Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lígia O. Martins | -- | 3855 | 2023-02-24 11:53:53 | | | |
| 2 | Jason Zhu | -99 word(s) | 3756 | 2023-02-27 04:41:37 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Rodrigues, C.F.; Borges, P.T.; Scocozza, M.F.; Silva, D.; Taborda, A.; Brissos, V.; Frazão, C.; Martins, L.O. Loops around Heme Pocket influence Bacillus subtilis's BsDyP. Encyclopedia. Available online: https://encyclopedia.pub/entry/41634 (accessed on 25 July 2026).
Rodrigues CF, Borges PT, Scocozza MF, Silva D, Taborda A, Brissos V, et al. Loops around Heme Pocket influence Bacillus subtilis's BsDyP. Encyclopedia. Available at: https://encyclopedia.pub/entry/41634. Accessed July 25, 2026.
Rodrigues, Carolina F., Patrícia T. Borges, Magali F. Scocozza, Diogo Silva, André Taborda, Vânia Brissos, Carlos Frazão, Lígia O. Martins. "Loops around Heme Pocket influence Bacillus subtilis's BsDyP" Encyclopedia, https://encyclopedia.pub/entry/41634 (accessed July 25, 2026).
Rodrigues, C.F., Borges, P.T., Scocozza, M.F., Silva, D., Taborda, A., Brissos, V., Frazão, C., & Martins, L.O. (2023, February 24). Loops around Heme Pocket influence Bacillus subtilis's BsDyP. In Encyclopedia. https://encyclopedia.pub/entry/41634
Rodrigues, Carolina F., et al. "Loops around Heme Pocket influence Bacillus subtilis's BsDyP." Encyclopedia. Web. 24 February, 2023.
Copy Citation
Bacillus subtilis BsDyP belongs to class I of the enzyme's dye-decolorizing peroxidase (DyP) family. It is an interesting biocatalyst due to its high redox potential, broad substrate spectrum, and thermostability. The engineering of the enzyme towards improved activity for phenolics revealed that loops close to the heme pocket could be modulated for tuning catalytic and stability DyP properties.
dye-decolorizing peroxidases
directed evolution
biorefineries
enzyme specificity
thermostability
structure–function relationships
soil bacteria
1. Introduction
Biocatalysis is both a green and sustainable technology and redox biocatalysts offer eco-friendly advantages in comparison with conventional chemical reactions due to the selectivity, controllability and economy of their reactions. Lignin is the largest reserve of aromatics on Earth and is a key renewable source of chemicals and materials [1]. Recent strategies developed for lignin depolymerization allowed the derivation of well-defined compounds in acceptable quantities, bringing the utilization of lignin as a feedstock for aromatic chemicals one step closer to reality [1][2][3]. At present, the challenge is the set-up of atom-economic and waste-free (bio)processes that allow the full implementation of a lignin-derived platform of chemicals, sustainable starting material for the production of drop-in chemicals, structural scaffolds exploitable in the field of medicinal chemistry, polymers and emerging functional materials [1][4][5][6][7]. In nature, white-rot fungi and certain bacteria are responsible for the depolymerization and conversion of lignin, and therefore, they are useful sources of ligninolytic enzymes, such as laccases and fungal lignin (LiP), versatile (VP), manganese peroxidases (MnP) and dye-decolorizing peroxidases (DyPs) [8][9][10][11].
DE is a powerful engineering tool that mimics the principles of natural selection through iterative rounds of mutagenesis, recombination and screening [12]. The properties of evolved variants based on their biochemical, kinetic and structural analysis are discussed. In DyPs, the oxidation of reduced substrates occurs in heme cavities and in tyrosine and tryptophan surface-exposed residues, similarly to LiP and VP enzymes, and then to transfer electrons to the heme using long-range electron transfer (LRET) pathways [13][14][15]. However, details of substrate binding and of molecular determinants of substrate specificity in DyPs remain open questions. This helps to understand the role of conserved loops around the heme pocket in substrate binding and catalysis and the interplay of catalytic and stability mechanisms of DyPs with implications in their industrial application and in the future design of enzymes.
2. Deletion of Tat-Signal Peptide from BsDyP
BsDyP has a 45-residue N-terminal twin-arginine signal peptide sequence (MSDEQKKPEQIHRRDILKWGAMAGAAVAIGASGLGGLAPLVQTA) with an Arg-Arg motif, recognized by the twin-arginine translocation (Tat) pathway, involved in the translocation of folded proteins from the cytoplasm and secretion to the extracellular milieu [16][17]. The recombinant production of BsDyP in Escherichia coli resulted in two forms with different molecular masses as assessed by SDS-PAGE (Figure S1a [18]): one upper band that most probably corresponds to the unprocessed cytoplasmic precursor containing the signal peptide, and a lower band that corresponds to the mature periplasmic enzyme without the signal peptide [16], similarly to what was observed in the heterologous expression of TfuDyP Thermobifida fusca [19] and E. coli YcdB [20], DyP members from class I also harboring an N-terminal twin-arginine sequence. Researchers deleted the N-terminal sequence of BsDyP to achieve homogeneous preparations of recombinant enzyme. The heterologous production of the truncated (mature) form of BsDyP resulted in one single band in the electrophoresis gel. The truncated enzyme shows spectroscopic and kinetic properties comparable to those of an intact enzyme [18]. This form (named hereafter wild-type BsDyP) was used.
3. Directed Evolution of BsDyP for Improved DMP Oxidation
Three rounds of evolution were performed and approximately 6000 clones were screened to identify a variant with improved catalytic efficiency for the lignin-related phenolic DMP (Figure 1). In the first round of evolution using epPCR, ~600 clones were screened using the qualitative “activity on-plate assay” followed by liquid activity screening in 96-well plates. Two variants, 3G5 (with mutations A330V, L166Q, V284A and T296S) and 1F9 (with the single S325P mutation), showed 2- and 5-fold higher activity for DMP, respectively, in comparison with the wild type. Variant 1F9 parented the next generation where ~3500 variants were screened. The top four variants inserted one additional mutation: K220R (variant 50F7), K317E (variant 54D6), E346V (variant 56C8), K248E (variant 51E9), and revealed very similar enzymatic activities (1.5 to 1.9-fold higher than 1F9), which impaired the selection of one clearly best variant to parent the following round of evolution. Therefore, to construct the third generation library of variants, random recombination by DNA shuffling of genes coding for 50F7, 51E9, 54D6 and 56C8 plus the gene coding for variant 3G5, found in the first round of evolution, was performed. A library of ~1980 mutants was screened and the top twenty-five variants were selected for further analysis. The DNA sequencing revealed that from the initial twenty-five phenotypes, only fifteen corresponded to different genotypes considering the non-synonymous substitutions; for example, the six top variants (5G5, 7E7, 6B8, 3C7, 2C5, 7E10) that reached 1.7 to 2.5-fold higher activity than the parent (considered 50F7) corresponded to only three genotypes. The highest activity for DMP was consistently measured for variant 5G5, which gathered mutations S325P and A330V from variants 1F9 and 3G5, respectively, from the first generation, and mutation K317E from variant 54D6 of the second generation.
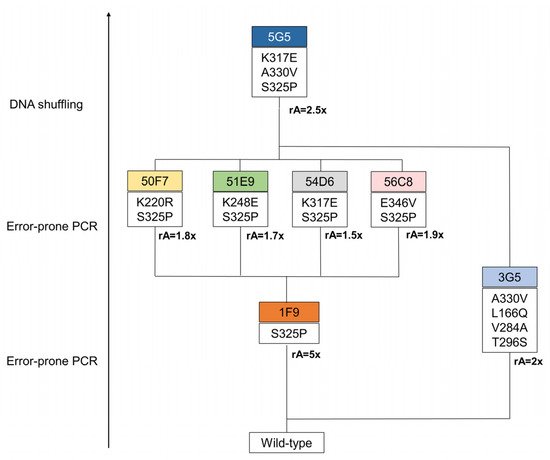
Figure 1. Lineage of BsDyP variants generated in this entry. In the first generation, a total of 614 variants, generated through error-prone PCR, were screened in 96-well plates using 1 mM of DMP. The 1F9 variant was selected on the basis of its 5-fold increased activity in crude extracts compared with the parent. Next, the second generation was evolved from the 1F9 variant as the parent. In this round, 365 variants, resulting from error-prone PCR, were screened in 96-well plates. The variants 50F7, 51E9, 54D6 and 56C8 were identified as having a slightly higher activity (between 1.5 and 1.9-fold) when compared to the parent. In the third generation, variants were constructed using DNA shuffling, where genes from variants of the second generation were recombined with the 3G5 gene, from the first round of evolution, with a 2-fold increased activity in crude extracts when compared with the wild type. In this round of evolution, 588 variants were screened in 96-well plates. From this process resulted one hit variant, 5G5, with 2-fold increased activity at pH 4. rA represents the relative activity to the parent.
4. Biochemical and Kinetic Characterization
To understand the role of the three mutations that originated the improved activity of BsDyP towards DMP, the wild type and variants 1F9, 3G5, 54D6 and 5G5 were overproduced, purified and characterized. The Reinheitszahl values of purified enzymes vary between 1.6 and 2.5 and the UV–visible absorption spectra of the purified enzymes revealed the characteristic Soret band at 406–407 nm (Table 1). The enzyme yields improved in the course of evolution from 8 mg L−1 in the wild type to 15 mg L−1 in hit variant 5G5 (Table 1), indicating the beneficial role of mutations acquired for BsDyP recombinant production. Furthermore, the incorporation of the heme cofactor almost doubled from 0.4 to 0.6–0.7 mol per mole of protein in the wild type and variants, respectively.
Table 1. Spectroscopic and redox properties of purified BsDyP wild type and variants.
| Enzyme | Production (mg L−1) | Rz | λmax (nm) | ε (mM−1 cm−1) | Heme Content |
|---|---|---|---|---|---|
| Wild Type | 8.3 ± 0.4 | 2.3 | 406 | 82 | 0.4 ± 0.1 |
| 1F9 | 11.1 ± 0.8 | 2.5 | 407 | 92 | 0.7 ± 0.2 |
| 3G5 | 6.5 ± 0.5 | 1.6 | 406 | 60 | 0.6 ± 0.2 |
| 54D6 | 16.0 ± 0.9 | 1.8 | 407 | 66 | 0.6 ± 0.1 |
| 5G5 | 15.0 ± 1.0 | 2.1 | 407 | 77 | 0.7 ± 0.1 |
An optimal pH around 4 was observed for DMP and for 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) for all tested enzymes. The activity for H2O2 and ABTS increased 2 to 4-fold in all variants, but as the Km values also slightly increased (2–3 fold) the catalytic efficiency (kcatapp/Kmapp) remained analogous in the variants as compared to the wild type (Table 2). The variants from the first and second round of evolution, 1F9, 3G5 and 54D6, showed similar kcatapp values for DMP, circa 2-fold of those of the wild type (Table 3). Noteworthy, the kcatapp of the hit 5G5 variant is ~7-fold higher for DMP, indicating a synergistic action of the three mutations S325P, A330V and K317E in the functional transition. The Km values for DMP of variants 54D6 and 5G5 (both containing the mutation K317E) increased 10-fold as compared to the wild type, which is reflected in a slightly lower catalytic efficiency (kcatapp/Km) for DMP as compared to the wild type. The lower Km indicates that mutation K317E introduced steric changes in the substrate-binding site(s) that negatively affected DMP binding to the enzyme. Note, however, that the turnover number (kcatapp) is considered the most important parameter for biotechnological applications since bioprocesses usually take place at high concentrations of substrate, i.e., the enzyme’s activity is not limited by substrate concentration but by the turnover number. In this respect, the hit variant 5G5 has a kcatapp for DMP 107-, 56- and 29-fold higher when compared, for example, with class I bacterial Thermobifida fusca TfuDyP (kcatapp = 0.026 s−1), class P P. putida PpDyP (kcatapp = 0.05 s−1) and class V Streptomyces avermitilis, SaDyP2 (kcatapp = 0.097 s−1), respectively [19][21][22]. 5G5 is also a promising candidate for further BsDyP evolution to increase the kcatapp parameter to values close to those of fungal counterparts, such as Auricularia auricula-judae AauDyP, which shows a paramount kcatapp of 89 s−1 for DMP [23].
Table 2. Apparent steady-state kinetic parameters of wild type and variants for hydrogen peroxide and ABTS, measured at 25 °C and pH 4.2.
| Enzyme | H2O2 | ABTS | ||||
|---|---|---|---|---|---|---|
| kcatapp (s−1) | Kmapp (mM) | kcatapp/Kmapp (M−1s−1) | Ki (mM) | Kmapp (mM) | kcatapp/Kmapp (M−1s−1) | |
| Wild Type | 14.2 ± 1.0 | 0.06 ± 0.01 | (2.2 ± 0.4) × 105 | 6.5 ± 1.9 | 0.42 ± 0.03 | (3.6 ± 0.3) × 104 |
| 1F9 | 50.4 ± 1.4 | 0.19 ± 0.05 | (2.7 ± 0.7) × 105 | 2.6 ± 0.2 | 0.89 ± 0.10 | (5.8 ± 0.7) × 104 |
| 3G5 | 28.1 ± 2.1 | 0.06 ± 0.01 | (4.9 ± 0.7) × 105 | 8.5 ± 0.4 | 1.37 ± 0.25 | (2.5 ± 0.5) × 104 |
| 54D6 | 47.7 ± 2.9 | 0.15 ± 0.02 | (3.3 ± 0.6) × 105 | 8.1 ± 0.7 | 0.80 ± 0.14 | (6.0 ± 1.1) × 104 |
| 5G5 | 56.1 ± 1.9 | 0.18 ± 0.02 | (3.3 ± 0.7) × 105 | 3.9 ± 0.6 | 1.29 ± 0.19 | (4.2 ± 0.6) × 104 |
Table 3. Apparent steady-state kinetic parameters of wild type and variants for DMP, measured at 25 °C in sodium phosphate buffer at pH 3.8 in the presence of 0.2 (wild type) and 0.6 mM (1F9, 3G5, 54D6 and 5G5) H2O2.
| Enzyme | kcatapp (s−1) | Kmapp (mM) | kcatapp/Kmapp (M−1s−1) |
|---|---|---|---|
| Wild Type | 0.42 ± 0.03 | 0.06 ± 0.01 | (7 ± 1) × 103 |
| 1F9 | 0.83 ± 0.04 | 0.15 ± 0.01 | (5.5 ± 0.4) × 103 |
| 3G5 | 0.8 ± 0.1 | 0.09 ± 0.01 | (9 ± 1) × 103 |
| 54D6 | 1.0 ± 0.1 | 0.60 ± 0.01 | (1.6 ± 0.5) × 103 |
| 5G5 | 2.8 ± 0.1 | 0.7 ± 0.1 | (3.8 ± 0.2) × 103 |
5. Role of Mutations in the Optimization of 5G5 for DMP Oxidation
To investigate the structural consequences of evolving the wild type to 5G5, the X-ray crystal structures of 5G5 and the wild type were solved at 2.10 and 2.49 Å resolution, respectively (Figure 2a and Table 4). The 5G5 variant crystal structure is very similar to the wild type, showing root-mean-square deviations (r.m.s.d.) between Cα atoms that range from 0.46 to 0.51 Å. The wild type crystallized in the trigonal P3121 space group with two molecules per asymmetric unit (Figure S6a). These two subunits have an r.m.s.d. of 0.31 Å between Cα atoms. The recently deposited BsDyP crystal structure (PDB 6KMN, [24]) belongs to the triclinic P1 space group, showing four subunits in the asymmetric unit. The crystal structure of the 5G5 variant belongs to the monoclinic P21 space group and four monomers in the asymmetric unit were identified. Chains A, B and C share a higher structural homology, with r.m.s.d. values ranging from 0.30–0.33 Å between Cα atoms, than chain D (r.m.s.d. = 0.43–0.45 Å); researchers opted to examine chain A for both the wild type and the 5G5 variant in most of the structural analysis.
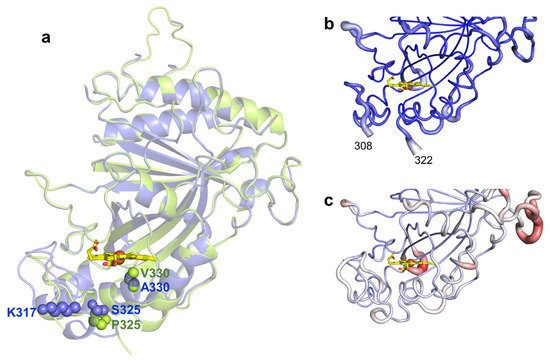
Figure 2. (a) Transparent secondary structure cartoon of superimposed wild type (blue) and 5G5 (green). The mutations P325 and V330 in 5G5 are shown as green spheres, and their homologs in the wild type as blue spheres. K317 is shown as blue spheres in the wild type and E317 is not visible in 5G5, and therefore is not represented. The heme is represented as sticks, with carbon, oxygen and nitrogen atoms colored as yellow, red and blue, respectively. The iron is shown as a brown sphere. Cartoon representation of the main-chain 5G5 (b) and wild-type (c) structures with thickness proportional to a.d.p. values, color coded from blue (21 Å2) to red (126 Å2). The full length enzymes contain 416 residues but the refined models consist of polypeptide chains with 349–354 amino acids; the first 45 residues, TAT signal, were deleted and the residues 46–55 (N-terminal) and 415–416 (C-terminal) are not visible in the electron density maps. A long loop region containing 32 residues (107–138) has high a.d.p.s, 55.4–109.8 Å2, and five residues (112–116) could not be modeled in the wild type due to lack of electron density. Additionally, the region 309–321 (boxed) becomes more flexible during evolution and is not visible in the electron density maps of 5G5 in chains A–C (b); in chain D, the missing region is between residues 317 and 321.
Table 4. X-ray data collection and refinement statistics. Values in parentheses belong to the highest resolution shell.
| BsDyP-Wild-Type | BsDyP-5G5 | |
|---|---|---|
| Data Collection | ||
| Beamline | BL13-XALOC | ID30A-3 |
| Wavelength (Å) | 0.97926 | 0.9680 |
| Space group | P 31 2 1 | P 1 21 1 |
| Unit cell parameters (Å) | a = 95.3, b = 95.3, c = 181.2 | a = 63.9, b = 114.8, c = 116.8 |
| Resolution (Å) | 75.09–2.49 (2.59–2.49) | 61.96–2.10 (2.20–2.10) |
| Number of observations | 215,157 (26,986) | 285,233 (46,534) |
| Unique reflections | 33,999 (5374) | 92,499 (15,038) |
| Completeness (%) | 99.8 (99.2) | 99.1 (98.0) |
| Multiplicity | 6.3 (5.0) | 3.1 (3.1) |
| Mosaicity (ᵒ) | 0.12 | 0.09 |
| CC1/2 (%) a | 99.7 (30.6) | 99.6 (47.8) |
| Rsym (%) b | 13.0 (78.8) | 8.8 (53.2) |
| Rmeas (%) c | 16.8 (243.4) | 12.2 (111.2) |
| Rpim (%) d | 5.6 (39.5) | 5.8 (35.2) |
| <I/σ(I)> | 10.37 (0.67) | 9.02 (1.39) |
| Wilson B-factor (Å2) | 61.4 | 42.4 |
| VM (Å3 Da−1) | 2.99 | 2.27 |
| Estimated solvent content (%) | 58.9 | 45.8 |
| Refinement | ||
| Rwork (%) e | 20.7 | 19.1 |
| Rfree (%) e | 23.2 | 21.5 |
| rmsd for bond lengths (Å) | 0.008 | 0.002 |
| rmsd for bond angles (°) | 0.922 | 0.627 |
| Average B-factor (Å2) | 68.3 (chain A), 68.2 (chain B) | 37.8 (chain A), 39.5 (chain B), 43.2 (chain C), 47.6 (chain D) |
| Ramachandran plot | ||
| Residues in favored regions (%) | 97.6 | 97.4 |
| Residues in allowed regions (%) | 2.4 | 2.6 |
| Residues in disallowed regions (%) | 0 | 0 |
| PDB code | 7PKX | 7PL0 |
The mutations present in 5G5 are located nearby the heme proximal side (Figure 2a). S325P is adjacent to the heme proximal histidine ligand, H326, and mutation A330V lines the cavity at ~3.5 Å to the heme. In the wild-type structure, the side chain of K317 is at the surface, located at ~10 Å from the heme propionate, but in 5G5, E317 is part of a region (309–321) that is not visible in the electron density maps (Figure 2b,c). When compared with the wild type, the 5G5 structure displays lower atomic displacement parameter (a.d.p.s) values, 68 Å2 and 38–48 Å2, respectively (Figure 2b,c and Table 4), but apparently the replacement of the positively charged K317 by the carboxylate glutamate resulted in the significantly higher flexibility of region 309–321 (see below).
In BsDyP, the heme cofactor is partially buried in a predominantly hydrophobic pocket lined by the conserved proximal ligand H326 and catalytic distal residues D240 and R339 (Figure 3a,b). It is coordinated by four nitrogen atoms of the porphyrin ring, at a mean distance of 2.0 Å, and by H326 at 2.3 Å. The side chains of D240 and R339 are oriented to the heme iron, with a D240OD1–Fe distance of 5.2–5.5 Å and R339NH2–Fe distance of 4.1–4.6 Å, similar to those measured in the reported BsDyP structure (PDB 6KMN). A water molecule is located approximately at 3.5 Å apart from the iron atom; in the heme pocket of the 6KMN structure, an electron density blob was fitted with molecular oxygen, where the closest oxygen atom to the iron atom is at a 3.4 Å distance [24]. Access to the heme from the surface, as defined using a 1.4-Å rolling probe, is made through a distal tunnel and an open wide cavity (Figure 3a). The tunnel, conserved in all characterized DyPs, is proposed to be the main entrance of H2O2 [25][26][27][28][29] and includes in BsDyP a 6 Å-wide side gallery. The catalytic D240 and R339 residues are part of the tunnel, and three water molecules were found in this pathway in the wild-type structure (and four in the 5G5 structure). The open wide cavity gives access to the solvent-exposed heme propionate p6 group and likely represents an electron transfer route from substrates to the porphyrin radical [29][30]. BsDyP structure PDB 6KMM unveiled the presence of two HEPES molecules: one is bound at 6 Å away from the propionate group in the heme cavity and another at 16 Å near the surface-exposed Y388 residue [24]. Moreover, the docking of HEPES and three different synthetic dyes using snapshots from molecular dynamics simulations of BsDyP also indicated two putative binding sites for reduced substrates [24]. BsDyP, similarly to other DyPs, is rich in tyrosines and tryptophans that can play a role in DyP catalysis by forming surface-exposed oxidation sites for bulky substrates, which are connected to the heme by long-range electron transfer (LRET) pathways [26][29][30][31][32][33][34][35][36][37]. Residue Y388 is the most exposed residue at the shortest distance to the heme and can hypothetically act as a second (radical) substrate-binding and -oxidation site in BsDyP.
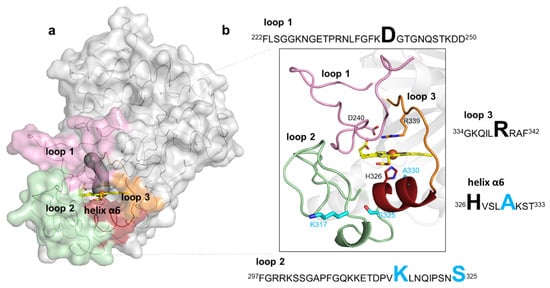
Figure 3. Representation of loops and the small helix delimiting the heme pocket in BsDyP. (a) The BsDyP monomeric form is shown as solvent-accessible surface colored in gray. The loops that surround the heme pocket are shown in pink (loop 1), green (loop 2) and orange (loop 3) and a small helix α6 is represented in dark red. The access to the heme, as defined using a 1.4-Å rolling probe, is made through a distal tunnel (in dark gray) and one cavity; (b) Zoomed view of the cartoon representation as shown in (a). The catalytic residues, D240 and R339, are shown as sticks with carbon atoms colored in pink and orange, respectively. The heme proximal ligand H326 is shown as sticks with carbon atoms colored in red. The residues K317, S325 and A330 that were replaced in the evolved variant 5G5 are shown as sticks with carbon atoms colored in cyan. The nitrogen and oxygen atoms are shown as sticks colored in blue and red, respectively. Highlighted in the text boxes are the catalytic residues (D240 and R339) and the heme proximal ligand (H326) in black, and the residues (K317, S325 and A330) that were replaced during the course of evolution in light blue.
The mutations in the course of evolution of BsDyP were inserted in flexible loops and in a small helix close to the heme pocket (Figure 3a,b). Flexible loops in the proximity of active sites frequently play roles in catalysis as they interact with solvent and substrates [38][39]; it is widely accepted that their pronounced conformational flexibility may contribute to their function, in substrate selectivity and recognition, and the facilitation of substrate binding, as well as protein evolution, as they represent molecular elements of variability [40]. Loop 1 (222–250) and loop 3 (334–342) are predominantly located at the distal side of the heme and comprise the catalytic residues D240 and R339, respectively. Loop 2 (297–325) and the small α-helix (326–333) are at the heme proximal side; loop 2 contains mutations K317E and S325P and the helix α6 comprises the adjacent heme proximal ligand H326 and the mutation A330V. A structural superposition of BsDyP–HEPES bound (PDB 6KMM) with BsDyP PDB 7PKX shows that a hydrogen bond network comprising solvent molecules and amino acids from loop 1, loop 2 and the small α6-helix extended from HEPES to the heme propionate that may facilitate electron transfer to the heme iron. Therefore, the insertion of mutations in these flexible elements during the evolutionary trajectory of 5G5 is expected to have changed the conformational and chemical environment of the catalytic active site, facilitating redox reactions.
K317 (and D314 from loop 2) establishes salt bridges with D250 (and K248) from symmetry-related molecules in the wild-type crystal structure (Figure 4a). E312 from loop 2 interacts through hydrogen bonds with K239, T242, R299 and G304, which are at the cavity entrance (Figure 4a,c). Therefore, the replacement of K317 with a glutamate disrupted the salt bridge to D250, hypothetically leading to loop 2 destabilization and structural rearrangements that resulted in the higher flexibility of region 309–321 (Figure 4b). For example, R299 in 5G5 is at a different conformation and is closer to K331 (~5.0 Å) than in the wild type (~7.0 Å) and is significantly more exposed to the solvent, similarly to residues K239, T242 and G304 that border the entry to the cavity (Figure 4c,d). Furthermore, the higher flexibility of the region 309–321 in 5G5 allowed an increase in the solvent ASA of the residue P325 (Figure 4c,d). The tunnel that gives access to the heme has a similar length (~10 Å) and diameter (~3 Å) in both the wild type and the 5G5 variant, but the open cavity not only has a higher volume and area, but also an entrance width that is 6 Å larger in 5G5 (~18.0 Å) than in the wild type (~12.0 Å). This results in an increased exposure of the heme group to the solvent: 9% ASA in 5G5 as compared to 4% in the wild type (Figure 4c,d). The results indicate that the variation in the loops’ flexibility and the widening of the active site entrance in the evolved variant 5G5 negatively affect substrate binding, as reflected in higher Michaelis constants, Km, but simultaneously facilitate electron transfer from substrates to the heme, as assessed by the increased catalytic rates of 5G5 for DMP (and also for ABTS). The comparative analysis of available DyPs structures reveals that the loops and α-helix regions delimiting the heme pocket are structurally conserved in all DyPs (Figure 5). Members of class V display longer loops, on average with more than 10 to 30 residues, than enzymes from P and I classes. These longer loops most likely contribute to the occluding of the respective heme cavities from contact with the solvent, as observed in the X-ray crystal structures. Nevertheless, in solution, the expected high flexibility of this region may promote solvent access of the heme and allow the oxidation of small substrates at the cavity surface; for example, aromatic molecules were observed to bind within the heme entry of AauDyP [14], and mutagenesis and kinetic analysis in this same enzyme indicated the involvement of a substrate-binding site close to the heme cavity [41].
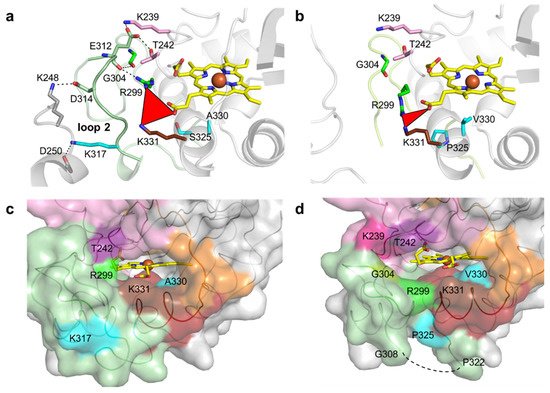
Figure 4. Residues surrounding the heme cavity in BsDyP wild type and 5G5. Stick representation of residues limiting the access to heme cavity and interacting with residues K317, D314 and E312 of loop 2 in wild type (a) and 5G5 variant (b). The carbon atoms of the mutated residues (S325P, A330V and K317E) are colored in cyan. The residues K239 and T242 (loop 1) and R299 and G304 (loop 2) are interacting through hydrogen bonds with E312 (loop 2) and are colored accordingly (Figure 3). The residues of the symmetry-related molecule K248 and D250 are represented with carbon atoms colored in gray. The hydrogen bonds between residues are shown as dashed lines. The red triangle represents the interatomic distances between R299, K331 and the heme propionate group. Solvent-accessible surface area (ASA) representation of the regions delimiting the cavity is colored as in Figure 3 for wild type (c) and 5G5 (d). The residues with higher ASA values (K239, T242, R299, G304 and K331) in 5G5 are highlighted. The mutated residues K317, P325 and A/V330 are colored as in (a) and (b). The missing region 309–321 is shown as a dashed line in (d).
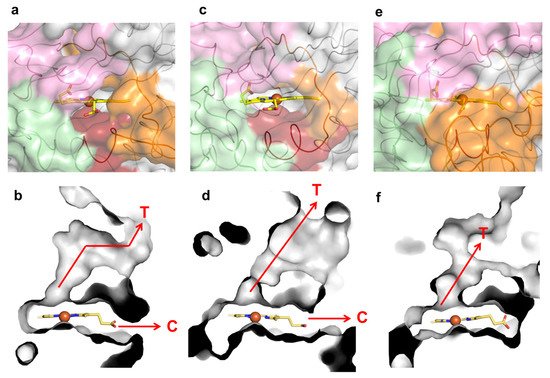
Figure 5. Loops, molecular tunnels and cavities in class P K. pneumoniae DyP (PDB 6FKS) (a,b), class I B. subtilis BsDyP (PDB 7PKX) (c,d) and class V A. auricula-judae DyP (PDB 4AU9) (e,f). The regions lining the cavities are colored in light pink (loop 1), green (loop 2), dark red (small α-helix) and orange (loop 3). The tunnels (T) and cavities (C) corresponding to panels (a,c,e) are represented as grey ASA in panels (b,d,f), respectively.
6. Conclusions
It is possible to tune the dynamics of catalytically relevant loops, which might be essential for the improvement or emergence of new catalytic properties in these enzymes. Researchers opens perspectives to further evolve BsDyP towards the conversion of phenolic compounds in green chemistry and biorefinery fields and to advance fundamental biochemical insights within the DyP-type peroxidase family of enzymes.
References
- Sun, Z.; Fridrich, B.; de Santi, A.; Elangovan, S.; Barta, K. Bright side of lignin depolymerization: Toward new platform chemicals. Chem. Rev. 2018, 118, 614–678.
- Hamalainen, V.; Gronroos, T.; Suonpaa, A.; Hekkila, M.W.; Romein, B.; Ihalainen, P.; Malandra, S.; Birikh, K.R. Enzymatic processes to unlock the lignin value. Front. Bioeng. Biotechnol. 2018, 6, 20.
- Van den Bosch, S.; Koelewijn, S.F.; Renders, T.; Van den Bossche, G.; Vangeel, T.; Schutyser, W.; Sels, B.F. Catalytic strategies towards lignin-derived chemicals. Top. Curr. Chem. 2018, 376, 36.
- Runeberg, P.A.; Brusentsev, Y.; Rendon, S.M.K.; Eklund, P.C. Oxidative transformations of lignans. Molecules 2019, 24, 300.
- Llevot, A.; Grau, E.; Carlotti, S.; Grelier, S.; Cramail, H. From Lignin-derived Aromatic compounds to novel biobased polymers. Macromol. Rapid Commun. 2016, 37, 9–28.
- Natte, K.; Narani, A.; Goyal, V.; Sarki, N.; Jagadeesh, R.V. Synthesis of functional chemicals from lignin-derived monomers by selective organic transformations. Adv. Synth. Catal. 2020, 362, 5143–5169.
- Zakzeski, J.; Bruijnincx, P.C.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599.
- Bugg, T.D.H.; Williamson, J.J.; Rashid, G.M.M. Bacterial enzymes for lignin depolymerisation: New biocatalysts for generation of renewable chemicals from biomass. Curr. Opin. Chem. Biol. 2020, 55, 26–33.
- de Gonzalo, G.; Colpa, D.I.; Habib, M.H.; Fraaije, M.W. Bacterial enzymes involved in lignin degradation. J. Biotechnol. 2016, 236, 110–119.
- Pollegioni, L.; Tonin, F.; Rosini, E. Lignin-degrading enzymes. FEBS J. 2015, 282, 1190–1213.
- Martinez, A.T.; Ruiz-Duenas, F.J.; Martinez, M.J.; del Rio, J.C.; Gutierrez, A. Enzymatic delignification of plant cell wall: From nature to mill. Curr. Opin. Biotechnol. 2009, 20, 348–357.
- Bornscheuer, U.T.; Hauer, B.; Jaeger, K.E.; Schwaneberg, U. Directed Evolution Empowered Redesign of Natural Proteins for the Sustainable Production of Chemicals and Pharmaceuticals. Angew. Chem. Int. Ed. 2019, 58, 36–40.
- Yoshida, T.; Sugano, Y. A structural and functional perspective of DyP-type peroxidase family. Arch. Biochem. Biophys. 2015, 574, 49–55.
- Strittmatter, E.; Serrer, K.; Liers, C.; Ullrich, R.; Hofrichter, M.; Piontek, K.; Schleicher, E.; Plattner, D.A. The toolbox of Auricularia auricula-judae dye-decolorizing peroxidase—Identification of three new potential substrate-interaction sites. Arch. Biochem. Biophys. 2015, 574, 75–85.
- Martinez, A.T.; Ruiz-Duenas, F.J.; Camarero, S.; Serrano, A.; Linde, D.; Lund, H.; Vind, J.; Tovborg, M.; Herold-Majumdar, O.M.; Hofrichter, M.; et al. Oxidoreductases on their way to industrial biotransformations. Biotechnol. Adv. 2017, 35, 815–831.
- Jongbloed, J.D.; Grieger, U.; Antelmann, H.; Hecker, M.; Nijland, R.; Bron, S.; van Dijl, J.M. Two minimal Tat translocases in Bacillus. Mol. Microbiol. 2004, 54, 1319–1325.
- van der Ploeg, R.; Mader, U.; Homuth, G.; Schaffer, M.; Denham, E.L.; Monteferrante, C.G.; Miethke, M.; Marahiel, M.A.; Harwood, C.R.; Winter, T.; et al. Environmental salinity determines the specificity and need for Tat-dependent secretion of the YwbN protein in Bacillus subtilis. PLoS ONE 2011, 6, e18140.
- Santos, A.; Mendes, S.; Brissos, V.; Martins, L.O. New dye-decolorizing peroxidases from Bacillus subtilis and Pseudomonas putida MET94: Towards biotechnological applications. Appl. Microbiol. Biotechnol. 2014, 98, 2053–2065.
- van Bloois, E.; Torres Pazmino, D.E.; Winter, R.T.; Fraaije, M.W. A robust and extracellular heme-containing peroxidase from Thermobifida fusca as prototype of a bacterial peroxidase superfamily. Appl. Microbiol. Biotechnol. 2010, 86, 1419–1430.
- Sturm, A.; Schierhorn, A.; Lindenstrauss, U.; Lilie, H.; Bruser, T. YcdB from Escherichia coli reveals a novel class of Tat-dependently translocated hemoproteins. J. Biol. Chem. 2006, 281, 13972–13978.
- Brissos, V.; Tavares, D.; Sousa, A.C.; Robalo, M.P.; Martins, L.O. Engineering a bacterial dyp-type peroxidase for enhanced oxidation of lignin-related phenolics at alkaline pH. ACS Catal. 2017, 7, 3454–3465.
- Sugawara, K.; Nishihashi, Y.; Narioka, T.; Yoshida, T.; Morita, M.; Sugano, Y. Characterization of a novel DyP-type peroxidase from Streptomyces avermitilis. J. Biosci. Bioeng. 2017, 123, 425–430.
- Liers, C.; Bobeth, C.; Pecyna, M.; Ullrich, R.; Hofrichter, M. DyP-like peroxidases of the jelly fungus Auricularia auricula-judae oxidize nonphenolic lignin model compounds and high-redox potential dyes. Appl. Microbiol. Biotechnol. 2010, 85, 1869–1879.
- Dhankhar, P.; Dalal, V.; Mahto, J.K.; Gurjar, B.R.; Tomar, S.; Sharma, A.K.; Kumar, P. Characterization of dye-decolorizing peroxidase from Bacillus subtilis. Arch. Biochem. Biophys. 2020, 693, 108590.
- Roberts, J.N.; Singh, R.; Grigg, J.C.; Murphy, M.E.; Bugg, T.D.; Eltis, L.D. Characterization of dye-decolorizing peroxidases from Rhodococcus jostii RHA1. Biochemistry 2011, 50, 5108–5119.
- Shrestha, R.; Huang, G.C.; Meekins, D.A.; Geisbrecht, B.V.; Li, P. Mechanistic insights into dye-decolorizing peroxidase revealed by solvent isotope and viscosity effects. Acs. Catal. 2017, 7, 6352–6364.
- Uchida, T.; Sasaki, M.; Tanaka, Y.; Ishimorit, K. A dye-decolorizing peroxidase from Vibrio cholerae. Biochemistry 2015, 54, 6610–6621.
- Yoshida, T.; Tsuge, H.; Konno, H.; Hisabori, T.; Sugano, Y. The catalytic mechanism of dye-decolorizing peroxidase DyP may require the swinging movement of an aspartic acid residue. FEBS J. 2011, 278, 2387–2394.
- Strittmatter, E.; Liers, C.; Ullrich, R.; Wachter, S.; Hofrichter, M.; Plattner, D.A.; Piontek, K. First crystal structure of a fungal high-redox potential dye-decolorizing peroxidase: Substrate interaction sites and long-range electron transfer. J. Biol. Chem. 2013, 288, 4095–4102.
- Yoshida, T.; Tsuge, H.; Hisabori, T.; Sugano, Y. Crystal structures of dye-decolorizing peroxidase with ascorbic acid and 2,6-dimethoxyphenol. FEBS Lett. 2012, 586, 4351–4356.
- Linde, D.; Ruiz-Duenas, F.J.; Fernandez-Fueyo, E.; Guallar, V.; Hammel, K.E.; Pogni, R.; Martinez, A.T. Basidiomycete DyPs: Genomic diversity, structural-functional aspects, reaction mechanism and environmental significance. Arch. Biochem. Biophys. 2015, 574, 66–74.
- Liers, C.; Aranda, E.; Strittmatter, E.; Piontek, K.; Plattner, D.A.; Zorn, H.; Ullrich, R.; Hofrichter, M. Phenol oxidation by DyP-type peroxidases in comparison to fungal and plant peroxidases. J. Mol. Cat B Enz. 2014, 103, 41–46.
- Strittmatter, E.; Wachter, S.; Liers, C.; Ullrich, R.; Hofrichter, M.; Plattner, D.A.; Piontek, K. Radical formation on a conserved tyrosine residue is crucial for DyP activity. Arch. Biochem. Biophys. 2013, 537, 161–167.
- Fernandez-Fueyo, E.; Linde, D.; Almendral, D.; Lopez-Lucendo, M.F.; Ruiz-Duenas, F.J.; Martinez, A.T. Description of the first fungal dye-decolorizing peroxidase oxidizing manganese(II). Appl. Microbiol. Biotechnol. 2015, 99, 8927–8942.
- Baratto, M.C.; Sinicropi, A.; Linde, D.; Saez-Jimenez, V.; Sorace, L.; Ruiz-Duenas, F.J.; Martinez, A.T.; Basosi, R.; Pogni, R. Redox-active sites in Auricularia auricula-judae dye-decolorizing peroxidase and several directed variants: A multifrequency EPR study. J. Phys. Chem. 2015, 119, 13583–13592.
- Linde, D.; Pogni, R.; Canellas, M.; Lucas, F.; Guallar, V.; Baratto, M.C.; Sinicropi, A.; Saez-Jimenez, V.; Coscolin, C.; Romero, A.; et al. Catalytic surface radical in dye-decolorizing peroxidase: A computational, spectroscopic and site-directed mutagenesis study. Biochem. J. 2015, 466, 253–262.
- Nys, K.; Furtmuller, P.G.; Obinger, C.; Van Doorslaer, S.; Pfanzagl, V. On the track of long-range electron transfer in b-type dye-decolorizing peroxidases: Identification of a tyrosyl radical by computational prediction and electron paramagnetic resonance spectroscopy. Biochemistry 2021, 60, 1226–1241.
- Nestl, B.M.; Hauer, B. Engineering of flexible loops in enzymes. ACS Catal. 2014, 4, 3201–3211.
- Heinemann, P.M.; Armbruster, D.; Hauer, B. Active-site loop variations adjust activity and selectivity of the cumene dioxygenase. Nat. Commun. 2021, 12, 1095.
- Pabis, A.; Risso, V.A.; Sanchez-Ruiz, J.M.; Kamerlin, S.C.L. Cooperativity and flexibility in enzyme evolution. Curr. Opin. Strut. Biol. 2018, 48, 83–92.
- Diederichs, K.; Karplus, P.A. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 1997, 4, 269–275.
More
Information
Subjects:
Biotechnology & Applied Microbiology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
717
Revisions:
2 times
(View History)
Update Date:
27 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No