Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrew Warrilow | -- | 1719 | 2023-02-26 04:20:57 | | | |
| 2 | Dean Liu | Meta information modification | 1719 | 2023-02-27 03:25:21 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Warrilow, A.; Pumpa, K.; Somerset, S.; Naumovski, N. Regulation of Dietary Energy Intake. Encyclopedia. Available online: https://encyclopedia.pub/entry/41669 (accessed on 25 July 2026).
Warrilow A, Pumpa K, Somerset S, Naumovski N. Regulation of Dietary Energy Intake. Encyclopedia. Available at: https://encyclopedia.pub/entry/41669. Accessed July 25, 2026.
Warrilow, Andrew, Kate Pumpa, Shawn Somerset, Nenad Naumovski. "Regulation of Dietary Energy Intake" Encyclopedia, https://encyclopedia.pub/entry/41669 (accessed July 25, 2026).
Warrilow, A., Pumpa, K., Somerset, S., & Naumovski, N. (2023, February 26). Regulation of Dietary Energy Intake. In Encyclopedia. https://encyclopedia.pub/entry/41669
Warrilow, Andrew, et al. "Regulation of Dietary Energy Intake." Encyclopedia. Web. 26 February, 2023.
Copy Citation
Obesity is one of the most important factors responsible for the marked increase in both the incidence and prevalence of type 2 diabetes mellitus (T2DM) in recent decades. Addressing the lifestyle factors associated with the progression to T2DM would present a potential rational early prevention strategy.
obesity
type 2 diabetes
satiation: satiety
fat
volume
1. Satiation and Satiety
The dietary energy intake is governed by a system that consists of two distinct phases—satiation and satiety. Whereas satiation refers to physiological responses to food intake during the consumption of food, which leads to a cessation of eating, satiety refers to the signals which inhibit eating prior to the subsequent meal [1]. The ability of foods to mediate satiation and satiety can be successfully determined in the controlled laboratory setting where potential confounders, such as palatability of the test food, may be well managed [2]. The methodology for measuring satiation is for the energy of food consumed to be calculated up to the point of satiation and after the food concerned has been consumed ab libitum in the fasted state. The ability of a food to mediate satiety may also be measured in the laboratory environment where a test food is given as a meal or preload, and the effect on energy intake of the subsequent meal is measured [1].
2. Physiological Regulation of Dietary Energy Intake
The gastrointestinal system continuously relays the information to the brain regarding the quality and quantity of ingested food and nutrients, which is important for satiation, meal termination, appetite and timing of meals. By acting on the brainstem and the hypothalamus, this stream of sensory information from the gut to the brain (and vice versa) generates sensations of satisfaction observed after a satiating meal [3]. The infundibular nucleus located at the base of the hypothalamus is the site of the receptors for many of the digestive hormones known to regulate food intake. Furthermore, the paraventricular nucleus, located in the anterior hypothalamus, interacts with the thyroid and the pituitary and adrenal glands to regulate metabolism [4]. The hypothalamus receives biochemical signals released from the gut in response to food, whereas the brain stem serves as a recipient of nerve signals travelling from the gut via the afferent vagus nerve [5]. The system for receiving feedback from the gut, which provides information crucial for regulating eating behaviour, therefore, is comprised of two separate systems interacting with each other (Figure 1).
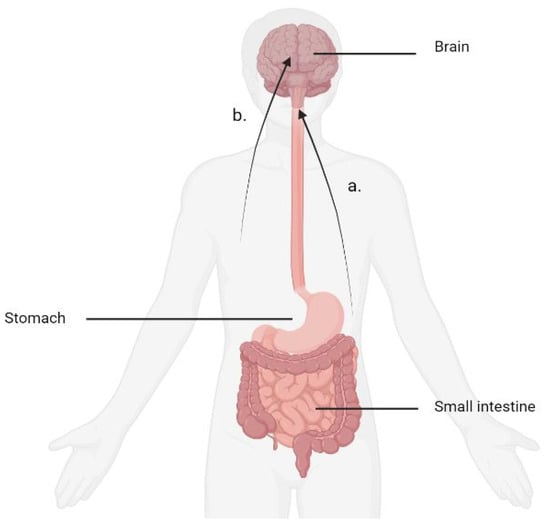
Figure 1. Mechanisms for digestive feedback. (a) The afferent vagus nerve delivers chemical signals produced by baroreceptors located in the stomach to the brainstem. (b) Biochemical signals emanating from the digestive tract are referred to the brain through the circulation. The infundibular nucleus located at the base of the hypothalamus is the site of many of these receptors.
Nerves known as vagal afferents transmit information to the relevant sections of the brain from a variety of organs including the gastrointestinal tract, pancreas and liver. Vagal efferent nerves travelling from the brain together with the sympathetic nervous system and hormonal mechanisms act as communication pathways for the brain to control digestion and other functions. These vagal sensory mechanisms play a crucial role in the neural mechanism of satiation [6].
When food is ingested, its presence and volume is detected by vagal afferent nerves in the external muscle layers sensitive to stretch and tension. Intra-ganglionic laminar vagal afferent endings are responsible for detecting muscle tension generated by both passive stretch and active contraction of the muscle layers [3]. Studies in animal models have uncovered an abundance of receptors of the digestive hormone cholecystokinin (CCK) in the vagal afferent nerves [7]. Therefore, digestive hormones may communicate with the brain directly through the bloodstream or via such receptors. Furthermore, it has been proposed that a digestive hormone could use different paths to produce physiological effects, such as changes in eating behaviour or interference with gastrointestinal functions. Nevertheless, for many pathways there is still conflicting evidence concerning which digestive hormone is responsible for what effect [3].
3. Biochemical Signalling
Biochemical signals play a crucial role in energy regulation and their function both has an influence on and is influenced by the development of obesity. Peptide tyrosine tyrosine (PYY) is a digestive hormone produced in the endocrine L cells of the ileum, large intestine and rectum [8]. PYY is found in the body in both PYY1-36 and PYY3-36 forms with the latter having an effect on energy intake and the former no effect [9]. The release of PYY peaks around 90 min postprandially and remains elevated for up to 6 h [10]. PYY is secreted in the distal areas of the gut and the amount of PYY released is proportional to the amount of food energy consumed [11], suggesting a role in satiety as opposed to satiation. In animal studies, increased feeding of protein to mice resulted in an increase in plasma PYY levels, decreased food intake and reduced adiposity [12]. Furthermore, PYY null mice have been shown to develop obesity, which was only reversed by the administration of endogenous PYY [12]. In humans, PYY has been shown to reduce food intake by 30% when administered before a subsequent meal [13].
PYY levels peak well before nutrients reach the location where the majority of secreting cells reside in the distal section of the digestive tract [14]. In research conducted on rats, vagotomy was performed completely halting the secretion of PYY in the distal gut. The idea that the signalling responsible for PYY secretion could occur via the vagus nerve is strongly suggestive of mechanisms for PYY release emanating not in the distal gut but in the stomach. Other influences on this transfer of signalling from the proximal to the distal gut include nicotinic synapses and nitric oxide (NO) release [14].
Ghrelin is the hormone responsible for the hunger sensation and is commonly referred to as the ‘hunger hormone’. The primary site of ghrelin secretion is in the stomach. The stomach contains ten to twenty times more ghrelin per gram of tissue than the duodenum, and the concentrations of ghrelin decline with increasing distance from stomach along the gastrointestinal tract [15]. Ghrelin secretion results in an increase in meal size [16]. Therefore, the process of satiation is linked to suppression of the secretion of ghrelin.
Insulin has been found to be is essential for meal-induced plasma ghrelin suppression [17] and, therefore, it plays a role in appetite regulation. Insulin is produced in the β cells of the pancreas and acts to regulate carbohydrate, lipid and protein metabolism [18]. Infusions of insulin have shown mixed results on appetite control with some early studies reporting an increase [19] or no increase in appetite [20]. There is evidence for endogenous insulin mediating a reduction in appetite [21]. However, this effect could potentially be due to the influence of glucose as no changes in appetite or food intake were apparent when glucose levels were maintained at constant levels [20].
Recently, the function of the incretin hormones in the suppression of appetite has become more fully elucidated in the scientific literature. Glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) are the two primary incretin hormones secreted from the intestine on the ingestion of glucose or nutrients to stimulate insulin secretion from pancreatic β cells [22]. GLP-1 is co-secreted with PYY in response to nutrients in the gut [4], and it plays a role in both peripheral and central pathways mediating satiation [23]. GLP-1 is produced in the ileum in the presence of carbohydrate and stimulates the pancreas to increase the production of insulin [24]. GLP-1 is proposed to play an important part in the ‘ileal brake’ mechanism, and this is believed to be directly related to the means by which GLP-1 influences appetite [25]. GLP-1 has been shown to reduce ad libitum energy intake [26] in a dose dependent manner [27], and its rapid degradation by dipeptidyl peptidase IV (DPP4) and short duration of action suggests a role in satiation as opposed to satiety.
The incretin hormone GIP is released in the presence of either glucose or fat [28]. Studies concerning GIP’s role in appetite regulation have shown mixed results with some studies demonstrating its secretion resulting in a reduction in appetite [29] and others failing to find an effect [30]. The insulinotropic effect of GIP is lost in persons living with T2DM [31].
CCK was one of the first digestive peptides to be discovered [32] and was subsequently shown to play an important role in appetite control in animal models [33]. Although CCK is released very quickly into the blood stream (it has been detected at 15 min postprandially) [34], it is also relatively quickly removed. The elimination half-life of the octapeptide variant of CCK is 18 min [35]; however, there are a multitude of variants and concentrations of bioactive CCK variants with larger molecules may remain elevated in the blood plasma for several hours after a meal in healthy humans [36]. Due to its rapid entrance into the blood plasma and short period of action, any effect of CCK on eating behaviour is more likely to be found within a meal rather than between meals and is, therefore, known as the digestive peptide most associated with satiation.
CCK is secreted by duodenal and ileal cells when fat and protein enter the lumen [32] and causes the release of digestive enzymes and bile from the pancreas and gallbladder, respectively [37]. The release of CCK into the blood stream acts on CCK receptors causing gastric emptying to slow [38]. The highest concentrations of CCK may be found in the proximal section [39] of the small intestine. CCK receptors are located in the pancreatic nerves, the gallbladder muscularis, the nerves and muscle along the gastrointestinal tract and in several areas of the brain [40].
The administration of exogenous CCK at pharmacological doses causes smaller meals to be consumed, whereas blocking the action of endogenous CCK or other satiety signals causes larger meals to be consumed [16]. At physiological doses, a preload consumed before the test meal was a necessary condition to achieve this result [41][42][43]. Initially, it was thought that the effect of CCK was due to the hormone’s ability to slow gastric emptying, but it was observed that the resultant reduction in food intake was greater than that expected from the reduction in gastric emptying, suggesting another potential mechanism of action. The enhancement of signals of stomach distention from the afferent vagus was proposed to be responsible for this effect [42][44][45][46]. CCK activates CCK-A-type receptors in the pyloric region of the stomach, and this signal is then transmitted via vagal afferent neurons to the nucleus of the tractus solitarius where it is relayed to the hypothalamus [47].
References
- Gerstein, D.E.; Woodward-Lopez, G.; Evans, A.E.; Kelsey, K.; Drewnowski, A. Clarifying concepts about macronutrients’ effects on satiation and satiety. J. Am. Diet. Assoc. 2004, 104, 1151–1153.
- Benelam, B. Satiation, satiety and their effects on eating behaviour. Nutr. Bull. 2009, 34, 126–173.
- Berthoud, H.R. Vagal and hormonal gut–brain communication: From satiation to satisfaction. Neurogastroenterol. Motil. 2008, 20, 64–72.
- Neary, N.M.; Goldstone, A.P.; Bloom, S.R. Appetite regulation: From the gut to the hypothalamus. Clin. Endocrinol. 2004, 60, 153–160.
- Blevins, J.E.; Baskin, D.G. Hypothalamic-brainstem circuits controlling eating. Forum. Nutr. 2010, 63, 133–140.
- Berthoud, H.R. The vagus nerve, food intake and obesity. Regul. Pept. 2008, 149, 15–25.
- Moriarty, P.; Dimaline, R.; Thompson, D.G.; Dockray, G.J. Characterization of cholecystokininA and cholecystokininB receptors expressed by vagal afferent neurons. Neuroscience 1997, 79, 905–913.
- Ekblad, E.; Sundler, F. Distribution of pancreatic polypeptide and peptide YY. Peptides 2002, 23, 251–261.
- Sloth, B.; Holst, J.J.; Flint, A.; Gregersen, N.T.; Astrup, A. Effects of PYY1–36 and PYY3–36 on appetite, energy intake, energy expenditure, glucose and fat metabolism in obese and lean subjects. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1062–E1068.
- Ueno, H.; Yamaguchi, H.; Mizuta, M.; Nakazato, M. The role of PYY in feeding regulation. Regul. Pept. 2008, 145, 12–16.
- Batterham, R.L.; Cowley, M.A.; Small, C.J.; Herzog, H.; Cohen, M.A.; Dakin, C.L.; Wren, A.M.; Brynes, A.E.; Low, M.J.; Ghatei, M.A. Gut hormone PYY3-36 physiologically inhibits food intake. Nature 2002, 418, 650–654.
- Batterham, R.L.; Heffron, H.; Kapoor, S.; Chivers, J.E.; Chandarana, K.; Herzog, H.; Le Roux, C.W.; Thomas, E.L.; Bell, J.D.; Withers, D.J. Critical role for peptide YY in protein-mediated satiation and body-weight regulation. Cell Metab. 2006, 4, 223–233.
- Batterham, R.L.; Cohen, M.A.; Ellis, S.M.; Le Roux, C.W.; Withers, D.J.; Frost, G.S.; Ghatei, M.A.; Bloom, S.R. Inhibition of food intake in obese subjects by peptide YY3–36. N. Engl. J. Med. 2003, 349, 941–948.
- Fu-Cheng, X.; Anini, Y.; Chariot, J.; Castex, N.; Galmiche, J.-P.; Rozé, C. Mechanisms of peptide YY release induced by an intraduodenal meal in rats: Neural regulation by proximal gut. Pflügers Arch. 1997, 433, 571–579.
- Higgins, S.C.; Gueorguiev, M.; Korbonits, M. Ghrelin, the peripheral hunger hormone. Ann. Med. 2007, 39, 116–136.
- Woods, S.C. Gastrointestinal Satiety Signals I. An overview of gastrointestinal signals that influence food intake. Am. J. Physiol.-Gastrointest. Liver Physiol. 2004, 286, G7–G13.
- Murdolo, G.; Lucidi, P.; Di Loreto, C.; Parlanti, N.; De Cicco, A.; Fatone, C.; Fanelli, C.G.; Bolli, G.B.; Santeusanio, F.; De Feo, P. Insulin is Required for Prandial Ghrelin Suppression in Humans. Diabetes 2003, 52, 2923–2927.
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39.
- Rodin, J.; Wack, J.; Ferrannini, E.; DeFronzo, R.A. Effect of insulin and glucose on feeding behavior. Metabolism 1985, 34, 826–831.
- Lavin, J.H.; Wittert, G.; Sun, W.M.; Horowitz, M.; Morley, J.E.; Read, N.W. Appetite regulation by carbohydrate: Role of blood glucose and gastrointestinal hormones. Am. J. Physiol.-Endocrinol. Metab. 1996, 271, E209–E214.
- Miller, S.; Petocz, P. Interrelationships among postprandial satiety, glucose and insulin responses and changes in subsequent food intake. Eur. J. Clin. Nutr. 1996, 50, 788–797.
- Yutaka, S.; Mitsuo, F.; Daisuke, Y. GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig. 2010, 1, 8–23.
- Holst, J.J. Incretin hormones and the satiation signal. Int. J. Obes. 2013, 37, 116.
- Müller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130.
- Zander, M.; Madsbad, S.; Madsen, J.L.; Holst, J.J. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and β-cell function in type 2 diabetes: A parallel-group study. Lancet 2002, 359, 824–830.
- Flint, A.; Raben, A.; Astrup, A.; Holst, J.J. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J. Clin. Investig. 1998, 101, 515–520.
- Gutzwiller, J.; Göke, B.; Drewe, J.; Hildebrand, P.; Ketterer, S.; Handschin, D.; Winterhalder, R.; Conen, D.; Beglinger, C. Glucagon-like peptide-1: A potent regulator of food intake in humans. Gut 1999, 44, 81–86.
- Baggio, L.L.; Drucker, D.J. Biology of Incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157.
- Verdich, C.; Toubro, S.; Buemann, B.; Lysgård Madsen, J.; Juul Holst, J.; Astrup, A. The role of postprandial releases of insulin and incretin hormones in meal-induced satiety—Effect of obesity and weight reduction. Int. J. Obes. 2001, 25, 1206–1214.
- Vozzo, R.; Baker, B.; Wittert, G.A.; Wishart, J.M.; Morris, H.; Horowitz, M.; Chapman, I. Glycemic, hormone, and appetite responses to monosaccharide ingestion in patients with type 2 diabetes. Metab. -Clin. Exp. 2002, 51, 949–957.
- Gautier, J.F.; Choukem, S.P.; Girard, J. Physiology of incretins (GIP and GLP-1) and abnormalities in type 2 diabetes. Diabetes Metab. 2008, 34, S65–S72.
- Rehfeld, J.F. Cholecystokinin. Best Pract. Res. Clin. Endocrinol. Metab. 2004, 18, 569–586.
- Gibbs, J.; Young, R.C.; Smith, G.P. Cholecystokinin decreases food intake in rats. J. Comp. Physiol. Psychol. 1973, 84, 488–495.
- Liddle, R.A.; Goldfine, I.D.; Rosen, M.S.; Taplitz, R.A.; Williams, J.A. Cholecystokinin bioactivity in human plasma. Molecular forms, responses to feeding, and relationship to gallbladder contraction. J. Clin. Investig. 1985, 75, 1144–1152.
- Koulischer, D.; Moroder, L.; Deschodt-Lanckman, M. Degradation of cholecystokinin octapeptide, related fragments and analogs by human and rat plasma in vitro. Regul. Pept. 1982, 4, 127–139.
- Rehfeld, J.F.; Sennels, H.P.; Jørgensen, H.L.; Fahrenkrug, J. Circadian variations in plasma concentrations of cholecystokinin and gastrin in man. Scand. J. Clin. Lab. Investig. 2020, 80, 546–551.
- Geary, N. Endocrine controls of eating: CCK, leptin, and ghrelin. Physiol. Behav. 2004, 81, 719–733.
- Schwizer, W.; Borovicka, J.; Kunz, P.; Fraser, R.; Kreiss, C.; D’Amato, M.; Fried, M. Role of cholecystokinin in the regulation of liquid gastric emptying and gastric motility in humans: Studies with the CCK antagonist loxiglumide. Gut 1997, 41, 500–504.
- Gilliam-Vigh, H.; Jorsal, T.; Rehfeld, J.F.; Pedersen, J.; Poulsen, S.S.; Vilsbøll, T.; Knop, F.K. Expression of Cholecystokinin and its Receptors in the Intestinal Tract of Type 2 Diabetes Patients and Healthy Controls. J. Clin. Endocrinol. Metab. 2021, 106, 2164–2170.
- Noble, F.; Wank, S.A.; Crawley, J.N.; Bradwejn, J.; Seroogy, K.B.; Hamon, M.; Roques, B.P. International Union of Pharmacology. XXI. Structure, distribution, and functions of cholecystokinin receptors. Pharm. Rev. 1999, 51, 745–781.
- Kissileff, H.R.; Pi-Sunyer, F.X.; Thornton, J.; Smith, G.P. C-terminal octapeptide of cholecystokinin decreases food intake in man. Am. J. Clin. Nutr. 1981, 34, 154–160.
- Muurahainen, N.E.; Kissileff, H.R.; Lachaussee, J.; Pi-Sunyer, F.X. Effect of a soup preload on reduction of food intake by cholecystokinin in humans. Am. J. Physiol. -Regul. Integr. Comp. Physiol. 1991, 260, R672–R680.
- Lieverse, R.J.; Jansen, J.B.M.J.; van de Zwan, A.; Samson, L.; Masclee, A.A.M.; Lamers, C.B.H.W. Effects of a physiological dose of cholecystokinin on food intake and postprandial satiation in man. Regul. Pept. 1993, 43, 83–89.
- Schwartz, G.J.; Berkow, G.; McHugh, P.R.; Moran, T.H. Gastric branch vagotomy blocks nutrient and cholecystokinin-induced suppression of gastric emptying. Am. J. Physiol. 1993, 3 Pt 2, R630–R637.
- Schwartz, G.J.; McHugh, P.R.; Moran, T.H. Gastric loads and cholecystokinin synergistically stimulate rat gastric vagal afferents. Am. J. Physiol 1993, 4 Pt 2, R872–R876.
- Melton, P.M.; Kissileff, H.R.; Pi-Sunyer, F.X. Cholecystokinin (CCK-8) affects gastric pressure and ratings of hunger and fullness in women. Am. J. Physiol. -Regul. Integr. Comp. Physiol. 1992, 263, R452–R456.
- Blundell, J.E.; MacDiarmid, J.I. Passive Overconsumption Fat Intake and Short-Term Energy Balancea. Ann. New York Acad. Sci. 1997, 827, 392–407.
More
Information
Subjects:
Health Care Sciences & Services
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
603
Revisions:
2 times
(View History)
Update Date:
28 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No