Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vishwanath Venketaraman | -- | 1630 | 2023-02-27 07:11:04 | | | |
| 2 | Dean Liu | -24 word(s) | 1606 | 2023-02-28 02:08:21 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mundra, A.; Yegiazaryan, A.; Karsian, H.; Alsaigh, D.; Bonavida, V.; Frame, M.; May, N.; Gargaloyan, A.; Abnousian, A.; Venketaraman, V. Type I Interferons in Mycobacterium tuberculosis Infection. Encyclopedia. Available online: https://encyclopedia.pub/entry/41682 (accessed on 28 July 2026).
Mundra A, Yegiazaryan A, Karsian H, Alsaigh D, Bonavida V, Frame M, et al. Type I Interferons in Mycobacterium tuberculosis Infection. Encyclopedia. Available at: https://encyclopedia.pub/entry/41682. Accessed July 28, 2026.
Mundra, Akaash, Aram Yegiazaryan, Haig Karsian, Dijla Alsaigh, Victor Bonavida, Mitchell Frame, Nicole May, Areg Gargaloyan, Arbi Abnousian, Vishwanath Venketaraman. "Type I Interferons in Mycobacterium tuberculosis Infection" Encyclopedia, https://encyclopedia.pub/entry/41682 (accessed July 28, 2026).
Mundra, A., Yegiazaryan, A., Karsian, H., Alsaigh, D., Bonavida, V., Frame, M., May, N., Gargaloyan, A., Abnousian, A., & Venketaraman, V. (2023, February 27). Type I Interferons in Mycobacterium tuberculosis Infection. In Encyclopedia. https://encyclopedia.pub/entry/41682
Mundra, Akaash, et al. "Type I Interferons in Mycobacterium tuberculosis Infection." Encyclopedia. Web. 27 February, 2023.
Copy Citation
Tuberculosis (TB) is a leading cause of mortality due to infectious disease and rates have increased during the emergence of COVID-19, but many of the factors determining disease severity and progression remain unclear. Type I Interferons (IFNs) have diverse effector functions that regulate innate and adaptive immunity during infection with microorganisms.
tuberculosis
mycobacteria
interferons
interleukins
1. Type I IFN Transcriptional Signature in M. tb Infection
Gene signatures in the blood of patients with active TB can be used as a marker for diagnosis, disease manifestation, and treatment monitoring. Berry et al. conducted a landmark study which demonstrated that patients with active TB have a predominantly type I IFN-inducible gene signature that correlates with lung radiographic disease severity and is downregulated following treatment [1]. These findings have since been replicated in numerous studies [2]. Bloom et al. and Wang et al. described that overexpressed IFN-inducible genes tended to be primarily in neutrophils and monocytes, hinting at overactivation and infection of these cell types during M. tb infection [3][4]. Studies have demonstrated early overexpression of type I IFN genes in patients with latent TB that eventually progressed to active infection. An enhanced type I IFN signature was correlated with progression to active TB up to 18 months prior to diagnosis [1][5][6][7][8][9]. This suggests that the type I IFN response can precede the onset of active disease and symptoms. Monitoring the type I IFN response to chemotherapy would thus be more advantageous compared to the detection of acid-fast bacilli and provide an earlier assessment in the clinical management of infected patients [10]. Multiple studies also described an increased resistance to TB in individuals with a mutation in IFNAR1 (IFN 1 receptor) that impaired type I IFN signaling [4][11][12]. In addition, patients with a deficiency in the gene encoding ISG15 displayed increased type I IFN responses and were seen to be more susceptible to M. tb [13]. Multiple studies have reported reactivation of TB in patients receiving IFN-α-based therapy for chronic viral hepatitis [14][15][16][17][18][19].
2. Mechanisms of Type I IFN Induction in M. tb Infection: ESX-1 Protein Secretion System and Pattern Recognition Receptors
The outcomes of an infection are determined by the dynamic interplay between virulence factors produced by a pathogen and the immune response mounted by the host. Immune responses are characterized by increased cytokines, chemokines, and other products normally meant to protect the host. Various signaling pathways have been described that induce type I IFN expression in response to M. tb infection. The M. tb virulence factor ESX-1 protein secretion system is seen to be a major contributor to increased levels of type I IFN. Studies have shown that type I IFN responses were diminished in human macrophages and mice models, although not completely, if the genomic region of difference-1 (RD1) of ESX-1 was deleted [20][21][22][23][24]. ESX-1 disrupts the phagosomal membrane leading to mitochondrial stress and resultant leakage of mitochondrial DNA and mycobacterial products (such as early secreted antigenic target-6 and culture-filtrate protein-10) into the cytosol [22]. These products are recognized by cyclic GMP-AMP synthase (cGAS), which then initiates the formation of a second messenger, cyclic-GMP-AMP (cGAMP) [Figure 1]. cGAMP and bacterial cyclic dinucleotides interact with STING or STING-accessory molecule DDX41. Activation of STING results in its relocation to the perinuclear Golgi, where it then initiates recruitment and activation of TANK-binding Kinase 1 (TBK1). TBK1 activates NF-kB, which mediates the induction of pro-inflammatory cytokines, as well as phosphorylates interferon regulatory factors IRF3 and IRF5, both of which facilitate type I IFN expression [9][21]. A study conducted by Wiens and Ernst demonstrated that the extent of mitochondrial DNA released during M. tb infection may determine the extent of type I IFN expression [25]. cGAMP can also access uninfected cells via gap junctions leading to STING activation and type I IFN expression in nearby bystander cells [24][26]. Both cGAS- and STING-deficient human and mouse macrophages are impaired in their ability to express IFN-β in response to M. tb infection [24][27][28][29]. Toll-like receptors (TLR) are transmembrane proteins that serve as pattern recognition receptors and can sense M. tb components [30]. The ligands of different M. tb strains were seen to dimerize different TLRs, resulting in intracellular signaling and varying macrophage responses that drive the production of type I IFN [31][32]. Other pattern recognition receptors including Nod-like receptor (NOD2) have also been linked to increased type I IFN expression [21][33]. NOD2 recognizes the mycobacterial product N-glycosylated muramyl dipeptide, which leads to RIP2-mediated activation of TBK1, resulting in downstream type I IFN expression via IRF5 dimerization and nuclear translocation. In a study performed by Pandey et al., this pathway was significantly diminished in infection with ESX-1-deficient M. tb [21]. In summation, extensive literature reveals that different M. tb strains induce varying type I IFN responses via multiple mechanisms and signaling pathways which may result in differing virulence.
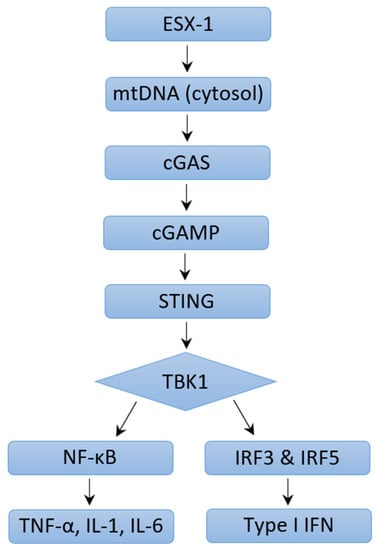
Figure 1. Mechanisms of Type I IFN Induction through ESX-1 pathway.
3. Mechanisms of Type I IFN Induction in M. tb Infection: ESX-1 Protein Secretion System Independent Mechanisms
As mentioned earlier, type I IFN expression is reduced but not eliminated in ESX-1-deficient M. tb. The attenuated Mycobacterium bovis strain Bacillus Calmette–Guérin (BCG) lacks the RD-1 region that encodes the ESX-1 secretion system, yet this strain still produces a type I IFN response in human macrophages [20][27]. This response was reduced in STING-cells, hinting that this is a STING-dependent process and that mycobacteria components can still gain access to the cytosol in the absence of ESX-1.
4. Pathogenic Effects of Type I IFN Signaling during M. tb Infection
How type I IFNs exacerbate M. tb infection is not yet fully understood, but many mechanisms have been described in various studies. Type I IFN inhibition of protective cytokines (IFN-γ, TFN-α, IL-12) and Th1 cell responsiveness in human cells was described in multiple studies and thought to be mediated by impairment of IL-12 production [20][34][35][36]. Mouse models infected with the hypervirulent HN878 M. tb strain were particularly seen to have reduced TH1 responses and lower IFN-y production secondary to a high type I IFN response [37][38][39]. Multiple studies have also shown that type I IFNs limited human and mouse mycobacteria-restricting monocyte/macrophage response to the antibacterial effects of IFN-γ during M. tb infections and this process was mediated by IL-10, a cytokine that can impair antimycobacterial immune responses during infection [34][35][40]. IL-10 inhibits the IFN-γ-driven vitamin D3/cathelicidin/defensin beta 4A antimicrobial defense pathway [35]. In addition, type I IFNs have also been shown to promote early cell death of alveolar macrophages and increase accumulation of myeloid cells which contribute to the spread of infection and lung inflammation [11][41]. Data from the study by Moreira-Teixeira et al. demonstrated that increased type I IFN signaling promoted pathogenesis of M. tb infection in mouse models via stimulation of neutrophils and a pathological neutrophil extracellular trap (NET) response [42]. NETs are networks of extracellular fibers composed of DNA from neutrophils which bind pathogens and are typically involved in an antimicrobial response. In M. tb infection, NETosis (NET activation and release) contributed to disease exacerbation. Type I IFNs have been reported to inhibit the production of IL-1α and IL-1β, which are important for host defense against M. tb infection in humans and mice [20][34][36][43][44][45][46]. This inhibition was shown to be dependent on NO synthase 2 and IL-10 [20][36][40]. NO interference with the NLRP3 inflammasome assembly impairs IL-1β processing by M. tb-infected macrophages [47]. Prostaglandin E2 (PGE2) serves as a mediator of IL-1-dependent host protection and is known to prevent necrosis of M. tb-infected macrophages by promoting apoptosis which limits pathogenic dissemination [48][49]. Type I IFNs are shown to inhibit PGE2, and this could thus promote necrosis and alter the ratio of host-protective PGE2 to host-detrimental 5-lipoxygenase products such as lipoxin A4 [50][51]. Animal model studies revealed evidence that suggests the degree of M. tb-induced type I IFN expression in mice is correlated with the virulence of M. tb strains [25][37]. In a study performed by Manca et al., administration of anti-IFNα/β antibodies to mice before and upon infection with M. tb was associated with a long-term survival benefit but no significant change in bacterial burden [38]. This was further affirmed in studies which demonstrated that administration of IFN-α/β in mice impaired host survival and worsened lung inflammation in the face of M. tb infection [38][41][50]. Dynamic cytokine interactions mediate the pathogenic and protective functions of various molecules in M. tb infection, and a greater understanding of these key mediators opens the door for host-directed therapeutic intervention in M. tb infection.
5. Potential Protective Functions of Type I IFN in M. tb Infection
There is emerging evidence that type I IFNs can display protective functions in M. tb infection under certain conditions. Ward et al. and Bax et al. demonstrated that patients with IFN-γ receptor signaling deficiencies who failed conventional TB antimycobacterial chemotherapy (consisting of isoniazid, rifampicin, ethambutol, and pyrazinamide) or had recurrent disease benefited from inhaled or subcutaneous coadministration of IFN-α in addition to standard TB antimycobacterial chemotherapy [52][53]. Further studies described that in the absence of IFN-γ signaling, type I IFNs may contribute to the survival of myeloid cells that help control M. tb pathogenesis [31][54]. These findings may explain some of the mechanisms underlying the beneficial effects of IFN-α treatment in patients with compromised IFN-γ responses. Rivas-Santiago and Guerrero showed that administration of IFN-α during BCG vaccination promoted production of protective cytokines (IFN-γ, TNF-α, IL-12) and provided increased protection against M. tb infection than that with BCG alone [55]. McNab et al. further demonstrated that basal type I IFN signaling is necessary for the maximum production of IL-12 and TNF-α by macrophages in response to M. tb [40]. The findings presented suggest that an understanding of balance between type I and type II IFNs is necessary to define the ability of the host to control M. tb infection. The interplay of these molecules could be harnessed to develop interventions against infection.
References
- Berry, M.P.R.; Graham, C.M.; McNab, F.W.; Xu, Z.; Bloch, S.A.A.; Oni, T.; Wilkinson, K.A.; Banchereau, R.; Skinner, J.; Wilkinson, R.J.; et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010, 466, 973–977.
- Ottenhoff, T.H.M.; Dass, R.H.; Yang, N.; Zhang, M.; Wong, H.E.E.; Sahiratmadja, E.; Khor, C.C.; Alisjahbana, B.; Van Crevel, R.; Marzuki, S.; et al. Genome-Wide Expression Profiling Identifies Type 1 Interferon Response Pathways in Active Tuberculosis. PLoS ONE 2012, 7, e45839.
- Bloom, C.I.; Graham, C.M.; Berry, M.P.R.; Rozakeas, F.; Redford, P.S.; Wang, Y.; Xu, Z.; Wilkinson, K.A.; Wilkinson, R.J.; Kendrick, Y.; et al. Transcriptional Blood Signatures Distinguish Pulmonary Tuberculosis, Pulmonary Sarcoidosis, Pneumonias and Lung Cancers. PLoS ONE 2013, 8, e70630.
- Wang, J.; Hussain, T.; Zhang, K.; Liao, Y.; Yao, J.; Song, Y.; Sabir, N.; Cheng, G.; Dong, H.; Li, M.; et al. Inhibition of type I interferon signaling abrogates early Mycobacterium bovis infection. BMC Infect. Dis. 2019, 19, 1031.
- Zak, D.E.; Penn-Nicholson, A.; Scriba, T.J.; Thompson, E.; Suliman, S.; Amon, L.M.; Mahomed, H.; Erasmus, M.; Whatney, W.; Hussey, G.D.; et al. A blood RNA signature for tuberculosis disease risk: A prospective cohort study. Lancet 2016, 387, 2312–2322.
- Scriba, T.J.; Penn-Nicholson, A.; Shankar, S.; Hraha, T.; Thompson, E.G.; Sterling, D.; Nemes, E.; Darboe, F.; Suliman, S.; Amon, L.M.; et al. Sequential inflammatory processes define human progression from M. tuberculosis infection to tuberculosis disease. PLoS Pathog. 2017, 13, e1006687.
- Singhania, A.; Verma, R.; Graham, C.M.; Lee, J.; Tran, T.; Richardson, M.; Lecine, P.; Leissner, P.; Berry, M.P.R.; Wilkinson, R.J.; et al. A modular transcriptional signature identifies phenotypic heterogeneity of human tuberculosis infection. Nat. Commun. 2018, 9, 2308.
- Moreira-Teixeira, L.; Mayer-Barber, K.; Sher, A.; O’Garra, A. Type I interferons in tuberculosis: Foe and occasionally friend. J. Exp. Med. 2018, 215, 1273–1285.
- Donovan, M.L.; Schultz, T.E.; Duke, T.J.; Blumenthal, A. Type I Interferons in the Pathogenesis of Tuberculosis: Molecular Drivers and Immunological Consequences. Front. Immunol. 2017, 8, 1633.
- Taneja, V.; Kalra, P.; Goel, M.; Khilnani, G.C.; Saini, V.; Prasad, G.B.K.S.; Gupta, U.D.; Prasad, H.K. Impact and prognosis of the expression of IFN-α among tuberculosis patients. PLoS ONE 2020, 15, e0235488.
- Dorhoi, A.; Yeremeev, V.; Nouailles, G.; Weiner, J.; Jörg, S.; Heinemann, E.; Oberbeck-Müller, D.; Knaul, J.K.; Vogelzang, A.; Reece, S.T.; et al. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur. J. Immunol. 2014, 44, 2380–2393.
- Zhang, G.; Deweerd, N.A.; Stifter, S.A.; Liu, L.; Zhou, B.; Wang, W.; Zhou, Y.; Ying, B.; Hu, X.; Matthews, A.Y.; et al. A proline deletion in IFNAR1 impairs IFN-signaling and underlies increased resistance to tuberculosis in humans. Nat. Commun. 2018, 9, 85.
- Bogunovic, D.; Byun, M.; Durfee, L.A.; Abhyankar, A.; Sanal, O.; Mansouri, D.; Salem, S.; Radovanovic, I.; Grant, A.V.; Adimi, P.; et al. Mycobacterial Disease and Impaired IFN-γ Immunity in Humans with Inherited ISG15 Deficiency. Science 2012, 337, 1684–1688.
- Sabbatani, S.; Manfredi, R.; Marinacci, G.; Pavoni, M.; Cristoni, L.; Chiodo, F. Reactivation of severe, acute pulmonary tuberculosis during treatment with pegylated interferon-alpha and ribavirin for chronic HCV hepatitis. Scand. J. Infect. Dis. 2006, 38, 205–208.
- Farah, R.; Awad, J. The association of interferon with the development of pulmonary tuberculosis. Int. J. Clin. Pharmacol. Ther. 2007, 45, 598–600.
- Telesca, C.; Angelico, M.; Piccolo, P.; Nosotti, L.; Morrone, A.; Longhi, C.; Carbone, M.; Baiocchi, L. Interferon-alpha treatment of hepatitis D induces tuberculosis exacerbation in an immigrant. J. Infect. 2007, 54, e223–e226.
- Belkahla, N.; Kchir, H.; Maamouri, N.; Ouerghi, H.; Hariz, F.; Chouaib, S.; Chaabouni, H.; Mami, N. Réactivation d’une tuberculose sous bithérapie interféron-pégylé et ribavirine pour une hépatite chronique C. Rev. Med. Interne 2010, 31, e1–e3.
- De Uehara, S.N.O.; Emori, C.T.; Perez, R.M.; Mendes-Correa, M.C.J.; Ferreira, A.D.S.P.; Feldner, A.C.D.C.A.; Silva, A.E.B.; Filho, R.J.C.; Silva, I.S.D.S.E.; Ferraz, M.L.C.G. High incidence of tuberculosis in patients treated for hepatitis C chronic infection. Braz. J. Infect. Dis. 2016, 20, 205–209.
- Matsuoka, S.; Fujikawa, H.; Hasegawa, H.; Ochiai, T.; Watanabe, Y.; Moriyama, M. Onset of Tuberculosis from a Pulmonary Latent Tuberculosis Infection during Antiviral Triple Therapy for Chronic Hepatitis C. Intern. Med. 2016, 55, 2011–2017.
- Novikov, A.; Cardone, M.; Thompson, R.; Shenderov, K.; Kirschman, K.D.; Mayer-Barber, K.D.; Myers, T.G.; Rabin, R.L.; Trinchieri, G.; Sher, A.; et al. Mycobacterium tuberculosis Triggers Host Type I IFN Signaling to Regulate IL-1β Production in Human Macrophages. J. Immunol. 2011, 187, 2540–2547.
- Pandey, A.K.; Yang, Y.; Jiang, Z.; Fortune, S.M.; Coulombe, F.; Behr, M.A.; Fitzgerald, K.; Sassetti, C.M.; Kelliher, M.A. NOD2, RIP2 and IRF5 Play a Critical Role in the Type I Interferon Response to Mycobacterium tuberculosis. PLoS Pathog. 2009, 5, e1000500.
- Stanley, S.A.; Johndrow, J.E.; Manzanillo, P.; Cox, J.S. The Type I IFN Response to Infection with Mycobacterium tuberculosis Requires ESX-1-Mediated Secretion and Contributes to Pathogenesis. J. Immunol. 2007, 178, 3143–3152.
- Manzanillo, P.S.; Shiloh, M.U.; Portnoy, D.A.; Cox, J.S. Mycobacterium tuberculosis Activates the DNA-Dependent Cytosolic Surveillance Pathway within Macrophages. Cell Host Microbe 2012, 11, 469–480.
- Wassermann, R.; Gulen, M.F.; Sala, C.; Perin, S.G.; Lou, Y.; Rybniker, J.; Schmid-Burgk, J.L.; Schmidt, T.; Hornung, V.; Cole, S.T.; et al. Mycobacterium tuberculosis Differentially Activates cGAS- and Inflammasome-Dependent Intracellular Immune Responses through ESX-1. Cell Host Microbe 2015, 17, 799–810.
- Wiens, K.E.; Ernst, J.D. The Mechanism for Type I Interferon Induction by Mycobacterium tuberculosis is Bacterial Strain-Dependent. PLoS Pathog. 2016, 12, e1005809.
- Ablasser, A.; Schmid-Burgk, J.L.; Hemmerling, I.; Horvath, G.L.; Schmidt, T.; Latz, E.; Hornung, V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 2013, 503, 530–534.
- Dey, B.; Dey, R.J.; Cheung, L.S.; Pokkali, S.; Guo, H.; Lee, J.-H.; Bishai, W.R. A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat. Med. 2015, 21, 401–406.
- Watson, R.O.; Bell, S.L.; MacDuff, D.A.; Kimmey, J.M.; Diner, E.J.; Olivas, J.; Vance, R.E.; Stallings, C.L.; Virgin, H.W.; Cox, J.S. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 2015, 17, 811–819.
- Collins, A.C.; Cai, H.; Li, T.; Franco, L.H.; Li, X.-D.; Nair, V.R.; Scharn, C.R.; Stamm, C.E.; Levine, B.; Chen, Z.J.; et al. Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium tuberculosis. Cell Host Microbe 2015, 17, 820–828.
- Stamm, C.E.; Collins, A.C.; Shiloh, M.U. Sensing of Mycobacterium tuberculosis and consequences to both host and bacillus. Immunol. Rev. 2015, 264, 204–219.
- Moreira-Teixeira, L.; Sousa, J.; McNab, F.W.; Torrado, E.; Cardoso, F.; Machado, H.; Castro, F.; Cardoso, V.; Gaifem, J.; Wu, X.; et al. Type I IFN Inhibits Alternative Macrophage Activation during Mycobacterium tuberculosis Infection and Leads to Enhanced Protection in the Absence of IFN-γ Signaling. J. Immunol. 2016, 197, 4714–4726.
- Carmona, J.; Cruz, A.; Moreira-Teixeira, L.; Sousa, C.; Sousa, J.; Osorio, N.S.; Saraiva, A.L.; Svenson, S.; Kallenius, G.; Pedrosa, J.; et al. Mycobacterium tuberculosis Strains Are Differentially Recognized by TLRs with an Impact on the Immune Response. PLoS ONE 2013, 8, e67277.
- Leber, J.H.; Crimmins, G.T.; Raghavan, S.; Meyer-Morse, N.P.; Cox, J.S.; Portnoy, D.A. Distinct TLR- and NLR-Mediated Transcriptional Responses to an Intracellular Pathogen. PLoS Pathog. 2008, 4, e6.
- de Paus, R.A.; van Wengen, A.; Schmidt, I.; Visser, M.; Verdegaal, E.M.; van Dissel, J.T.; van de Vosse, E. Inhibition of the type I immune responses of human monocytes by IFN-α and IFN-β. Cytokine 2013, 61, 645–655.
- Teles, R.M.B.; Graeber, T.G.; Krutzik, S.R.; Montoya, D.; Schenk, M.; Lee, D.J.; Komisopoulou, E.; Kelly-Scumpia, K.; Chun, R.; Iyer, S.S.; et al. Type I Interferon Suppresses Type II Interferon-Triggered Human Anti-Mycobacterial Responses. Science 2013, 339, 1448–1453.
- Mayer-Barber, K.D.; Andrade, B.B.; Barber, D.L.; Hieny, S.; Feng, C.; Caspar, P.; Oland, S.; Gordon, S.; Sher, A. Innate and Adaptive Interferons Suppress IL-1α and IL-1β Production by Distinct Pulmonary Myeloid Subsets during Mycobacterium tuberculosis Infection. Immunity 2011, 35, 1023–1034.
- Manca, C.; Tsenova, L.; Bergtold, A.; Freeman, S.; Tovey, M.; Musser, J.M.; Barry, C.E.; Freedman, V.H.; Kaplan, G. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-α/β. Proc. Natl. Acad. Sci. USA 2001, 98, 5752–5757.
- Manca, C.; Tsenova, L.; Freeman, S.; Barczak, A.K.; Tovey, M.; Murray, P.J.; Barry, C.; Kaplan, G. Hypervirulent M. tuberculosis W/Beijing Strains Upregulate Type I IFNs and Increase Expression of Negative Regulators of the Jak-Stat Pathway. J. Interf. Cytokine Res. 2005, 25, 694–701.
- Ordway, D.; Henao-Tamayo, M.; Harton, M.; Palanisamy, G.; Troudt, J.; Shanley, C.; Basaraba, R.J.; Orme, I.M. The Hypervirulent Mycobacterium tuberculosis Strain HN878 Induces a Potent TH1 Response followed by Rapid Down-Regulation. J. Immunol. 2007, 179, 522–531.
- McNab, F.W.; Ewbank, J.; Howes, A.; Moreira-Teixeira, L.; Martirosyan, A.; Ghilardi, N.; Saraiva, M.; O’Garra, A. Type I IFN Induces IL-10 Production in an IL-27–Independent Manner and Blocks Responsiveness to IFN-γ for Production of IL-12 and Bacterial Killing in Mycobacterium tuberculosis–Infected Macrophages. J. Immunol. 2014, 193, 3600–3612.
- Antonelli, L.R.; Rothfuchs, A.; Gonçalves, R.; Roffê, E.; Cheever, A.W.; Bafica, A.; Salazar, A.M.; Feng, C.; Sher, A. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J. Clin. Investig. 2010, 120, 1674–1682.
- Moreira-Teixeira, L.; Stimpson, P.J.; Stavropoulos, E.; Hadebe, S.; Chakravarty, P.; Ioannou, M.; Aramburu, I.V.; Herbert, E.; Priestnall, S.L.; Suarez-Bonnet, A.; et al. Type I IFN exacerbates disease in tuberculosis-susceptible mice by inducing neutrophil-mediated lung inflammation and NETosis. Nat. Commun. 2020, 11, 5566.
- Fremond, C.M.; Togbe, D.; Doz, E.; Rose, S.; Vasseur, V.; Maillet, I.; Jacobs, M.; Ryffel, B.; Quesniaux, V.F.J. IL-1 Receptor-Mediated Signal Is an Essential Component of MyD88-Dependent Innate Response to Mycobacterium tuberculosis Infection. J. Immunol. 2007, 179, 1178–1189.
- Mayer-Barber, K.D.; Barber, D.L.; Shenderov, K.; White, S.D.; Wilson, M.S.; Cheever, A.; Kugler, D.; Hieny, S.; Caspar, P.; Núñez, G.; et al. Cutting Edge: Caspase-1 Independent IL-1β Production Is Critical for Host Resistance to Mycobacterium tuberculosis and Does Not Require TLR Signaling In Vivo. J. Immunol. 2010, 184, 3326–3330.
- Jayaraman, P.; Sada-Ovalle, I.; Nishimura, T.; Anderson, A.C.; Kuchroo, V.K.; Remold, H.G.; Behar, S.M. IL-1β Promotes Antimicrobial Immunity in Macrophages by Regulating TNFR Signaling and Caspase-3 Activation. J. Immunol. 2013, 190, 4196–4204.
- Zhang, G.; Zhou, B.; Li, S.; Yue, J.; Yang, H.; Wen, Y.; Zhan, S.; Wang, W.; Liao, M.; Zhang, M.; et al. Allele-Specific Induction of IL-1β Expression by C/EBPβ and PU.1 Contributes to Increased Tuberculosis Susceptibility. PLoS Pathog. 2014, 10, e1004426.
- Mishra, B.B.; Rathinam, V.A.K.; Martens, G.W.; Martinot, A.J.; Kornfeld, H.; A Fitzgerald, K.; Sassetti, C.M. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome–dependent processing of IL-1β. Nat. Immunol. 2012, 14, 52–60.
- Chen, M.; Divangahi, M.; Gan, H.; Shin, D.S.; Hong, S.; Lee, D.M.; Serhan, C.N.; Behar, S.M.; Remold, H.G. Lipid mediators in innate immunity against tuberculosis: Opposing roles of PGE2 and LXA4 in the induction of macrophage death. J. Exp. Med. 2008, 205, 2791–2801.
- Divangahi, M.; Chen, M.; Gan, H.; Desjardins, D.; Hickman, T.T.; Lee, D.M.; Fortune, S.; Behar, S.M.; Remold, H.G. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 2009, 10, 899–906.
- Mayer-Barber, K.D.; Andrade, B.B.; Oland, S.D.; Amaral, E.P.; Barber, D.L.; Gonzales, J.; Derrick, S.C.; Shi, R.; Kumar, N.P.; Wei, W.; et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 2014, 511, 99–103.
- Mayer-Barber, K.D.; Sher, A. Cytokine and lipid mediator networks in tuberculosis. Immunol. Rev. 2015, 264, 264–275.
- Ward, C.M.; Jyonouchi, H.; Kotenko, S.V.; Smirnov, S.V.; Patel, R.; Aguila, H.; McSherry, G.; Dashefsky, B.; Holland, S.M. Adjunctive treatment of disseminated Mycobacterium avium complex infection with interferon alpha-2b in a patient with complete interferon-gamma receptor R1 deficiency. Eur. J. Pediatr. 2006, 166, 981–985.
- Bax, H.I.; Freeman, A.F.; Ding, L.; Hsu, A.P.; Marciano, B.; Kristosturyan, E.; Jancel, T.; Spalding, C.; Pechacek, J.; Olivier, K.N.; et al. Interferon Alpha Treatment of Patients with Impaired Interferon Gamma Signaling. J. Clin. Immunol. 2013, 33, 991–1001.
- Desvignes, L.; Wolf, A.J.; Ernst, J.D. Dynamic Roles of Type I and Type II IFNs in Early Infection with Mycobacterium tuberculosis. J. Immunol. 2012, 188, 6205–6215.
- Rivas-Santiago, C.E.; Guerrero, G.G. IFN-α Boosting of Mycobacterium bovis Bacillus Calmette Güerin-Vaccine Promoted Th1 Type Cellular Response and Protection against M. tuberculosis Infection. Biomed. Res. Int. 2017, 2017, 8796760.
More
Information
Subjects:
Infectious Diseases
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
859
Revisions:
2 times
(View History)
Update Date:
01 Mar 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No