunitinib was used as a control having IC50 values of 6.48, 7.25, 20.66, and 34.55 µM against the cancer cell lines A-41, MCF-7, PC-3, and MCF-10A, respectively [47].
While evaluating the above reported quinoline derivatives (Figure 4 (12–21)), most of the compounds were evaluated with their cytotoxic activity commonly against HeLa, MCF-7, and HCT-116 cells. Compounds 13, 15, and 18 were reported to be potent anticancer compounds but the most potent one among them was compound 13 with the lowest IC50 value of 0.08 µM on HeLa cells, 0.12 µM on MDA-MB-231, and 0.34 µM on SMMC-7721, having a steroid nucleus directly attached with quinoline along with a hydrazine chain. This also demonstrated the ability to cause apoptosis in HeLa cells.
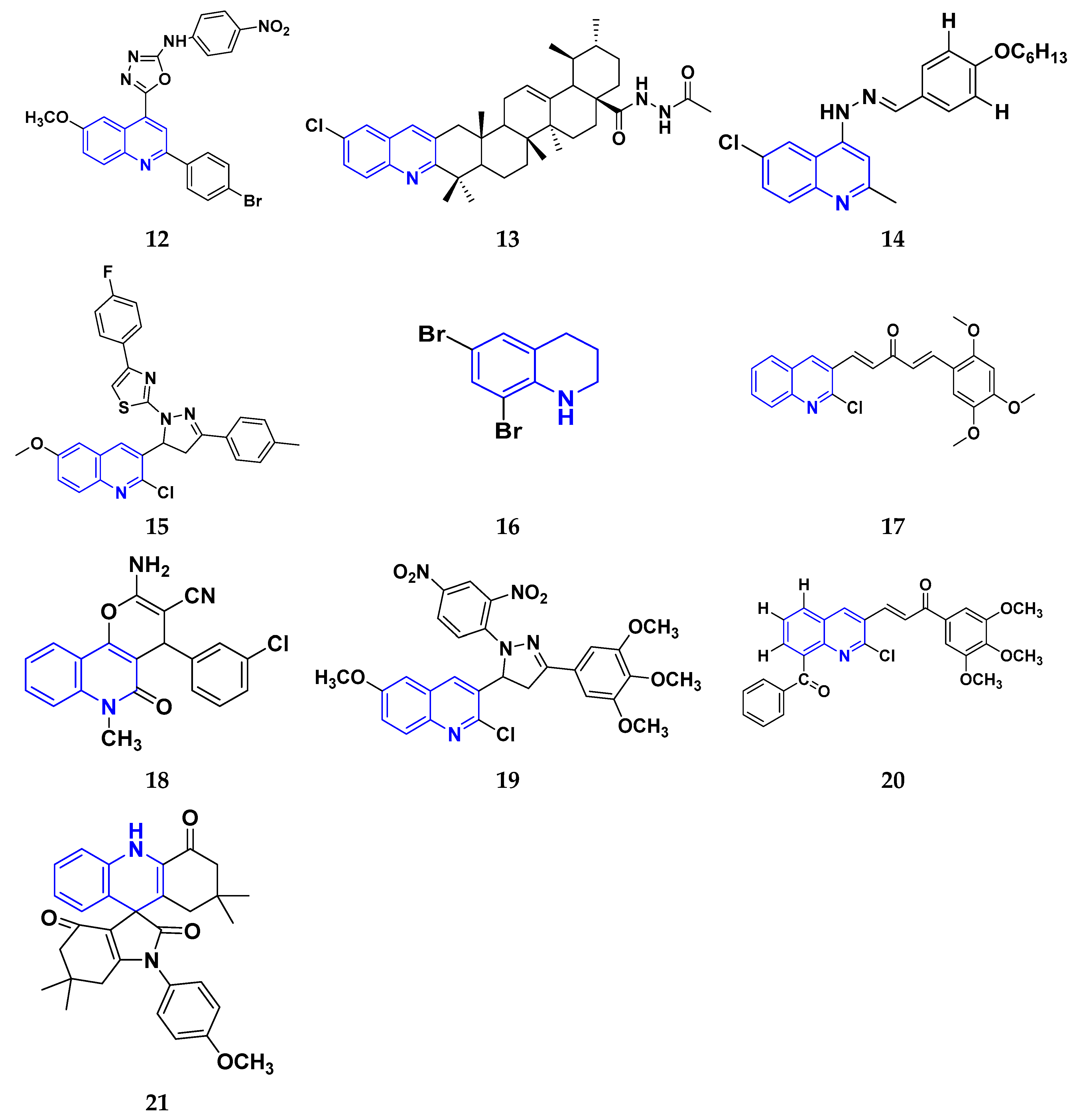
Figure 4. Quinoline derivatives (12–21) as anticancer agents.
2.3. Carbazole Derivatives as Anticancer Agents
A polycyclic aromatic hydrocarbon called carbazole has a broad aromatic system and a central nitrogen atom that exhibits substantial electron delocalization. It consists of a five-membered nitrogen-containing ring sandwiched between two six-membered benzene rings. It has an indole-like structure, but at the indole position 2–3, a second benzene ring is fused to the five-membered ring. When creating electron donor-electron acceptor (D-A) chemical dyes, carbazole is frequently used as a conjugated bridge [48].
Murali et al. (2017) studied the creation of hetero annulated isoxazolo-, pyrido-, and pyrimido carbazoles, also screening them for in vitro anticancer activity. By cyclo condensation with the appropriate reactants (hydroxylamine hydrochloride, malononitrile, and guanidine nitrate), the newly synthesized heterocycles isoxazolo-, pyrido-, and pyrimidocarbazoles were produced from the readily available 2-(3′-bromo-4′-methoxybenzylidene)-2,3,4,9-tetrahydro-1H-carbazol. All the synthesized substances were tested for in vitro cytotoxicity against the A-549 and MCF-7 human cancer cell lines. Compound 22 demonstrated substantial activity against MCF-7 with the IC50 value of 20 µM as compared to cisplatin IC50 value of 18 µM. All other compounds showed moderate to powerful activity and consequent apoptotic cell death, which was demonstrated by AO/EB and DAPI of fluorescence microscopy analysis [49]. Wang et al. (2011) designed and synthesized substituted 11H-benzo[a] carbazole-5-carboxamides as novel anticancer agents and tested them against human cancer A549 and HCT-116 cell lines. The most potent derivative was compound 23 with IC50 value 8.2, 9.5 µM against the cancer cell lines A549, and HCT-116 respectively. Amonafide was used as the standard with IC50 values 8.1 µM and 15.3 µM, respectively [50]. Debray et al. (2010) synthesized N-ethoxycarbonyl-N-arylguanidines by the montmorillonite K-10 catalyzed cyclization, which provides access to pyrimido[4,5-c]carbazole and pyrimidoo[5,4-b]indole derivatives. Further, 3-aminocarbazole and 3-aminoindole were converted into two novel heterocycles, pyrimido[4,5-c]carbazole and pyrimido[5,4-b]indole, respectively. With the aid of montmorillonite K-10 clay as a catalyst and microwave irradiation, the essential Friedel–Crafts intramolecular cyclization was accomplished. The micromolar IC50 of the pyrimido[4,5-c]carbazole derivative was significant against cancer cell lines. The most potent compound was found to be compound 24 with the IC50 values 17 and 17 µM against the cancer cell lines HL60 N and HL60 MX2, respectively. Etoposide is used as a reference with IC50 values of 1.3 µM and 11.9 µM [51].
Sun et al. (2017) synthesized novel carbazole sulfonamide derivatives and evaluated their anti-proliferative activity and aqueous solubility as an antitumor agent. A number of novel carbazole sulfonamide derivatives were produced by the current optimization of IG-105 on the carbazole-ring. Each compound’s anti-proliferative effectiveness was tested on HepG2 cells. When tested for anti-proliferative activity against MIA PaCa-2 (pancreatic cancer), MCF-7 (breast cancer), and Bel-7402 (hepatocellular/liver cancer), compounds that had shown action superior or equivalent to that of IG-105 against HepG2 were found to be effective. Five of the seven substances were chosen for further investigation and discovered to have IC50 values against the four cell lines that were comparable to those for IG-105. The activity of two compounds, compound 25a and 25b against HepG2 and MCF-7 (IC50 values of 0.01 and 0.07 µM) was close to that of the positive controls podophyllotoxin and CA-4. The most potent compound out of these two was Compound 25a with IC50 value against HEPG2 was 0.012 µM as compared to the controls podophyllotoxin with IC50 value 0.003 µM and CA-4 with IC50 value of 0.002 µM respectively [52]. Arya et al. (2018) performed eco-compatible synthesis of highly functionalized pyrido[2,3-a] carbazole derivatives and tested them for their cytotoxic effects on cancer cell lines MCF-7 and A549. The result revealed compound 26 as the potent anticancer compound with lowest IC50 values of 45 µM and 50 µM against MCF-7 and A549 while the control drug Ellipticine presented IC50 values of 73 µM and 65 µM, respectively [53]. Padmaja et al. (2014) tested the anticancer activity of novel pyrano[3,2-c]carbazole derivatives which induce cell death by the inhibition of tubulin polymerization. A facile one-pot, three-component reaction using malononitrile-ethyl cyanoacetate, aromatic aldehydes, and 4-hydroxycarbazoles, catalyzed by trimethylamine was used to synthesize pyrano[3,2-c]carbazole derivatives. Investigations were performed to test their ability to inhibit the proliferation of several cancer cell lines, including K562, MDA-MB-231, HeLa, and A549. The most potent anticancer agent was compound 27 with IC50 values of 0.43, 1.13, 3.41, 6.12, and 69.24 μM against the cell lines MDA-MB 231, K562, A549, HeLa, and L929, respectively. Combrestatin A-4 (CA-4) was used as the control. This compound causes apoptosis by preventing tubulin polymerization and G2/M phase arrest of the cell cycle [54]. Patel et al. (2021) synthesized and characterized coumarin carbazole based functionalized pyrimidines, evaluated the anticancer effect, and performed molecular docking studies. The synthesized compounds were evaluated against three cell lines (HeLa, NCI-H520, and NRK-52E). Compound 28a and 28b were reported to be most active because of their ability to cause apoptosis and cell cycle arrest. Both of these molecules demonstrated high binding affinity towards CDK2 protein. Compound 28a was the most potent anticancer agent with IC50 values of 12.59 µM, 11.26 µM, and 28.37 µM against the cell lines HeLa, NCI-H520, and NRK-52E respectively. It was compared to the control Cisplatin with IC50 values of 7.75, 10.41, and 12.93 µM and 5-Flurouracil with IC50 values of 55.72, 8.36, and 46.68 µM, respectively [55]. Huang et al. (2021) performed the synthesis of novel carbazole derivatives as selective and potent anticancer drugs, as well as performed their biological evaluation and structure–activity relationship studies. The in vitro cytotoxic effects of two series of carbazole compounds against the three cell lines A875, HepG2, and MARC145 were assessed. Compared to the control 5-fluorouracil, the results showed that some of these carbazole derivatives had much better cytotoxic effects against the examined cell lines. Particularly, the carbazole acylhydrazone compounds 29a and 29b showed strong inhibitory effect against cancer cells, but essentially no activity against normal cells. Particularly, compound 29a showed significantly selective proliferation inhibition on cancer and normal cell lines with the selectivity index up to 13, and it had a stronger inhibitory effect on the A875 with IC50 value of 7.65 µM and HepG2 cell lines with IC50 value of 8.16 µM compared to the normal cells line MARC145 with IC50 value of > 105 µM. The control 5-Flurouracil had IC50 values of 72.33, 81.94, and 77.56 µM against A875, HepG2, and MARC145 cell lines, respectively [56].
Chen et al. (2018) synthesized racemic and chiral carbazole aminoalcohols and tested their anticancer potential. It was found that the topoisomerase I was inhibited due to a number of reasons, e.g., substitution site, heterocycle, chirality, and length of alkyl chain. MDA-MB-231, HCT116, and A549 cancer cell lines were used as test subjects and carbazole amino alcohols with potent topo I inhibitory activities, such as pyrrolidine derivative and propyl- to pentyl- amine derivatives, demonstrated good anticancer activities. The most potent compound was compound 30 with broad spectrum anti-tumor activity against 15 cancer cell lines. The IC50 values were 7.9, 4.6, 2.8, 7.5, 3.9, 3.4, 3.8, 2, 4.8, 1.6, 1.5, 1.7, 1.7, 1.7, and 2.2 μM against A549, HCT116, MDA-MB-231, HeLa, H3122, BT549, BEL7402, 3AO, HO-8910, Rh30, Pfeiffer, Molm13, OC1-AML2, Jurkat, and HL60 cell lines, respectively [57].
Vairavelu et al. (2014) performed solvent-free synthesis of heteroannulated carbazoles using grinding conditions and a number of new carbazole analogues. Templates for isoxazolo, pyrido, pyrazolo, and pyrimido were developed and synthesized in high yield. The synthesized compounds were tested in vitro for their anticancer potential. Compound 31 was the most potent anticancer compound with IC50 values of 0.37 µM and 15.12 µM against HeLa and AGSb cell lines respectively. Ellipticine was used as the control with IC50 values of 4.12 and 7.33 µM, respectively [58].
Among the above-mentioned carbazole derivatives (Figure 5 (22–31)), the most potent one was compound 25a having a pyridine ring joined by sulphonamide linkage and substituted with 2,5-dimethoxy group, having the lowest IC50 value of 0.012 µM against the HEPG2 as compared to the control podophyllotoxin with IC50 value of 0.003 µM and CA-4 with IC50 value of 0.002 µM.
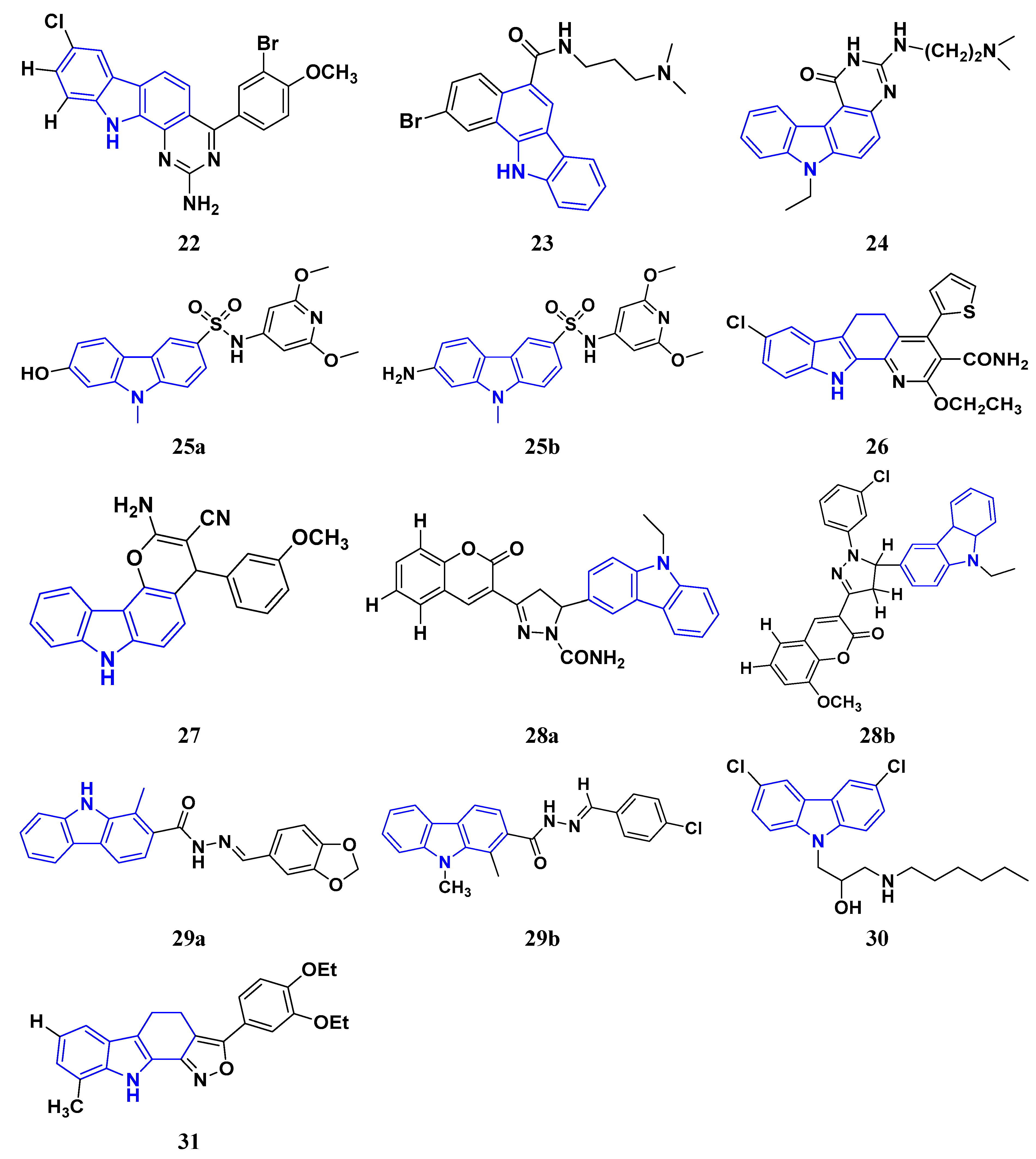
Figure 5. Carbazole derivatives (22–31) as anticancer agents.
2.4. Pyridine Derivatives as Anti-Cancer Agents
Pyridine has the chemical formula C5H5N and is a fundamental heterocyclic organic molecule. The word “pyridine” is taken from Greek and combines the words “idine” and “pyr,” which both refer to aromatic bases. Picoline, the first pyridine base, was discovered by Anderson in 1846. It took quite some time for Wilhelm Korner and James Dewar to discover its structure. It resembles the well-known and fundamental aromatic molecule benzene in many ways, but with one C-H group replaced by an atom of Nitrogen. Like benzene, pyridine possesses a conjugated system of six delocalized electrons distributed around the heterocyclic ring. The molecule satisfies the Huckel requirements for aromaticity and is planar in nature [59].
Gomha et al. (2018) performed the studies on anti-tumor activity through the Synthesis of some new pyridine-based heterocyclic compounds. By the reaction of 7-(pyridin-4-yl)-2-thioxo-2,3-dihydropyrido[2,3-d] pyrimidin-4(1H)-one and 2-cyano-N-(1-(pyridin-4-yl)ethylidene)-acetohydrazide with hydrazonoyl halides, 5-amino-N-(1-(pyridin-4-yl)ethylidene)-1H-pyrazole-4-carbohydrazides and 8-(pyridine-4-yl)pyrido[2,3-d][1,2,4]triazolo[4,3-a]pyrimidin-5(1H)-ones were synthesized. The anti-cancer activity of the synthesized compounds was tested against the HEPG2 cell line. The most potent compound was compound 32, with an IC50 value of 0.97 µM against the HEPG2 cell line Doxorubicin is used as the standard with an IC50 value of 0.74 µM [60]. Fayed et al. (2019) designed, synthesized, and performed molecular modeling studies of coumarin derivatives. The synthesized compounds were evaluated for their cytotoxic activity against different cell lines; HCT-116, MCF-7, A549, and HepG-2. The most potent compound was compound 33 with IC50 values of 1.11, 6.44, 4.51, and 7.18 µM against the cancer cell lines MCF-7, HCT-116, HepG-2, and A549, respectively. 5-Flurouracil was the standard with IC50 values of 7.76, 8.78, 8.15, and 7.65 µM, respectively. Compound 33 induced cell cycle arrest in the G2/M phase and apoptosis. Caspase-3 is used for stimulating apoptosis [61]. Nagender et al. (2016) synthesized novel hydrazone and azole-functionalized pyrazolo[3,4-b]pyridine derivatives. With the key intermediate ethyl 2-(3-amino-6-(trifluoromethyl)-1H-pyrazolo[3,4-b]pyridin-1-yl)acetate, a variety of pyrazolo[3,4-b]pyridine compounds were synthesized. The reaction was carried out with hydrazine hydrate followed by isothiocyanate, acid chloride, and different aldehydes to form 1,2,4 triazoles, oxadiazoles, hydrazones, and thiadiazoles. All the synthesized compounds were screened for anti-cancer activity. With an IC50 value of 3.2 µM, compound 34 with the trifluoromethylthio group demonstrated excellent action against the lung cancer cell line (A549) compared with the control 5-Fluorouracil with an IC50 value of 1.7 µM [62].
El-Naggar et al. (2018) conducted the synthesis and biological evaluation of pyridine-urea derivatives as anti-cancer drugs. All the synthesized compounds were tested for their in vitro antiproliferative activity in breast cancer against the MCF-7 cell line. Two compounds, 35a with IC50 values of 0.22 µM (48 h) and 1.88 µM (72 h) and 35b with IC50 values of 0.11 µM (48 h) and 0.80 µM (72 h), were reported to present potent anti-cancer activity against the MCF-7 cell line. The most potent compound was compound 35b with the lowest IC50 value, while compared to the reference drugs, doxorubicin presented IC50 values of 1.93 µM (48 h) and 1.07 µM (72 h) and sorafenib with IC50 values of 4.50 µM (48 h) and 1.71 µM (72 h) respectively [63].
Dinda et al. (2014) carried out a study on the cytotoxicity of pyridine-wingtip substituted annelated N-heterocyclic carbene complexes of silver (I), gold (I), and gold (III). Three new compounds were synthesized from 1-methyl-2-pyridin-2-yl-2H-imidazo[1,5-a]pyridin-4-ylium chloride. The synthesized complexes were tested for their cytotoxicity against HepG2 (human hepatocellular carcinoma), A549 (human lung adenocarcinoma), HCT 116 (human colorectal carcinoma), and MCF-7 (human breast adenocarcinoma) cells. Compound 36 was the most potent antiproliferative agent with IC50 values of 4.91, 5.08, 5.23, and 5.18 µM against the cell lines HepG2, HCT-116, A549, and MCF-7, respectively. Cisplatin was used here with IC50 values of 4.31, 4.89, 6.12, and 4.42 µM, respectively [64]. El-Gohary et al. (2019) synthesized novel pyrazolo[3, 4-b]pyridine analogs, screened their anti-cancer potential, and performed molecular modeling. An in vitro anti-cancer assay toward MCF-7, HepG2, and Hela cancer cells and in vivo anti-cancer assay over E.A.C. in mice were carried out. The most potent compound was compound 37, with IC50 values of 3.63, 3.11, and 4.91 µM against the cell lines HepG2, MCF-7, and Hela, respectively. Doxorubicin was used as the reference with IC50 values of 4.3, 3.97, and 5.17 µM, respectively. According to molecular modeling studies, it was reported that the synthesized compounds bind to DNA through intercalation similar to doxorubicin [65].
Sangani et al. (2014) designed, synthesized, and evaluated the molecular modeling of pyrazole-quinoline-pyridine hybrids as a potential class of antibacterial and anti-cancer drugs. A one-pot multicomponent reaction was carried out using a base-catalyzed cyclo condensation reaction, and a new series of pyrazole-quinoline-pyridine hybrids were synthesized. All the compounds synthesized tested for in-vitro anti-cancer and antimicrobial activity. The most potent compound with the lowest cytotoxicity was compound 38, compared to reference drug Erlotinib with IC50 values of 0.13, 0.12, and 0.032 µM, against the cell lines A549, HepG2, and E.G.F.R. respectively. Compound 38 showed three hydrogen bonds and one -π cation interaction with a minimum binding energy of −54.6913 kcal/mol in the molecular docking studies with the catalytic pocket of protein [66]. Elzahabi (2011) synthesized and characterized some benzazoles bearing pyridine moiety in a search for novel anti-cancer agents. Thirteen new benzazole compounds were synthesized as potential anti-cancer agents. The National Cancer Institute (NCI), U.S.A. chose four derivatives of the produced compounds to be tested for their anti-cancer potential against a panel of 60 cancer cell lines at a single high dose. Compounds 4-[p-chlorophenyl]pyridine and 4-[pmethoxyphenyl]pyridine were chosen for further testing at five doses after exhibiting a broad and moderate anti-cancer activity against 41 tumor cell lines belonging to the nine subpanels employed. The compounds 39a and 39b showed excellent activity in HOP-92 cells with GI50 values of 0.275 µM and 2.65 µM, respectively. Regarding colon cancer, compound 39a’s GI50 value against HCT116 was 3.47 µM, and the most sensitive cell line in this subpanel was KM12 towards compound 39b with GI50 4.79 µM. The most sensitive lines for compounds 39a and 39b were SF-539 and SNB-75, belonging to C.N.S. cancer cell lines with GI50 2.07 µM and 2.36 µM, respectively [67].
Zheng (2014) researched the design, synthesis, and biological evaluation of novel combretastatin-A4 pyridine-bridged analogs as anti-cancer drugs. The synthesized compounds potentially suppressed cell growth and survival, stopped the cell cycle, and prevented angiogenesis and the development of blood vessels, similar to CA-4 (combretastatin-A4). The IC50 values of compound 40a against three cancer cell lines were 0.0031, 0.089, and 0.0038 µM against MDA-MB-231, A549, and HeLa, respectively, and for compound 40b were 0.0046, 0.044, and 0.0014 µM respectively. The reference compound used was combretastatin-A4 with IC50 values of 0.0028, 0.0038, and 0.0009 µM, respectively [68]. Abbas et al. (2015) synthesized novel pyridines having an imidazole moiety. The one-pot multicomponent reaction of 5-acetyl imidazole, substituted benzaldehyde (or terephthaldehyde), malonitrile (or ethyl cyanoacetate or diethyl malonate), and ammonium acetate was carried out to produce a novel series of pyridine and bipyridine derivatives. Some recently synthesized compounds were tested for their anti-cancer activities against the human breast cell line (MCF-7) and liver carcinoma cell line (HEPG2) with the reference Doxorubicin IC50 values of 0.46 µM and 0.42 µM, respectively. The most potent compound was compound 41, with IC50 values of 1.7 and 6 µM, respectively [69]. Ivasechko (2022) synthesized novel pyridine-thiazole hybrid molecules as potential anti-cancer agents and evaluated their anti-cancer activity against several kinds of tumors, e.g., carcinomas of the breast, lung, colon, glioblastoma, and leukemia. Two compounds showed the highest anti-cancer activity. The most potent compound reported was compound 42, with an IC50 value of 2.79 µM against the MCF-7 cell line. Doxorubicin was standard with an IC50 value of 1.04 µM against the MCF-7 cell line [70].
Among the above-reported potent anti-cancer compounds (Figure 6 (32–42)) having a pyridine moiety, the most active compound which showed the best cytotoxic activity was compound 40a (a diaryl compound joined by pyridine ring. Diaryl rings were substituted with methoxy group on different positions). It showed the lowest IC50 value of 0.0031 µM, 0.089 µM, and 0.0038 µM against the three human cancer cell lines MDA-MB-23, A549, and HeLa, respectively.
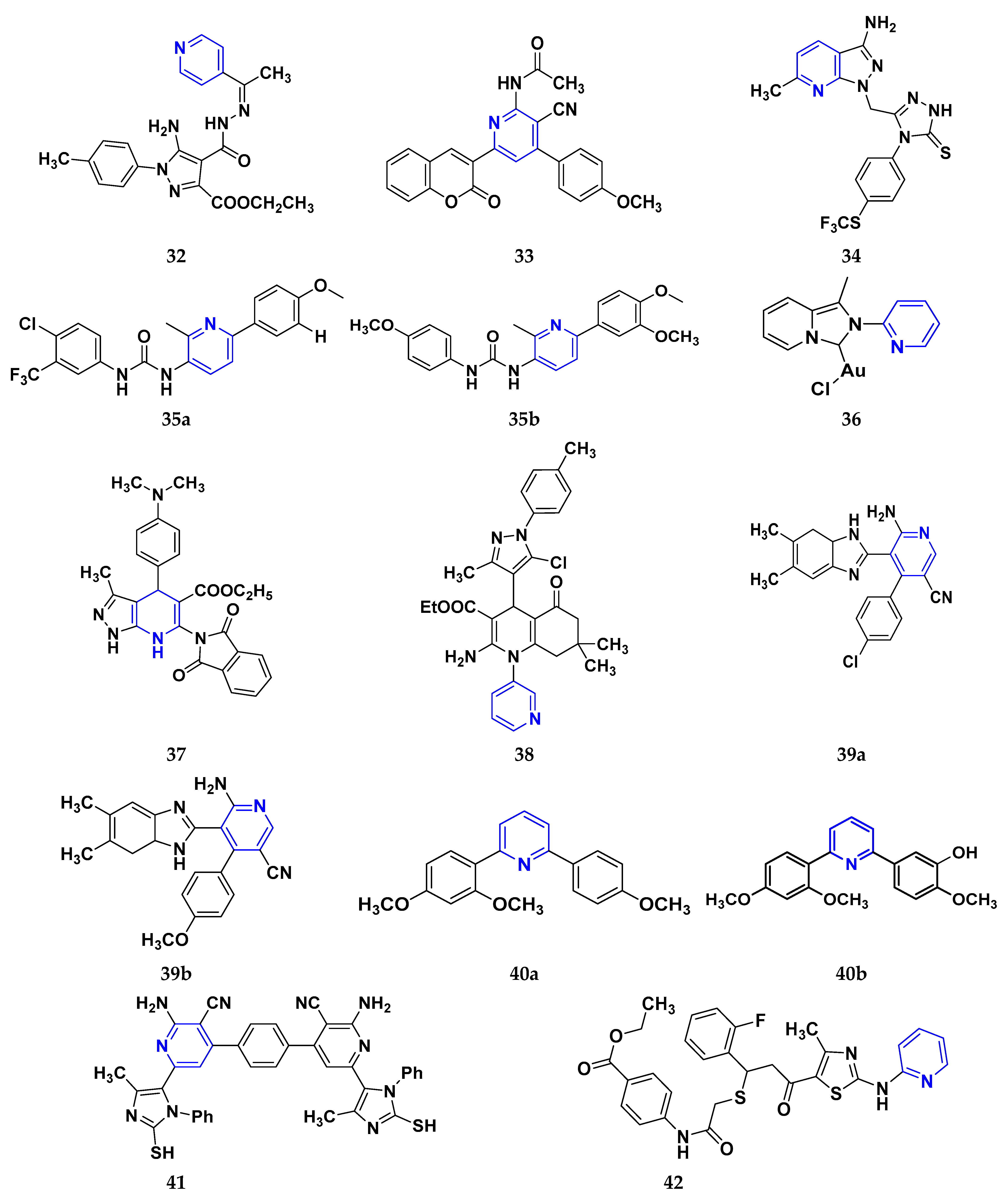
Figure 6. Pyridine derivatives (32–42) as anticancer agents.
References
- Angre, T.; Kumar, A.; Singh, A.K.; Thareja, S.; Kumar, P. Role of Collagen Regulators in Cancer Treatment: A Comprehensive Review. Anti-Cancer Agents Med. Chem. 2022, 22, 2956–2984.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 2021, 71, 209–249.
- Gu, L.; Shan, T.; Ma, Y.-X.; Tay, F.R.; Niu, L. Novel biomedical applications of crosslinked collagen. Trends Biotechnol. 2019, 37, 464–491.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 2018, 68, 394–424.
- Singh, A.K.; Kumar, A.; Thareja, S.; Kumar, P. Current insights into the role of BRAF inhibitors in treatment of melanoma. Anti-Cancer Agents Med. Chem. 2022, 1–7.
- Rang, H.; Dale, M.; Ritter, J.; Moore, P. Edinburgh, Scotland, Pharmacology, 5th ed.; Churchill Livingstone: London, UK, 2003.
- Williams David, A.; Lemke Thomson, A. Foyes Principle of Medicinal Chemistry; Lippincott, Williams & Wilkins: Philadelphia, FL, USA, 2002.
- Stefanowicz, P.; Jaremko, Ł.; Jaremko, M.; Lis, T. Crystal-state studies on p-toluenesulfonates of N-oxyimides—A possible structural basis of serine proteases inhibition. New J. Chem. 2006, 30, 258–265.
- Alvárez-Builla, J.; Barluenga, J. Heterocyclic compounds: An introduction. Mod Heterocycl Chem 2011, 1, 1–9.
- Ferreira, P.M.; Maia, H.L.; Monteiro, L.s.S. Synthesis of 2, 3, 5-substituted pyrrole derivatives. Tetrahedron. Lett. 2002, 43, 4491–4493.
- Kijewska, M.; Sharfalddin, A.A.; Jaremko, Ł.; Cal, M.; Setner, B.; Siczek, M.; Stefanowicz, P.; Hussien, M.A.; Emwas, A.-H.; Jaremko, M. Lossen Rearrangement of p-Toluenesulfonates of N-Oxyimides in Basic Condition, Theoretical Study, and Molecular Docking. Front. Chem. 2021, 9, 662533.
- Shukla, P.K.; Verma, A.; Mishra, P. Significance of nitrogen heterocyclic nuclei in the search of pharmacological active compounds. In New Perspective in Agricultural and Human Health; Shukla, R.P., Mishra, R.S., Tripathi, A.D., Yadav, A.K., Tiwari, M., Mishra, R.R., Eds.; Bharti Publications: Delhi, India, 2017; pp. 100–126.
- Martins, P.; Jesus, J.; Santos, S.; Raposo, L.R.; Roma-Rodrigues, C.; Baptista, P.V.; Fernandes, A.R. Heterocyclic anticancer compounds: Recent advances and the paradigm shift towards the use of nanomedicine’s tool box. Molecules 2015, 20, 16852–16891.
- Ajani, O.; Audu, O.; Aderohunmu, D.; Owolabi, F.; Olomieja, A. Undeniable pharmacological potentials of quinazoline motifs in therapeutic medicine. Am. J. Drug Discov. Dev. 2017, 7, 1–24.
- Özkay, Y.; Işıkdağ, İ.; İncesu, Z.; Akalın, G. Synthesis of 2-substituted-N- acetamide derivatives and evaluation of their anticancer activity. Eur. J. Med. Chem. 2010, 45, 3320–3328.
- Jangale, A.D.; Dalal, D.S. Green synthetic approaches for biologically relevant organic compounds. Synth. Commun. 2017, 47, 2139–2173.
- Yang, D.; An, B.; Wei, W.; Tian, L.; Huang, B.; Wang, H. Copper-catalyzed domino synthesis of nitrogen heterocycle-fused benzoimidazole and 1, 2, 4-benzothiadiazine 1, 1-dioxide derivatives. ACS Comb. Sci. 2015, 17, 113–119.
- Srivastava, M.; Singh, J.; Singh, S.B.; Tiwari, K.; Pathak, V.K.; Singh, J. Synthesis of novel fused heterocycle-oxa-aza-phenanthrene and anthracene derivatives via sequential one-pot synthesis in aqueous micellar system. Green Chem. 2012, 14, 901–905.
- Czarnik, A.W. Peer Reviewed: Combinatorial Chemistry. Anal. Chem. 1998, 70, 378–386.
- García-Valverde, M.; Torroba, T. Sulfur-Nitrogen Heterocycles. Guest editorial; Molecules 2005, 10, 318–320.
- Al-Ghorbani, M.; Bushra, B.A.; Mamatha, S.; Khanum, S.A. Piperazine and morpholine: Synthetic preview and pharmaceutical applications. Res. J. Pharm. Technol. 2015, 8, 611–628.
- Heravi, M.M.; Zadsirjan, V. Prescribed drugs containing nitrogen heterocycles: An overview. RSC Adv. 2020, 10, 44247–44311.
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, 930–940.
- Lagoja, I.M. Pyrimidine as constituent of natural biologically active compounds. Chem. Biodivers. 2005, 2, 1–50.
- Albratty, M.; Alhazmi, H.A. Novel pyridine and pyrimidine derivatives as promising anticancer agents: A review. Arab. J. Chem. 2022, 15, 103846.
- Fathalla, O.A.; Mohamed, N.A.; El-Serwy, W.S.; AbdelHamid, H.F.; El-Moez, A.; Sherein, I.; Soliman, A.-m.M. Synthesis of some new pyrimidine derivatives and evaluation of their anticancer and antibacterial activities. Res. Chem. Intermed. 2013, 39, 821–841.
- Ahmed, M.H.; El-Hashash, M.A.; Marzouk, M.I.; El-Naggar, A.M. Synthesis and antitumor activity of some nitrogen heterocycles bearing pyrimidine moiety. J. Heterocycl. Chem. 2020, 57, 3412–3427.
- Gupta, S.; Bartwal, G.; Singh, A.; Tanwar, J.; Khurana, J. Design, synthesis and biological evaluation of spiro-isoquinoline-pyrimidine derivatives as anticancer agents against MCF-7 cancer cell lines. Results Chem. 2022, 4, 100386.
- Al-Issa, S. Synthesis and anticancer activity of some fused pyrimidines and related heterocycles. Saudi Pharm. J. 2013, 21, 305–316.
- Osmaniye, D.; Hıdır, A.; Sağlık, B.N.; Levent, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis of New Pyrimidine-Triazole Derivatives and Investigation of Their Anticancer Activities. Chem. Biodivers. 2022, 19, e202200216.
- Qin, W.-W.; Sang, C.-Y.; Zhang, L.-L.; Wei, W.; Tian, H.-Z.; Liu, H.-X.; Chen, S.-W.; Hui, L. Synthesis and biological evaluation of 2, 4-diaminopyrimidines as selective Aurora A kinase inhibitors. Eur. J. Med. Chem. 2015, 95, 174–184.
- Venturini Filho, E.; Pina, J.W.; Antoniazi, M.K.; Loureiro, L.B.; Ribeiro, M.A.; Pinheiro, C.B.; Guimarães, C.J.; de Oliveira, F.C.; Pessoa, C.; Taranto, A.G. Synthesis, docking, machine learning and antiproliferative activity of the 6-ferrocene/heterocycle-2-aminopyrimidine and 5-ferrocene-1H-Pyrazole derivatives obtained by microwave-assisted Atwal reaction as potential anticancer agents. Bioorg. Med. Chem. Lett. 2021, 48, 128240.
- Mohi El-Deen, E.M.; Anwar, M.M.; El-Gwaad, A.A.A.; Karam, E.A.; El-Ashrey, M.K.; Kassab, R.R. Novel Pyridothienopyrimidine Derivatives: Design, Synthesis and Biological Evaluation as Antimicrobial and Anticancer Agents. Molecules 2022, 27, 803.
- Al-Anazi, M.; Khairuddean, M.; Al-Najjar, B.O.; Alidmat, M.M.; Kamal, N.N.S.N.M.; Muhamad, M. Synthesis, anticancer activity and docking studies of pyrazoline and pyrimidine derivatives as potential epidermal growth factor receptor (EGFR) inhibitors. Arab. J. Chem. 2022, 15, 103864.
- El-Metwally, S.A.; Abou-El-Regal, M.M.; Eissa, I.H.; Mehany, A.B.; Mahdy, H.A.; Elkady, H.; Elwan, A.; Elkaeed, E.B. Discovery of thieno pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents. Bioorg. Chem. 2021, 112, 104947.
- Madia, V.N.; Nicolai, A.; Messore, A.; De Leo, A.; Ialongo, D.; Tudino, V.; Saccoliti, F.; De Vita, D.; Scipione, L.; Artico, M.; et al. Design, synthesis and biological evaluation of new pyrimidine derivatives as anticancer agents. Molecules 2021, 26, 771.
- Marella, A.; Tanwar, O.P.; Saha, R.; Ali, M.R.; Srivastava, S.; Akhter, M.; Shaquiquzzaman, M.; Alam, M.M. Quinoline: A versatile heterocyclic. Saudi Pharm. J. 2013, 21, 1–12.
- Hamdy, R.; Elseginy, S.A.; Ziedan, N.I.; Jones, A.T.; Westwell, A.D. New quinoline-based heterocycles as anticancer agents targeting bcl-2. Molecules 2019, 24, 1274.
- Jin, X.-Y.; Chen, H.; Li, D.-D.; Li, A.-L.; Wang, W.-Y.; Gu, W. Design, synthesis, and anticancer evaluation of novel quinoline derivatives of ursolic acid with hydrazide, oxadiazole, and thiadiazole moieties as potent MEK inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 955–972.
- Katariya, K.D.; Shah, S.R.; Reddy, D. Anticancer, antimicrobial activities of quinoline based hydrazone analogues: Synthesis, characterization and molecular docking. Bioorg. Chem. 2020, 94, 103406.
- George, R.F.; Samir, E.M.; Abdelhamed, M.N.; Abdel-Aziz, H.A.; Abbas, S.E. Synthesis and anti-proliferative activity of some new quinoline based 4, 5-dihydropyrazoles and their thiazole hybrids as EGFR inhibitors. Bioorg. Chem. 2019, 83, 186–197.
- Köprülü, T.K.; Ökten, S.; Atalay, V.E.; Tekin, Ş.; Çakmak, O. Biological activity and molecular docking studies of some new quinolines as potent anticancer agents. Med. Oncol. 2021, 38, 1–11.
- Ramya, P.S.; Guntuku, L.; Angapelly, S.; Karri, S.; Digwal, C.S.; Babu, B.N.; Naidu, V.; Kamal, A. Curcumin inspired 2-chloro/phenoxy quinoline analogues: Synthesis and biological evaluation as potential anticancer agents. Bioorg. Med. Chem. Lett. 2018, 28, 892–898.
- Upadhyay, K.D.; Dodia, N.M.; Khunt, R.C.; Chaniara, R.S.; Shah, A.K. Synthesis and biological screening of pyrano quinoline analogues as anti-inflammatory and anticancer agents. ACS Med. Chem. Lett. 2018, 9, 283–288.
- Hagras, M.; El Deeb, M.A.; Elzahabi, H.S.; Elkaeed, E.B.; Mehany, A.B.; Eissa, I.H. Discovery of new quinolines as potent colchicine binding site inhibitors: Design, synthesis, docking studies, and anti-proliferative evaluation. J. Enzym. Inhib. Med. Chem. 2021, 36, 640–658.
- Mirzaei, S.; Hadizadeh, F.; Eisvand, F.; Mosaffa, F.; Ghodsi, R. Synthesis, structure-activity relationship and molecular docking studies of novel quinoline-chalcone hybrids as potential anticancer agents and tubulin inhibitors. J. Mol. Struct. 2020, 1202, 127310.
- Chate, A.V.; Kamdi, S.P.; Bhagat, A.N.; Jadhav, C.K.; Nipte, A.; Sarkate, A.P.; Tiwari, S.V.; Gill, C.H. Design, Synthesis and SAR Study of Novel Spiro Quinoline-10,5′-PyrroloPyrimidine] Derivatives as Promising Anticancer Agents. J. Heterocycl. Chem. 2018, 55, 2297–2302.
- Ziarani, G.; Moradi, R.; Lashgari, N.; Kruger, H.G. Metal-Free Synthetic Organic Dyes; Elsevier: Amsterdam, The Netherlands, 2018.
- Murali, K.; Sparkes, H.A.; Prasad, K.J.R. Synthesis of hetero annulated isoxazolo-, pyrido-and pyrimido carbazoles: Screened for in vitro antitumor activity and structure activity relationships, a novel 2-amino-4-(3′-bromo-4′-methoxyphenyl)-8-chloro-11H-pyrimido carbazole as an antitumor agent. Eur. J. Med. Chem. 2017, 128, 319–331.
- Wang, Y.-Q.; Li, X.-H.; He, Q.; Chen, Y.; Xie, Y.-Y.; Ding, J.; Miao, Z.-H.; Yang, C.-H. Design, synthesis and biological evaluation of substituted 11H-benzo carbazole-5-carboxamides as novel antitumor agents. Eur. J. Med. Chem. 2011, 46, 5878–5884.
- Debray, J.; Zeghida, W.; Baldeyrou, B.; Mahieu, C.; Lansiaux, A.; Demeunynck, M. Montmorillonite K-10 catalyzed cyclization of N-ethoxycarbonyl-N′-arylguanidines: Access to pyrimido carbazole and pyrimido indole derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 4244–4247.
- Sun, L.; Wu, Y.; Liu, Y.; Chen, X.; Hu, L. Novel carbazole sulfonamide derivatives of antitumor agent: Synthesis, antiproliferative activity and aqueous solubility. Bioorg. Med. Chem. Lett. 2017, 27, 261–265.
- Arya, K.R.; Rajendra Prasad, K.J. Rational eco-compatible synthesis and biological screening of new highly functionalized pyrido carbazole derivatives: A novel class of antioxidant and anticancer agents. Synth. Commun. 2018, 48, 1465–1481.
- Padmaja, P.; Rao, G.K.; Indrasena, A.; Reddy, B.V.S.; Patel, N.; Shaik, A.B.; Reddy, N.; Dubey, P.K.; Bhadra, M.P. Synthesis and biological evaluation of novel pyrano carbazole derivatives as anti-tumor agents inducing apoptosis via tubulin polymerization inhibition. Org. Biomol. Chem. 2015, 13, 1404–1414.
- Patel, M.; Pandey, N.; Timaniya, J.; Parikh, P.; Chauhan, A.; Jain, N.; Patel, K. Coumarin–carbazole based functionalized pyrazolines: Synthesis, characterization, anticancer investigation and molecular docking. RSC Adv. 2021, 11, 27627–27644.
- Huang, W.; Gao, Z.; Zhang, Z.; Fang, W.; Wang, Z.; Wan, Z.; Shi, L.; Wang, K.; Ke, S. Selective and effective anticancer agents: Synthesis, biological evaluation and structure–activity relationships of novel carbazole derivatives. Bioorg. Chem. 2021, 113, 104991.
- Singh, H.; Walker, A.J.; Amiri-Kordestani, L.; Cheng, J.; Tang, S.; Balcazar, P.; Barnett-Ringgold, K.; Palmby, T.R.; Cao, X.; Zheng, N. US Food and Drug Administration Approval: Neratinib for the Extended Adjuvant Treatment of Early-Stage HER2-Positive Breast CancerFDA Approval Summary: Neratinib. Clin. Cancer Res. 2018, 24, 3486–3491.
- Vairavelu, L.; Zeller, M.; Prasad, K.R. Solvent-free synthesis of heteroannulated carbazoles: A novel class of anti-tumor agents. Bioorg. Chem. 2014, 54, 12–20.
- Altaf, A.A.; Shahzad, A.; Gul, Z.; Rasool, N.; Badshah, A.; Lal, B.; Khan, E. A review on the medicinal importance of pyridine derivatives. J. Drug Des. Med. Chem. 2015, 1, 1–11.
- Gomha, S.M.; Abdelrazek, F.M.; Abdelrahman, A.H.; Metz, P. Synthesis of some new pyridine-based heterocyclic compounds with anticipated antitumor activity. J. Heterocycl. Chem. 2018, 55, 1729–1737.
- Fayed, E.A.; Sabour, R.; Harras, M.F.; Mehany, A. Design, synthesis, biological evaluation and molecular modeling of new coumarin derivatives as potent anticancer agents. Med. Chem. Res. 2019, 28, 1284–1297.
- Nagender, P.; Kumar, R.N.; Reddy, G.M.; Swaroop, D.K.; Poornachandra, Y.; Kumar, C.G.; Narsaiah, B. Synthesis of novel hydrazone and azole functionalized pyrazolo pyridine derivatives as promising anticancer agents. Bioorg. Med. Chem. Lett. 2016, 26, 4427–4432.
- El-Naggar, M.; Almahli, H.; Ibrahim, H.S.; Eldehna, W.M.; Abdel-Aziz, H.A. Pyridine-ureas as potential anticancer agents: Synthesis and in vitro biological evaluation. Molecules 2018, 23, 1459.
- Dinda, J.; Nandy, A.; Rana, B.K.; Bertolasi, V.; Saha, K.D.; Bielawski, C.W. Cytotoxicity of silver (I), gold (I) and gold (III) complexes of a pyridine wingtip substituted annelated N-heterocyclic carbene. RSC Adv. 2014, 4, 60776–60784.
- El-Gohary, N.; Gabr, M.; Shaaban, M. Synthesis, molecular modeling and biological evaluation of new pyrazolo pyridine analogs as potential antimicrobial, antiquorum-sensing and anticancer agents. Bioorg. Chem. 2019, 89, 102976.
- Sangani, C.B.; Makawana, J.A.; Zhang, X.; Teraiya, S.B.; Lin, L.; Zhu, H.-L. Design, synthesis and molecular modeling of pyrazole–quinoline–pyridine hybrids as a new class of antimicrobial and anticancer agents. Eur. J. Med. Chem. 2014, 76, 549–557.
- Elzahabi, H.S. Synthesis, characterization of some benzazoles bearing pyridine moiety: Search for novel anticancer agents. Eur. J. Med. Chem. 2011, 46, 4025–4034.
- Zheng, S.; Zhong, Q.; Mottamal, M.; Zhang, Q.; Zhang, C.; LeMelle, E.; McFerrin, H.; Wang, G. Design, synthesis, and biological evaluation of novel pyridine-bridged analogues of combretastatin-A4 as anticancer agents. J. Med. Chem. 2014, 57, 3369–3381.
- Abbas, I.; Gomha, S.; Elaasser, M.; Bauomi, M. Synthesis and biological evaluation of new pyridines containing imidazole moiety as antimicrobial and anticancer agents. Turk. J. Chem. 2015, 39, 334–346.
- Ivasechko, I.; Yushyn, I.; Roszczenko, P.; Senkiv, J.; Finiuk, N.; Lesyk, D.; Holota, S.; Czarnomysy, R.; Klyuchivska, O.; Khyluk, D. Development of Novel Pyridine-Thiazole Hybrid Molecules as Potential Anticancer Agents. Molecules 2022, 27, 6219.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.0K
Revisions:
2 times
(View History)
Update Date:
02 Mar 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No