Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Wei Huang | -- | 2658 | 2023-02-25 05:38:31 | | | |
| 2 | Peter Tang | + 2 word(s) | 2660 | 2023-02-25 08:07:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Liu, S.; Luo, W.; Szatmary, P.; Zhang, X.; Lin, J.; Chen, L.; Liu, D.; Sutton, R.; Xia, Q.; Jin, T.; et al. Monocytic HLA-DR Expression in Acute Pancreatitis and COVID-19. Encyclopedia. Available online: https://encyclopedia.pub/entry/41654 (accessed on 10 August 2026).
Liu S, Luo W, Szatmary P, Zhang X, Lin J, Chen L, et al. Monocytic HLA-DR Expression in Acute Pancreatitis and COVID-19. Encyclopedia. Available at: https://encyclopedia.pub/entry/41654. Accessed August 10, 2026.
Liu, Shiyu, Wenjuan Luo, Peter Szatmary, Xiaoying Zhang, Jing-Wen Lin, Lu Chen, Dan Liu, Robert Sutton, Qing Xia, Tao Jin, et al. "Monocytic HLA-DR Expression in Acute Pancreatitis and COVID-19" Encyclopedia, https://encyclopedia.pub/entry/41654 (accessed August 10, 2026).
Liu, S., Luo, W., Szatmary, P., Zhang, X., Lin, J., Chen, L., Liu, D., Sutton, R., Xia, Q., Jin, T., Liu, T., & Huang, W. (2023, February 25). Monocytic HLA-DR Expression in Acute Pancreatitis and COVID-19. In Encyclopedia. https://encyclopedia.pub/entry/41654
Liu, Shiyu, et al. "Monocytic HLA-DR Expression in Acute Pancreatitis and COVID-19." Encyclopedia. Web. 25 February, 2023.
Copy Citation
Acute pancreatitis is a common gastrointestinal disease with increasing incidence worldwide. COVID-19 is a potentially life-threatening contagious disease spread throughout the world, caused by severe acute respiratory syndrome coronavirus 2. More severe forms of both diseases exhibit commonalities with dysregulated immune responses resulting in amplified inflammation and susceptibility to infection. Human leucocyte antigen (HLA)-DR, expressed on antigen-presenting cells, acts as an indicator of immune function. Research advances have highlighted the predictive values of monocytic HLA-DR (mHLA-DR) expression for disease severity and infectious complications in both acute pancreatitis and COVID-19 patients.
acute pancreatitis
COVID-19
HLA-DR
monocytes
immune response
immunosuppression
1. Introduction
New insights into the mechanisms of pathology can sometimes arise from similarities between fundamentally different diseases. This effect can be most pronounced during the emergence of a new infectious disease, such as the recent COVID-19 pandemic. One such unlikely pairing is acute pancreatitis (AP) and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection.
AP is a sterile inflammatory disorder of the pancreas with an increasing global incidence [1] affecting around 2.8 million patients annually [2]. The etiology of AP is diverse and includes gallstones, alcohol excess, hypertriglyceridemia, endoscopic retrograde cholangiopancreatography, certain medicines, and other rarer causes [3]. Most cases of AP patients are mild and uneventful given that supportive care is in time and appropriate. However, some are more severe, which involve local complications (acute pancreatic necrosis or fluid collection; moderately severe acute pancreatitis, MSAP) and/or persistent organ failure (SOFA score of respiratory, circulatory, and renal system equal or more than 2 lasting > 48 h; severe acute pancreatitis, SAP) [4]. Feed-forward auto-amplification of the initial cellular injury in SAP [5][6] results in persistent systemic inflammatory response syndrome (SIRS), multiple organ dysfunction syndrome (MODS), infection, and death. Persistent organ failure [7][8][9][10] and infected pancreatic necrosis [11][12], alone or in combination, are key determinants of severity in AP and contribute to an immune anergy, secondary infections, and a mortality of > 30%. Currently, there are no specific therapies effectively targeting the initial cellular injury or determinants that resulting in MODS [13].
COVID-19, on the other hand, is a potentially lethal infectious disease caused by the enveloped, positive-strand RNA, SARS-CoV-2, affecting over 600 million cases globally [14]. The disease spectrum of COVID-19 is also highly variable, ranging from asymptomatic (test-positive) disease to critical illness (respiratory failure, septic shock, and/or MODS) [15][16]. SARS-CoV-2 mainly utilizes the angiotensin-converting enzyme 2 (ACE2) as the human host cell entry receptor [17], which is ubiquitously expressed in the nasal epithelium, lung, heart, intestine, and kidney and rarely expressed on immune cells [18]. ACE2 is also expressed on pancreatic ductal cells, acinar cells, and islet cells, making the pancreas vulnerable to viral infection [19].
2. Pathogenesis and Immunopathology in AP and COVID-19
2.1. Pathophysiological Mechanisms in AP and COVID-19
Diverse stimuli evoke inflammatory cascades with apparently analogous patterns and clinical manifestations, implying similarities in the pathogenesis and symptomatology of AP and COVID-19 [20]. Cytokines and damage-associated molecular patterns (DAMPs), such as histones, high-mobility group box-1 protein, hyaluronan fragments, mitochondrial DNA, and heat-shock proteins are released from dying or injured cells in the injured pancreas or SARS-CoV-2 infected tissues—particularly lungs. This is associated with and results from a series of molecular events, including premature trypsinogen activation, calcium overload, mitochondria failure, endoplasmic reticulum stress, impaired autophagy, or by SARS-CoV-2 proliferation and release, respectively [6][21][22][23][24]. Interaction of DAMPs with pattern-recognition receptors (PRRs), including Toll-like receptors and NLRP3 inflammasome of the adjacent parenchymal cells or immune cells, promotes the production of various pro-inflammatory cytokines and chemokines [22][25][26][27]. Of note, cell death pathways (e.g., autophagy, NETosis, pyroptosis, apoptosis, necroptosis, and ferroptosis) in surrounding immune cells and stromal cells are activated, fueling the cytokine storm and cultivating a positive cell death-inflammation feedback loop [21][28][29]. In COVID-19, virus particles themselves act as pathogen-associated molecular patterns (PAMPs), which could also be identified by PRRs and activate local inflammation and an innate immune response, evoking the cytokine storm and assembling those induced by DAMPs [20][30]. Activated circulating leukocytes, particularly monocytes, are then recruited to the inflamed pancreas or infected lungs, provoking systemic inflammation and organ failure in AP and COVID-19 alike [20][31][32][33][34]. Moreover, monocytes/macrophages could be infected by SARS-CoV-2, triggering massive inflammatory responses in COVID-19 [35].
The involvement of adaptive immunity in AP has been recognized, but its precise role in the sterile inflammatory response seen in AP remains poorly characterized [36]. In contrast, SARS-CoV-2 directly activates specific T cell subsets, initiating an adaptive immune response [37]. Persistent viral stimulation, however, leads to T cell exhaustion, with reduced effector functions and proliferative capacity [38]. This T cell exhaustion phenomenon can also be observed in AP patients [39].
Levels of several circulating pro-inflammatory cytokines are dramatically elevated and closely correlate with the development of SAP or severe/critical COVID-19 [40][41][42][43]. Patterns of cytokine alterations in AP and COVID-19 were shown to be remarkably similar in a recent meta-analysis, with tumor necrosis factor-alpha (TNF-α), interleukin (IL)-6, IL-8, and IL-10 concentrations significantly higher in more severe forms than non-severe forms of the two diseases [44]. The crosstalk between excessive inflammatory cytokines, platelet activation, complement activation, and endothelial injury forms a deleterious hyper-inflammatory and hyper-coagulopathy environment which is associated with life-threatening complications (i.e., coagulopathy and vascular immune-thrombosis) of AP and COVID-19 [42][45][46][47][48][49].
Systemic lipotoxicity deserves to be highlighted in this context. In severe/critical COVID-19, lipotoxicity can trigger multiple organ failure and mortality resembling SAP [50]. SARS-CoV-2 can directly infect adipose tissue and promotes the release of several inflammatory cytokines [51]. The pancreas itself is a target of SARS-CoV-2, resulting in the interstitial leakage of pancreatic lipase which induces lipolysis of intrapancreatic adipose tissue and release of excess unsaturated fatty acids (UFAs). These toxic UFAs in turn further directly lead to parenchymal cell injury and provoke the release of pro-inflammatory mediators, driving the cytokine storm and organ failure in SAP and severe/critical COVID-19 [50][52][53]. Lipase inhibitors have been shown to ameliorate lipolysis-induced cytokine storms and mortality [52][53][54][55].
In summary, the pathophysiological mechanisms of AP and COVID-19 share many similarities including cell death-inflammation cascade, cytokine storms, enhanced lipolysis, and dysregulated immune responses. These immune responses will be discussed in the next section.
2.2. Altered Immune Responses in AP and COVID-19
Immune anergy, evidenced by the failure of delayed hypersensitivity responses, correlates with the development of sepsis and mortality in trauma and surgical patients [56][57][58], as well as in SAP [59]. In the first stage of SAP, an excessive pro-inflammatory burst is rapidly followed by an anti-inflammatory reaction that may result in a generalized inflammatory response in sites remote from the initial pancreatic injury site and gives rise to SIRS [60][61][62]. There is a compensatory response to counteract the overwhelming pro-inflammatory state [63], which may ultimately result in immune suppression [64]. In 1996, Bone termed this immunological phenomenon as “compensatory anti-inflammatory response syndrome” (CARS) [56][57][63].
Unlike SIRS, which is clearly defined by clinical parameters, CARS lacks clinical manifestations and can only be defined molecularly by a combination of immunological alterations. In the landmark paper of Volk’s group in 1997, it was described that many septic patients who died from nosocomial infections had associated downregulation of mHLA-DR [65]. Monocytes from these patients had reduced capacity to act in a pro-inflammatory manner by producing TNF-α following stimulation of lipopolysaccharide (LPS) in vitro, termed “immunoparalysis” [65][66]. Where CARS was once thought to follow sequentially from SIRS, current thinking views CARS responses as concomitant to SIRS; balance in both responses restores homeostasis, but an overshoot of the mechanisms of either SIRS or CARS leads to further injury by excessive inflammation or secondary infection and, ultimately, organ failure and death [58][67][68][69][70][71][72][73][74]. Development of CARS results in lymphocyte apoptosis, T lymphocyte anergy, and deactivation of monocytes resulting in reduced mHLA-DR expression. Furthermore, CARS is associated with elevated levels of circulating IL-10, transforming growth factor-beta (TGF-β) and other anti-inflammatory cytokines, which contribute to the risk of secondary infection.
Immune response to SARS-CoV-2 is characterized by the failure of robust type I and type III interferon response and high expression of pro-inflammatory cytokines and chemokines [17]. Like AP, immune alterations, including severe lymphopenia and functional monocyte deactivation, are indicative of immunosuppression in severe/critical COVID-19 patients [75]. Indeed, monocytes exhibit heterogeneous, dynamic, and severity-dependent alterations of transcription and immune phenotype upon acute pathological insults which appear similar in both SAP and severe/critical COVID-19 patients (Figure 1).
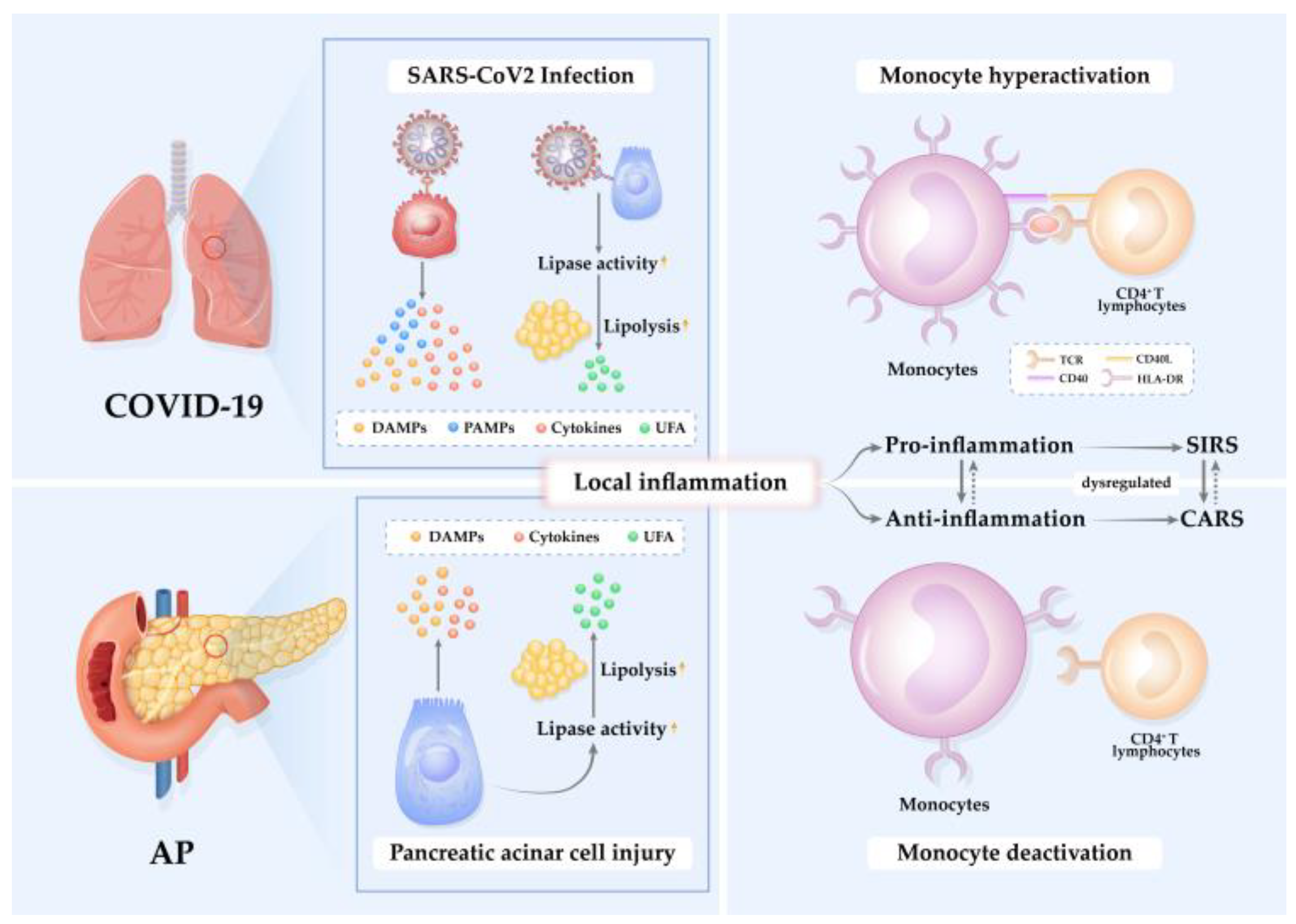
Figure 1. Pathogenesis of inflammation in AP and COVID-19. Acute pathological insults of SARS-CoV-2 infection and pancreatic acinar cell injury elicit local inflammation mediated by cytokines, unsaturated fatty acids (UFAs), damage-associated molecular patterns (DAMPs), and/or pathogen-associated molecular patterns (PAMPs). The pro-inflammatory reaction induces an anti-inflammatory response to restrict inflammation. When the pro-/anti-inflammation is unbalanced and dysregulated, systemic inflammatory response syndrome (SIRS) or compensatory anti-inflammatory response syndrome (CARS) occurs. During SIRS, monocytes are hyperactivated in response to high levels of pro-inflammatory cytokines and chemokines. In contrast, during CARS, monocytes are deactivated, exhibit reduced mHLA-DR expression, and are incapable of presenting antigens to activate CD4+ T lymphocytes.
Inflammatory monocytes are enriched in the lungs of severe/critical COVID-19 patients and are also the most altered pancreatic immune cells during progression and recovery of AP [76][77]. Decreased monocytic expression of HLA-DR has a predictive value for the poor prognosis of patients with sepsis [78][79], and the level of mHLA-DR expression may identify patients who are susceptible to the development of infectious complications after trauma [80], major surgery [81], and burns [82]. Here, the researchers review the utility of mHLA-DR in assessing the state of the immune response in AP and COVID-19 and detail-relevant implications for therapy.
3. Structure and Expression of mHLA-DR
HLA-DR is a type of major histocompatibility complex (MHC) II molecule [83]. It is a heterodimeric glycoprotein composed of the 33–35 kD heavy/α chain and the 27–29 kD light/β chain, assembling into a structure comprising a peptide binding site on top of two immunoglobulin domains [83]. Encoded by adjacent genes, the β chain is polymorphic around the amino acid residues of the peptide-binding site in contrast to the invariant α chain [84].
HLA-DR is mostly expressed on antigen-presenting cells (APCs) such as monocytes, macrophages, dendritic cells, and B cells. The primary function of HLA-DR is to present peptide antigens to the immune system for the purpose of eliciting or suppressing T-(helper)-cell responses, eventually leading to the production of antibodies against the same peptide antigen. HLA-D/DR-controlled antigens play an essential role in the cell-to-cell interactions required to generate an immune response [85][86].
The biosynthesis, trafficking, and recycling of HLA-DR are regulated by multiple factors affecting cell surface expression. Consequently, the tightly regulated level of HLA-DR expression on the surface of monocytes is thought to be an indicator for monocyte function and the state of the immune response, with high levels of mHLA-DR associated with enhanced antigen presenting capacity and immune activation, and low levels associated with immune suppression.
3.1. Measurement of mHLA-DR
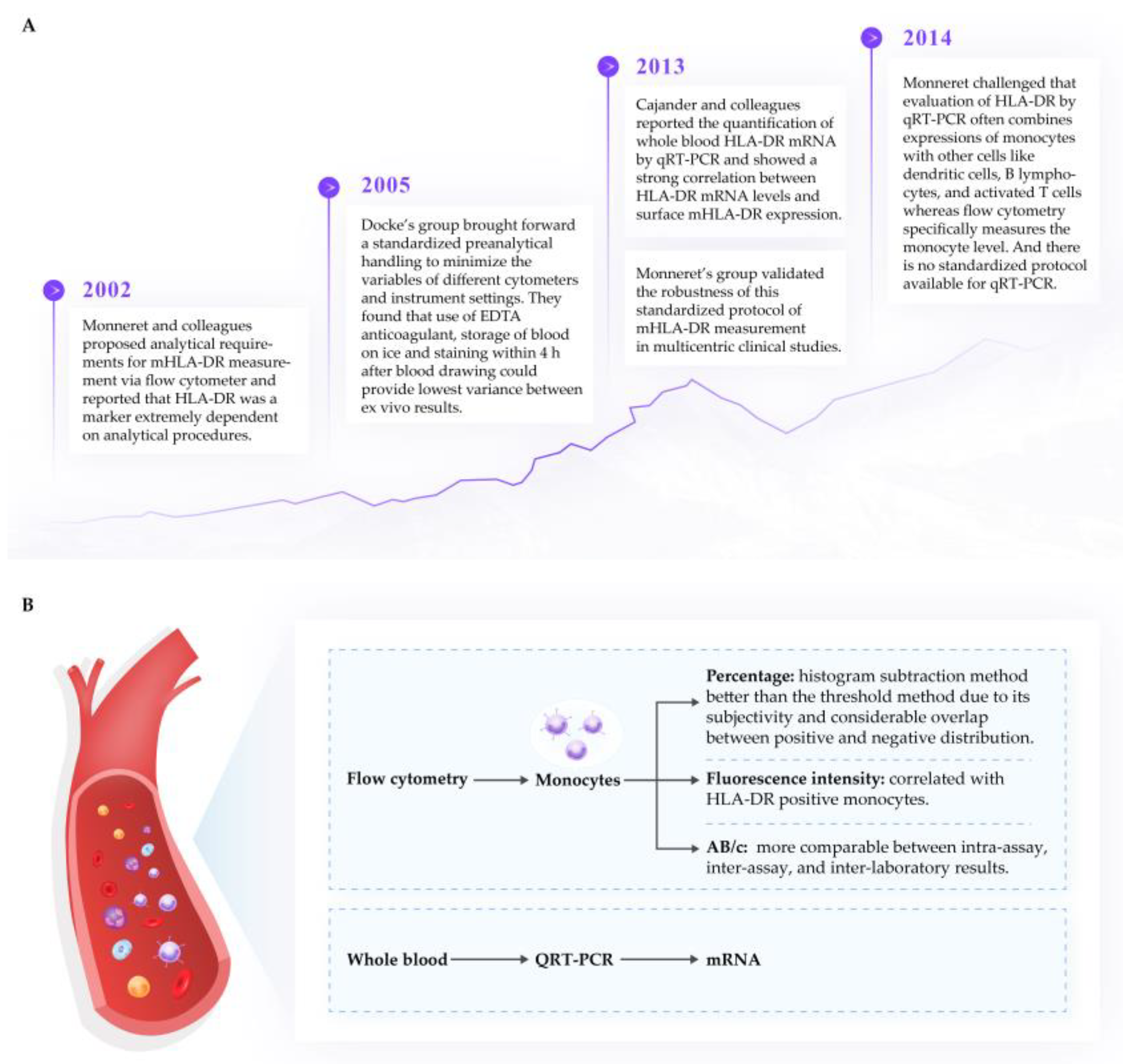
Several reviews [58][87][88] have emphasized the importance of flow cytometry as an indicator of immune function in clinical practice. The unit of measurement of HLA-DR via flow cytometer can be the percentage of HLA-DR positive monocytes (%), the mean fluorescence intensity (MFI), the fluorescence unit relative to the monocyte population (RFU), or antibodies per cell (AB/c). Due to the dynamic nature of HLA-DR expression and recycling, it is critical that measurement of expression is standardized. The researchers support the process published by Docke’s and Monnaret’s groups [89][90][91], which have been widely tested and published and appear to result in a strong correlation between transcription and cell surface expression of mHLA-DR. It should be highlighted that a percentage of HLA-DR+ monocytes less than 30% or values of AB/c below 5000 represents immunoparalysis, and values greater than 80% or 15 000 AB/c indicate immunocompetence [90]. The critical features for the sampling and measurement of mHLA-DR from human plasma samples are summarized in Figure 2.
3.2. Regulation of mHLA-DR Expression
The transcription of mHLA-DR is complex and heterogeneous, mediated by a series of conserved cis-acting regulatory promoter elements and interacting transcription factors [93]. Among these, class II transactivator (CIITA) is the master regulator of HLA-DR transcription [94]. Polymorphisms of CIITA promoter are associated with decreased mHLA-DR expression in patients with septic shock [95]. Besides biosynthesis, the expression of mHLA-DR can be post-translationally regulated by exocytosis, stability, and recycling. The class II-associated Ii peptide (CLIP), generated from cleavage of CD74 (MHC class II invariant chain, Ii) via members of the cathepsin family, is critical for the transport of HLA-DR to the cell surface [96]. In CD74 knockout mice, MHC II molecules are mainly retained in endoplasmic reticulum with reduced levels on the cell surface [97]. Reducing CLIP generation by blocking cysteine protease activity reduced surface MHC II expression, including HLA-DR to 60% on human monocytes in steady state [98]. HLA-DM, the key accessory molecules in the MHC class II loading compartment, catalyzes the dissociation of CLIP in exchange for more stably binding peptides [99]. MHC II molecules on the cell surface are normal in amounts but mainly loaded with CLIP in HLA-DM-deficient mice [100]. HLA-DR loaded with high-affinity peptides are postulated to be more stable than those with CLIP, indicating the role of HLA-DM in regulating mHLA-DR expression [98]. Of note, surface HLA-DR could be internalized, exchanged from lower affinity peptides into other peptides, and rapidly recycled back to the cell surface [101]. In summary, expression of mHLA-DR is finely regulated by multiple steps, including biosynthesis, peptide-loading via cathepsin-induced CLIP and HLA-DM, vesicular transport to the cell surface, and recycling.
Multiple pro- and anti-inflammatory cytokines are reported to dynamically control the expression of mHLA-DR [102].
4. The Role of mHLA-DR in AP and COVID-19
Monocytic HLA-DR expression alters dynamically in response to the variation of immune responses in the body during the disease course of AP and COVID-19. Evaluating the dynamic expression of mHLA-DR provides indicative information for diagnosis and prediction of disease severity, infectious complications, and prognosis.
The expression of mHLA-DR on admission was downregulated in AP patients compared to healthy controls; it further decreased on days 1 and 2 with differential degrees depending on severity [103][104][105]. While mHLA-DR expression recovered rapidly at day 3 and became normal after day 7 in less severe patients, it persisted at low levels for 1–2 weeks in more severe cases [104][106]. Indeed, mHLA-DR expression displays an inverse relationship with severity throughout at least the first three weeks of disease [107], with the lowest expression of mHLA-DR in SAP consistently recorded between 48 and 72 h of disease onset [107][108].
Overall, mHLA-DR expression either increases or decreases slightly in mild COVID-19 patients compared with healthy controls [109][110]. However, a marked and persistent decrease in expression is described in severe/critical COVID-19 patients in most studies [109][111][112][113][114][115][116][117][118][119][120]. The immune response to severe COVID-19 can be categorized into three groups according to the kinetics of mHLA-DR expression: (i) hyperactivated monocytes/macrophage phenotype (persistently high mHLA-DR > 30,000 AB/c)—strongly associated with mortality; (ii) prolonged immunodepression (persistently low mHLA-DR < 15,000 AB/c after days 5–7)—strongly correlating with secondary infection; (iii) transient immunodepression (early mHLA-DR < 15,000 AB/c, rising above 15,000 AB/c after 5–7 days)—at risk of secondary infection [121]. Patients with acute respiratory distress syndrome (ARDS) secondary to COVID-19 exhibit either immune dysregulation evidenced by very low mHLA-DR expression (i.e., lower than 5000 AB/c) and depletion of lymphocytes, or macrophage activation syndrome characterized by elevated ferritin, where associated HLA-DR levels might be reduced [122], or comparable to healthy controls [123]. Expression of mHLA-DR may be able to provide some information on disease course and has been observed to normalize upon recovery from critical illness in patients with COVID-19 (from 1–3 days to over 10 days after admission), but continued to fall in a patient who died [116]. Critically ill COVID-19 patients with long hospital stays (>25 days) presented with a more profound reduction in mHLA-DR expression than patients with short hospital stays (<25 days) [120]. Furthermore, convalescent COVID-19 patients exhibit mHLA-DR levels which are higher than those of healthy controls at 6 months, and equal to healthy controls at 9 and 12 months following discharge from the hospital [120][124].
The reduction of HLA-DR expression in COVID-19 patients has been reported in both classical monocytes [124][125], as well as intermediate monocytes and/or non-classical monocytes [112][126][127], although usually in one group or the other, depending on the respective study. Classical monocytes are the first peripheral immune cell type to recover HLA-DR positivity during the recovery of critically ill COVID-19 patients [128].
References
- Iannuzzi, J.P.; King, J.A.; Leong, J.H.; Quan, J.; Windsor, J.W.; Tanyingoh, D.; Coward, S.; Forbes, N.; Heitman, S.J.; Shaheen, A.A.; et al. Global incidence of acute pancreatitis is increasing over time: A systematic review and meta-analysis. Gastroenterology 2022, 162, 122–134.
- Li, C.L.; Jiang, M.; Pan, C.Q.; Li, J.; Xu, L.G. The global, regional, and national burden of acute pancreatitis in 204 countries and territories, 1990–2019. BMC Gastroenterol. 2021, 21, 332.
- Szatmary, P.; Grammatikopoulos, T.; Cai, W.; Huang, W.; Mukherjee, R.; Halloran, C.; Beyer, G.; Sutton, R. Acute pancreatitis: Diagnosis and treatment. Drugs 2022, 82, 1251–1276.
- Banks, P.A.; Bollen, T.L.; Dervenis, C.; Gooszen, H.G.; Johnson, C.D.; Sarr, M.G.; Tsiotos, G.G.; Vege, S.S.; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis--2012: Revision of the Atlanta classification and definitions by international consensus. Gut 2013, 62, 102–111.
- Linkermann, A.; Stockwell, B.R.; Krautwald, S.; Anders, H.J. Regulated cell death and inflammation: An auto-amplification loop causes organ failure. Nat. Rev. Immunol. 2014, 14, 759–767.
- Barreto, S.G.; Habtezion, A.; Gukovskaya, A.; Lugea, A.; Jeon, C.; Yadav, D.; Hegyi, P.; Venglovecz, V.; Sutton, R.; Pandol, S.J. Critical thresholds: Key to unlocking the door to the prevention and specific treatments for acute pancreatitis. Gut 2021, 70, 194–203.
- Guo, Q.; Li, A.; Xia, Q.; Liu, X.; Tian, B.; Mai, G.; Huang, Z.; Chen, G.; Tang, W.; Jin, X.; et al. The role of organ failure and infection in necrotizing pancreatitis: A prospective study. Ann. Surg. 2014, 259, 1201–1207.
- Sternby, H.; Bolado, F.; Canaval-Zuleta, H.J.; Marra-López, C.; Hernando-Alonso, A.I.; Del-Val-Antoñana, A.; García-Rayado, G.; Rivera-Irigoin, R.; Grau-García, F.J.; Oms, L.; et al. Determinants of severity in acute pancreatitis: A nation-wide multicenter prospective cohort study. Ann. Surg. 2019, 270, 348–355.
- Schepers, N.J.; Bakker, O.J.; Besselink, M.G.; Ahmed Ali, U.; Bollen, T.L.; Gooszen, H.G.; van Santvoort, H.C.; Bruno, M.J. Impact of characteristics of organ failure and infected necrosis on mortality in necrotising pancreatitis. Gut 2019, 68, 1044–1051.
- Shi, N.; Liu, T.; de la Iglesia-Garcia, D.; Deng, L.; Jin, T.; Lan, L.; Zhu, P.; Hu, W.; Zhou, Z.; Singh, V.; et al. Duration of organ failure impacts mortality in acute pancreatitis. Gut 2020, 69, 604–605.
- Petrov, M.S.; Shanbhag, S.; Chakraborty, M.; Phillips, A.R.; Windsor, J.A. Organ failure and infection of pancreatic necrosis as determinants of mortality in patients with acute pancreatitis. Gastroenterology 2010, 139, 813–820.
- Werge, M.; Novovic, S.; Schmidt, P.N.; Gluud, L.L. Infection increases mortality in necrotizing pancreatitis: A systematic review and meta-analysis. Pancreatology 2016, 16, 698–707.
- Moggia, E.; Koti, R.; Belgaumkar, A.P.; Fazio, F.; Pereira, S.P.; Davidson, B.R.; Gurusamy, K.S. Pharmacological interventions for acute pancreatitis. Cochrane Database Syst. Rev. 2017, 4, Cd011384.
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 20 September 2022).
- Coronavirus Disease 2019 (COVID-19) Treatment Guidelines; National Institutes of Health (US): Bethesda, MD, USA, 2021.
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999.
- Carvalho, T.; Krammer, F.; Iwasaki, A. The first 12 months of COVID-19: A timeline of immunological insights. Nat. Rev. Immunol. 2021, 21, 245–256.
- Chung, J.Y.; Thone, M.N.; Kwon, Y.J. COVID-19 vaccines: The status and perspectives in delivery points of view. Adv. Drug Deliv. Rev. 2021, 170, 1–25.
- Liu, F.; Long, X.; Zhang, B.; Zhang, W.; Chen, X.; Zhang, Z. ACE2 expression in pancreas may cause pancreatic damage after SARS-CoV-2 infection. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2020, 18, 2128–2130.e2.
- Karki, R.; Kanneganti, T.D. The ‘cytokine storm’: Molecular mechanisms and therapeutic prospects. Trends Immunol. 2021, 42, 681–705.
- Guéant, J.L.; Guéant-Rodriguez, R.M.; Fromonot, J.; Oussalah, A.; Louis, H.; Chery, C.; Gette, M.; Gleye, S.; Callet, J.; Raso, J.; et al. Elastase and exacerbation of neutrophil innate immunity are involved in multi-visceral manifestations of COVID-19. Allergy 2021, 76, 1846–1858.
- de Vries, F.; Huckriede, J.; Wichapong, K.; Reutelingsperger, C.; Nicolaes, G.A.F. The role of extracellular histones in COVID-19. J. Intern. Med. 2022, 293, 275–292.
- Lee, P.J.; Papachristou, G.L. New insights into acute pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 479–496.
- Szatmary, P.; Huang, W.; Criddle, D.; Tepikin, A.; Sutton, R. Biology, role and therapeutic potential of circulating histones in acute inflammatory disorders. J. Cell Mol. Med. 2018, 22, 4617–4629.
- Vora, S.M.; Lieberman, J.; Wu, H. Inflammasome activation at the crux of severe COVID-19. Nat. Rev. Immunol. 2021, 21, 694–703.
- Naqvi, I.; Giroux, N.; Olson, L.; Morrison, S.A.; Llanga, T.; Akinade, T.O.; Zhu, Y.; Zhong, Y.; Bose, S.; Arvai, S.; et al. DAMPs/PAMPs induce monocytic TLR activation and tolerance in COVID-19 patients; nucleic acid binding scavengers can counteract such TLR agonists. Biomaterials 2022, 283, 121393.
- Zheng, L.; Xue, J.; Jaffee, E.M.; Habtezion, A. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology 2013, 144, 1230–1240.
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 2021, 184, 149–168.e17.
- Li, X.; Zhang, Z.; Wang, Z.; Gutiérrez-Castrellón, P.; Shi, H. Cell deaths: Involvement in the pathogenesis and intervention therapy of COVID-19. Signal Transduct. Target. Ther. 2022, 7, 186.
- Yamada, T.; Takaoka, A. Innate immune recognition against SARS-CoV-2. Inflamm. Regen. 2023, 43, 7.
- Habtezion, A.; Gukovskaya, A.S.; Pandol, S.J. Acute pancreatitis: A multifaceted set of organelle and cellular interactions. Gastroenterology 2019, 156, 1941–1950.
- Garg, P.K.; Singh, V.P. Organ failure due to systemic injury in acute pancreatitis. Gastroenterology 2019, 156, 2008–2023.
- Xue, J.; Sharma, V.; Habtezion, A. Immune cells and immune-based therapy in pancreatitis. Immunol. Res. 2014, 58, 378–386.
- Liu, S.; Szatmary, P.; Lin, J.W.; Wang, Q.; Sutton, R.; Chen, L.; Liu, T.; Huang, W.; Xia, Q. Circulating monocytes in acute pancreatitis. Front. Immunol. 2022, 13, 1062849.
- Felsenstein, S.; Herbert, J.A.; McNamara, P.S.; Hedrich, C.M. COVID-19: Immunology and treatment options. Clin. Immunol. 2020, 215, 108448.
- Fonteh, P.; Smith, M.; Brand, M. Adaptive immune cell dysregulation and role in acute pancreatitis disease progression and treatment. Arch. Immunol. Et Ther. Exp. 2018, 66, 199–209.
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P.; Pan, P.; Wang, W.; Hu, D.; Liu, X.; et al. Coronavirus infections and immune responses. J. Med. Virol. 2020, 92, 424–432.
- Anka, A.U.; Tahir, M.I.; Abubakar, S.D.; Alsabbagh, M.; Zian, Z.; Hamedifar, H.; Sabzevari, A.; Azizi, G. Coronavirus disease 2019 (COVID-19): An overview of the immunopathology, serological diagnosis and management. Scand. J. Immunol. 2021, 93, e12998.
- Pietruczuk, M.; Dabrowska, M.I.; Wereszczynska-Siemiatkowska, U.; Dabrowski, A. Alteration of peripheral blood lymphocyte subsets in acute pancreatitis. World J. Gastroenterol. 2006, 12, 5344–5351.
- Norman, J. The role of cytokines in the pathogenesis of acute pancreatitis. Am. J. Surg. 1998, 175, 76–83.
- Makhija, R.; Kingsnorth, A.N. Cytokine storm in acute pancreatitis. J. Hepato-Biliary-Pancreat. Surg. 2002, 9, 401–410.
- To, K.K.; Sridhar, S.; Chiu, K.H.; Hung, D.L.; Li, X.; Hung, I.F.; Tam, A.R.; Chung, T.W.; Chan, J.F.; Zhang, A.J.; et al. Lessons learned 1 year after SARS-CoV-2 emergence leading to COVID-19 pandemic. Emerg. Microbes Infect. 2021, 10, 507–535.
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469.
- Hegyi, P.; Szakács, Z.; Sahin-Tóth, M. Lipotoxicity and cytokine storm in severe acute pancreatitis and COVID-19. Gastroenterology 2020, 159, 824–827.
- Pena, A.L.B.; Oliveira, R.A.; Severo, R.G.; Simões, E.S.A.C. COVID-19 related coagulopathy: What is Known Up to Now. Curr. Med. Chem. 2021, 28, 4207–4225.
- Subramaniam, S.; Kothari, H.; Bosmann, M. Tissue factor in COVID-19-associated coagulopathy. Thromb. Res. 2022, 220, 35–47.
- Bettac, L.; Denk, S.; Seufferlein, T.; Huber-Lang, M. Complement in pancreatic disease-perpetrator or savior? Front. Immunol. 2017, 8, 15.
- Dumnicka, P.; Maduzia, D.; Ceranowicz, P.; Olszanecki, R.; Drożdż, R.; Kuśnierz-Cabala, B. The interplay between inflammation, coagulation and endothelial injury in the early phase of acute pancreatitis: Clinical implications. Int. J. Mol. Sci. 2017, 18, 354.
- Kakafika, A.; Papadopoulos, V.; Mimidis, K.; Mikhailidis, D.P. Coagulation, platelets, and acute pancreatitis. Pancreas 2007, 34, 15–20.
- El-Kurdi, B.; Khatua, B.; Rood, C.; Snozek, C.; Cartin-Ceba, R.; Singh, V.P. Mortality from coronavirus disease 2019 increases with unsaturated fat and may be reduced by early calcium and albumin supplementation. Gastroenterology 2020, 159, 1015–1018.e4.
- Saccon, T.D.; Mousovich-Neto, F.; Ludwig, R.G.; Carregari, V.C.; Dos Anjos Souza, A.B.; Dos Passos, A.S.C.; Martini, M.C.; Barbosa, P.P.; de Souza, G.F.; Muraro, S.P.; et al. SARS-CoV-2 infects adipose tissue in a fat depot- and viral lineage-dependent manner. Nat. Commun. 2022, 13, 5722.
- Patel, K.; Trivedi, R.N.; Durgampudi, C.; Noel, P.; Cline, R.A.; DeLany, J.P.; Navina, S.; Singh, V.P. Lipolysis of visceral adipocyte triglyceride by pancreatic lipases converts mild acute pancreatitis to severe pancreatitis independent of necrosis and inflammation. Am. J. Pathol. 2015, 185, 808–819.
- de Oliveira, C.; Khatua, B.; Noel, P.; Kostenko, S.; Bag, A.; Balakrishnan, B.; Patel, K.S.; Guerra, A.A.; Martinez, M.N.; Trivedi, S.; et al. Pancreatic triglyceride lipase mediates lipotoxic systemic inflammation. J. Clin. Investig. 2020, 130, 1931–1947.
- Navina, S.; Acharya, C.; DeLany, J.P.; Orlichenko, L.S.; Baty, C.J.; Shiva, S.S.; Durgampudi, C.; Karlsson, J.M.; Lee, K.; Bae, K.T.; et al. Lipotoxicity causes multisystem organ failure and exacerbates acute pancreatitis in obesity. Sci. Transl. Med. 2011, 3, 107ra110.
- Baek, Y.B.; Kwon, H.J.; Sharif, M.; Lim, J.; Lee, I.C.; Ryu, Y.B.; Lee, J.I.; Kim, J.S.; Lee, Y.S.; Kim, D.H.; et al. Therapeutic strategy targeting host lipolysis limits infection by SARS-CoV-2 and influenza A virus. Signal Transduct. Target. Ther. 2022, 7, 367.
- Meakins, J.L.; Pietsch, J.B.; Bubenick, O.; Kelly, R.; Rode, H.; Gordon, J.; MacLean, L.D. Delayed hypersensitivity: Indicator of acquired failure of host defenses in sepsis and trauma. Ann. Surg. 1977, 186, 241–250.
- MacLean, L.D.; Meakins, J.L.; Taguchi, K.; Duignan, J.P.; Dhillon, K.S.; Gordon, J. Host resistance in sepsis and trauma. Ann. Surg. 1975, 182, 207–217.
- Monneret, G.; Gossez, M.; Aghaeepour, N.; Gaudilliere, B.; Venet, F. How clinical flow cytometry rebooted sepsis immunology. Cytom. Part A J. Int. Soc. Anal. Cytol. 2019, 95, 431–441.
- Garcia-Sabrido, J.L.; Valdecantos, E.; Bastida, E.; Tellado, J.M. The anergic state as a predictor of pancreatic sepsis. Zent. Fur Chir. 1989, 114, 114–120.
- Mayerle, J.; Dummer, A.; Sendler, M.; Malla, S.R.; van den Brandt, C.; Teller, S.; Aghdassi, A.; Nitsche, C.; Lerch, M.M. Differential roles of inflammatory cells in pancreatitis. J. Gastroenterol. Hepatol. 2012, 27 (Suppl. 2), 47–51.
- Jain, S.; Midha, S.; Mahapatra, S.J.; Gupta, S.; Sharma, M.K.; Nayak, B.; Jacob, T.G.; Shalimar; Garg, P.K. Interleukin-6 significantly improves predictive value of systemic inflammatory response syndrome for predicting severe acute pancreatitis. Pancreatology 2018, 18, 500–506.
- Bhatia, M. Acute pancreatitis as a model of SIRS. Front. Biosci. (Landmark Ed.) 2009, 14, 2042–2050.
- Bone, R.C. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit. Care Med. 1996, 24, 1125–1128.
- Zhulai, G.A.; Oleinik, E.K.; Ostrovskii, K.A.; Oleinik, V.M.; Kravchenko, P.N.; Churov, A.V. Alterations of lymphocyte subsets and indicators of immune suppression in patients with acute pancreatitis. Eksp. Klin. Gastroenterol. 2014, 9, 21–25.
- Döcke, W.D.; Randow, F.; Syrbe, U.; Krausch, D.; Asadullah, K.; Reinke, P.; Volk, H.D.; Kox, W. Monocyte deactivation in septic patients: Restoration by IFN-gamma treatment. Nat. Med. 1997, 3, 678–681.
- Volk, H.D.; Reinke, P.; Krausch, D.; Zuckermann, H.; Asadullah, K.; Müller, J.M.; Döcke, W.D.; Kox, W.J. Monocyte deactivation--rationale for a new therapeutic strategy in sepsis. Intensive Care Med. 1996, 22 (Suppl. 4), S474–S481.
- Venet, F.; Lepape, A.; Monneret, G. Clinical review: Flow cytometry perspectives in the ICU-from diagnosis of infection to monitoring of injury-induced immune dysfunctions. Crit. Care 2011, 15, 231–239.
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Immunosuppression in sepsis: A novel understanding of the disorder and a new therapeutic approach. Lancet Infect. Dis. 2013, 13, 260–268.
- Leentjens, J.; Kox, M.; van der Hoeven, J.G.; Netea, M.G.; Pickkers, P. Immunotherapy for the adjunctive treatment of sepsis: From immunosuppression to immunostimulation. Time for a paradigm change? Am. J. Respir. Crit. Care Med. 2013, 187, 1287–1293.
- Giamarellos-Bourboulis, E.J. What is the pathophysiology of the septic host upon admission? Int. J. Antimicrob. Agents 2010, 36 (Suppl. 2), S2–S5.
- Minkov, G.A.; Halacheva, K.S.; Yovtchev, Y.P.; Gulubova, M.V. Pathophysiological mechanisms of acute pancreatitis define inflammatory markers of clinical prognosis. Pancreas 2015, 44, 713–717.
- Zhuang, Y.G.; Peng, H.; Chen, Y.Z.; Zhou, S.Q.; Chen, Y.Q. Dynamic monitoring of monocyte HLA-DR expression for the diagnosis, prognosis, and prediction of sepsis. Front. Biosci.-Landmrk 2017, 22, 1344–1354.
- Li, J.; Yang, W.-J.; Huang, L.-M.; Tang, C.-W. Immunomodulatory therapies for acute pancreatitis. World J. Gastroenterol. 2014, 20, 16935–16947.
- Misra, A.K.; Levy, M.M.; Ward, N.S. Biomarkers of Immunosuppression. Crit. Care Clin. 2020, 36, 167–176.
- Benlyamani, I.; Venet, F.; Coudereau, R.; Gossez, M.; Monneret, G. Monocyte HLA-DR measurement by flow cytometry in COVID-19 patients: An interim review. Cytom. Part A J. Int. Soc. Anal. Cytol. 2020, 97, 1217–1221.
- Manohar, M.; Jones, E.K.; Rubin, S.J.S.; Subrahmanyam, P.B.; Swaminathan, G.; Mikhail, D.; Bai, L.; Singh, G.; Wei, Y.; Sharma, V.; et al. Novel circulating and tissue monocytes as well as macrophages in pancreatitis and recovery. Gastroenterology 2021, 161, 2014–2029.e14.
- Napoli, C.; Benincasa, G.; Criscuolo, C.; Faenza, M.; Liberato, C.; Rusciano, M. Immune reactivity during COVID-19: Implications for treatment. Immunol. Lett. 2021, 231, 28–34.
- Volk, H.D.; Thieme, M.; Heym, S.; Döcke, W.D.; Ruppe, U.; Tausch, W.; Manger, D.; Zuckermann, S.; Golosubow, A.; Nieter, B.; et al. Alterations in function and phenotype of monocytes from patients with septic disease--predictive value and new therapeutic strategies. Behring. Inst. Mitt. 1991, 88, 208–215.
- Lin, R.Y.; Astiz, M.E.; Saxon, J.C.; Rackow, E.C. Altered leukocyte immunophenotypes in septic shock. Studies of HLA-DR, CD11b, CD14, and IL-2R expression. Chest 1993, 104, 847–853.
- Ditschkowski, M.; Kreuzfelder, E.; Rebmann, V.; Ferencik, S.; Majetschak, M.; Schmid, E.N.; Obertacke, U.; Hirche, H.; Schade, U.F.; Grosse-Wilde, H. HLA-DR expression and soluble HLA-DR levels in septic patients after trauma. Ann. Surg. 1999, 229, 246–254.
- Wakefield, C.H.; Carey, P.D.; Foulds, S.; Monson, J.R.; Guillou, P.J. Changes in major histocompatibility complex class II expression in monocytes and T cells of patients developing infection after surgery. Br. J. Surg. 1993, 80, 205–209.
- Sachse, C.; Prigge, M.; Cramer, G.; Pallua, N.; Henkel, E. Association between reduced human leukocyte antigen (HLA)-DR expression on blood monocytes and increased plasma level of interleukin-10 in patients with severe burns. Clin. Chem. Lab. Med. 1999, 37, 193–198.
- Kaufman, J.F.; Auffray, C.; Korman, A.J.; Shackelford, D.A.; Strominger, J. The class II molecules of the human and murine major histocompatibility complex. Cell 1984, 36, 1–13.
- Unanue, E.R.; Turk, V.; Neefjes, J. Variations in MHC Class II antigen processing and presentation in health and disease. Annu. Rev. Immunol. 2016, 34, 265–297.
- Crux, N.B.; Elahi, S. Human leukocyte antigen (HLA) and immune regulation: How do classical and non-classical HLA alleles modulate immune response to human immunodeficiency virus and hepatitis C virus infections? Front. Immunol. 2017, 8, 832.
- Mosaad, Y.M. Clinical role of human leukocyte antigen in health and disease. Scand. J. Immunol. 2015, 82, 283–306.
- Monneret, G.; Venet, F. Sepsis-induced immune alterations monitoring by flow cytometry as a promising tool for individualized therapy. Cytom. B Clin. Cytom. 2016, 90, 376–386.
- Monneret, G.; Venet, F. Monocyte HLA-DR in sepsis: Shall we stop following the flow? Crit. Care 2014, 18, 102.
- Monneret, G.; Elmenkouri, N.; Bohe, J.; Debard, A.L.; Gutowski, M.C.; Bienvenu, J.; Lepape, A. Analytical requirements for measuring monocytic human lymphocyte antigen DR by flow cytometry: Application to the monitoring of patients with septic shock. Clin. Chem. 2002, 48, 1589–1592.
- Docke, W.D.; Hoflich, C.; Davis, K.A.; Rottgers, K.; Meisel, C.; Kiefer, P.; Weber, S.U.; Hedwig-Geissing, M.; Kreuzfelder, E.; Tschentscher, P.; et al. Monitoring temporary immunodepression by flow cytometric measurement of monocytic HLA-DR expression: A multicenter standardized study. Clin. Chem. 2005, 51, 2341–2347.
- Demaret, J.; Walencik, A.; Jacob, M.C.; Timsit, J.F.; Venet, F.; Lepape, A.; Monneret, G. Inter-laboratory assessment of flow cytometric monocyte HLA-DR expression in clinical samples. Cytom. B Clin. Cytom. 2013, 84, 59–62.
- Cajander, S.; Backman, A.; Tina, E.; Stralin, K.; Soderquist, B.; Kallman, J. Preliminary results in quantitation of HLA-DRA by real-time PCR: A promising approach to identify immunosuppression in sepsis. Crit. Care 2013, 17, R223.
- van den Elsen, P.J. Expression regulation of major histocompatibility complex class I and class II encoding genes. Front. Immunol. 2011, 2, 48.
- Steimle, V.; Otten, L.A.; Zufferey, M.; Mach, B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell 1993, 75, 135–146.
- Miatello, J.; Lukaszewicz, A.C.; Carter, M.J.; Faivre, V.; Hua, S.; Martinet, K.Z.; Bourgeois, C.; Quintana-Murci, L.; Payen, D.; Boniotto, M.; et al. CIITA promoter polymorphism impairs monocytes HLA-DR expression in patients with septic shock. iScience 2022, 25, 105291.
- Pishesha, N.; Harmand, T.J.; Ploegh, H.L. A guide to antigen processing and presentation. Nat. Rev. Immunol. 2022, 22, 751–764.
- Viville, S.; Neefjes, J.; Lotteau, V.; Dierich, A.; Lemeur, M.; Ploegh, H.; Benoist, C.; Mathis, D. Mice lacking the MHC class II-associated invariant chain. Cell 1993, 72, 635–648.
- Wolk, K.; Kunz, S.; Crompton, N.E.; Volk, H.D.; Sabat, R. Multiple mechanisms of reduced major histocompatibility complex class II expression in endotoxin tolerance. J. Biol. Chem. 2003, 278, 18030–18036.
- Alfonso, C.; Karlsson, L. Nonclassical MHC class II molecules. Annu. Rev. Immunol. 2000, 18, 113–142.
- Fung-Leung, W.P.; Surh, C.D.; Liljedahl, M.; Pang, J.; Leturcq, D.; Peterson, P.A.; Webb, S.R.; Karlsson, L. Antigen presentation and T cell development in H2-M-deficient mice. Science 1996, 271, 1278–1281.
- Pinet, V.; Vergelli, M.; Martin, R.; Bakke, O.; Long, E.O. Antigen presentation mediated by recycling of surface HLA-DR molecules. Nature 1995, 375, 603–606.
- Koppelman, B.; Neefjes, J.J.; de Vries, J.E.; de Waal Malefyt, R. Interleukin-10 down-regulates MHC class II alphabeta peptide complexes at the plasma membrane of monocytes by affecting arrival and recycling. Immunity 1997, 7, 861–871.
- Kylanpaa-Back, M.L.; Takala, A.; Kemppainen, E.; Puolakkainen, P.; Kautiainen, H.; Jansson, S.E.; Haapiainen, R.; Repo, H. Cellular markers of systemic inflammation and immune suppression in patients with organ failure due to severe acute pancreatitis. Scand. J. Gastroenterol. 2001, 36, 1100–1107.
- Richter, A.; Nebe, T.; Wendl, K.; Schuster, K.; Klaebisch, G.; Quintel, M.; Lorenz, D.; Post, S.; Trede, M. HLA-DR expression in acute pancreatitis. Eur. J. Surg. 1999, 165, 947–951.
- Turunen, A.; Kuuliala, A.; Penttilä, A.; Kaukonen, K.M.; Mustonen, H.; Pettilä, V.; Puolakkainen, P.; Kylänpää, L.; Kuuliala, K. Time course of signaling profiles of blood leukocytes in acute pancreatitis and sepsis. Scand. J. Clin. Lab. Investig. 2020, 80, 114–123.
- Li, J.P.; Yang, J.; Huang, J.R.; Jiang, D.L.; Zhang, F.; Liu, M.F.; Qiang, Y.; Gu, Y.L. Immunosuppression and the infection caused by gut mucosal barrier dysfunction in patients with early severe acute pancreatitis. Front. Biosci. (Landmark Ed.) 2013, 18, 892–900.
- Mentula, P.; Kylanpaa, M.L.; Kemppainen, E.; Jansson, S.E.; Sarna, S.; Puolakkainen, P.; Haapiainen, R.; Repo, H. Plasma anti-inflammatory cytokines and monocyte human leucocyte antigen-DR expression in patients with acute pancreatitis. Scand. J. Gastroenterol. 2004, 39, 178–187.
- Yu, W.K.; Li, W.Q.; Li, N.; Li, J.S. Mononuclear histocompatibility leukocyte antigen-DR expression in the early phase of acute pancreatitis. Pancreatology 2004, 4, 233–243.
- Walter, L.O.; Cardoso, C.C.; Santos-Pirath, Í.M.; Costa, H.Z.; Gartner, R.; Werle, I.; Mohr, E.T.B.; da Rosa, J.S.; Felisberto, M.; Kretzer, I.F.; et al. The relationship between peripheral immune response and disease severity in SARS-CoV-2-infected subjects: A cross-sectional study. Immunology 2022, 165, 481–496.
- Yang, Q.; Wen, Y.; Qi, F.; Gao, X.; Chen, W.; Xu, G.; Wei, C.; Wang, H.; Tang, X.; Lin, J.; et al. Suppressive monocytes impair MAIT cells response via IL-10 in patients with severe COVID-19. J. Immunol. 2021, 207, 1848–1856.
- Gatti, A.; Radrizzani, D.; Viganò, P.; Mazzone, A.; Brando, B. Decrease of non-classical and intermediate monocyte subsets in severe acute SARS-CoV-2 infection. Cytom. Part A J. Int. Soc. Anal. Cytol. 2020, 97, 887–890.
- Mudd, P.A.; Crawford, J.C.; Turner, J.S.; Souquette, A.; Reynolds, D.; Bender, D.; Bosanquet, J.P.; Anand, N.J.; Striker, D.A.; Martin, R.S.; et al. Distinct inflammatory profiles distinguish COVID-19 from influenza with limited contributions from cytokine storm. Sci. Adv. 2020, 6, eabe3024.
- Xu, G.; Qi, F.; Li, H.; Yang, Q.; Wang, H.; Wang, X.; Liu, X.; Zhao, J.; Liao, X.; Liu, Y.; et al. The differential immune responses to COVID-19 in peripheral and lung revealed by single-cell RNA sequencing. Cell Discov. 2020, 6, 73.
- Bonnet, B.; Cosme, J.; Dupuis, C.; Coupez, E.; Adda, M.; Calvet, L.; Fabre, L.; Saint-Sardos, P.; Bereiziat, M.; Vidal, M.; et al. Severe COVID-19 is characterized by the co-occurrence of moderate cytokine inflammation and severe monocyte dysregulation. EBioMedicine 2021, 73, 103622.
- Christensen, E.E.; Jørgensen, M.J.; Nore, K.G.; Dahl, T.B.; Yang, K.; Ranheim, T.; Huse, C.; Lind, A.; Nur, S.; Stiksrud, B.; et al. Critical COVID-19 is associated with distinct leukocyte phenotypes and transcriptome patterns. J. Intern. Med. 2021, 290, 677–692.
- Qin, S.; Jiang, Y.; Wei, X.; Liu, X.; Guan, J.; Chen, Y.; Lu, H.; Qian, J.; Wang, Z.; Lin, X. Dynamic changes in monocytes subsets in COVID-19 patients. Hum. Immunol. 2021, 82, 170–176.
- Zenarruzabeitia, O.; Astarloa-Pando, G.; Terrén, I.; Orrantia, A.; Pérez-Garay, R.; Seijas-Betolaza, I.; Nieto-Arana, J.; Imaz-Ayo, N.; Pérez-Fernández, S.; Arana-Arri, E.; et al. T cell activation, highly armed cytotoxic cells and a shift in monocytes CD300 receptors expression is characteristic of patients with severe COVID-19. Front. Immunol. 2021, 12, 655934.
- Cizmecioglu, A.; Emsen, A.; Sumer, S.; Ergun, D.; Akay Cizmecioglu, H.; Turk Dagi, H.; Artac, H. Reduced monocyte subsets, their HLA-DR expressions, and relations to acute phase reactants in severe COVID-19 cases. Viral Immunol. 2022, 35, 273–282.
- Hammad, R.; Kotb, H.G.; Eldesoky, G.A.; Mosaad, A.M.; El-Nasser, A.M.; Abd El Hakam, F.E.; Eldesoky, N.A.; Mashaal, A.; Farhoud, H. Utility of monocyte expression of HLA-DR versus T lymphocyte frequency in the assessment of COVID-19 outcome. Int. J. Gen. Med. 2022, 15, 5073–5087.
- Venet, F.; Gossez, M.; Bidar, F.; Bodinier, M.; Coudereau, R.; Lukaszewicz, A.C.; Tardiveau, C.; Brengel-Pesce, K.; Cheynet, V.; Cazalis, M.A.; et al. T cell response against SARS-CoV-2 persists after one year in patients surviving severe COVID-19. EBioMedicine 2022, 78, 103967.
- Marais, C.; Claude, C.; Semaan, N.; Charbel, R.; Barreault, S.; Travert, B.; Piloquet, J.E.; Demailly, Z.; Morin, L.; Merchaoui, Z.; et al. Myeloid phenotypes in severe COVID-19 predict secondary infection and mortality: A pilot study. Ann. Intensive Care 2021, 11, 111.
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe 2020, 27, 992–1000.e3.
- Moser, D.; Biere, K.; Han, B.; Hoerl, M.; Schelling, G.; Choukér, A.; Woehrle, T. COVID-19 impairs immune response to candida albicans. Front. Immunol. 2021, 12, 640644.
- Utrero-Rico, A.; González-Cuadrado, C.; Chivite-Lacaba, M.; Cabrera-Marante, O.; Laguna-Goya, R.; Almendro-Vazquez, P.; Díaz-Pedroche, C.; Ruiz-Ruigómez, M.; Lalueza, A.; Folgueira, M.D.; et al. Alterations in circulating monocytes predict COVID-19 severity and include chromatin modifications still detectable six months after recovery. Biomedicines 2021, 9, 1253.
- Mairpady Shambat, S.; Gómez-Mejia, A.; Schweizer, T.A.; Huemer, M.; Chang, C.C.; Acevedo, C.; Bergada-Pijuan, J.; Vulin, C.; Hofmaenner, D.A.; Scheier, T.C.; et al. Hyperinflammatory environment drives dysfunctional myeloid cell effector response to bacterial challenge in COVID-19. PLoS Pathog. 2022, 18, e1010176.
- Boumaza, A.; Gay, L.; Mezouar, S.; Bestion, E.; Diallo, A.B.; Michel, M.; Desnues, B.; Raoult, D.; La Scola, B.; Halfon, P.; et al. Monocytes and macrophages, targets of severe acute respiratory syndrome coronavirus 2: The clue for coronavirus disease 2019 immunoparalysis. J. Infect. Dis. 2021, 224, 395–406.
- Laing, A.G.; Lorenc, A.; Del Molino Del Barrio, I.; Das, A.; Fish, M.; Monin, L.; Muñoz-Ruiz, M.; McKenzie, D.R.; Hayday, T.S.; Francos-Quijorna, I.; et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020, 26, 1623–1635.
- Penttilä, P.A.; Van Gassen, S.; Panovska, D.; Vanderbeke, L.; Van Herck, Y.; Quintelier, K.; Emmaneel, A.; Filtjens, J.; Malengier-Devlies, B.; Ahmadzadeh, K.; et al. High dimensional profiling identifies specific immune types along the recovery trajectories of critically ill COVID19 patients. Cell Mol. Life Sci. 2021, 78, 3987–4002.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
782
Revisions:
2 times
(View History)
Update Date:
25 Feb 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No