Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Christine Blattner | -- | 8008 | 2023-02-24 09:48:53 | | | |
| 2 | Catherine Yang | Meta information modification | 8008 | 2023-02-27 03:14:03 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Pan, M.; Blattner, C. E3s That Target Wild-Type p53. Encyclopedia. Available online: https://encyclopedia.pub/entry/41623 (accessed on 26 July 2026).
Pan M, Blattner C. E3s That Target Wild-Type p53. Encyclopedia. Available at: https://encyclopedia.pub/entry/41623. Accessed July 26, 2026.
Pan, Mengwu, Christine Blattner. "E3s That Target Wild-Type p53" Encyclopedia, https://encyclopedia.pub/entry/41623 (accessed July 26, 2026).
Pan, M., & Blattner, C. (2023, February 24). E3s That Target Wild-Type p53. In Encyclopedia. https://encyclopedia.pub/entry/41623
Pan, Mengwu and Christine Blattner. "E3s That Target Wild-Type p53." Encyclopedia. Web. 24 February, 2023.
Copy Citation
p53 plays a role in different biological processes such as proliferation, invasion, pluripotency, metabolism, cell cycle control, ROS (reactive oxygen species) production, apoptosis, inflammation and autophagy. In the nucleus, p53 functions as a bona-fide transcription factor which activates and represses transcription of a number of target genes. In the cytoplasm, p53 can interact with proteins of the apoptotic machinery and by this also induces cell death. Despite being so important for the fate of the cell, expression levels of p53 are kept low in unstressed cells and the protein is largely inactive.
p53
E3s
tumor
1. E3s That Ubiquitinate p53 and Target. It for Degradation
Most E3s that have been reported to modify p53 target p53 for proteasomal degradation (Table 1, Figure 1). The largest group of them are E3s with a RING domain.
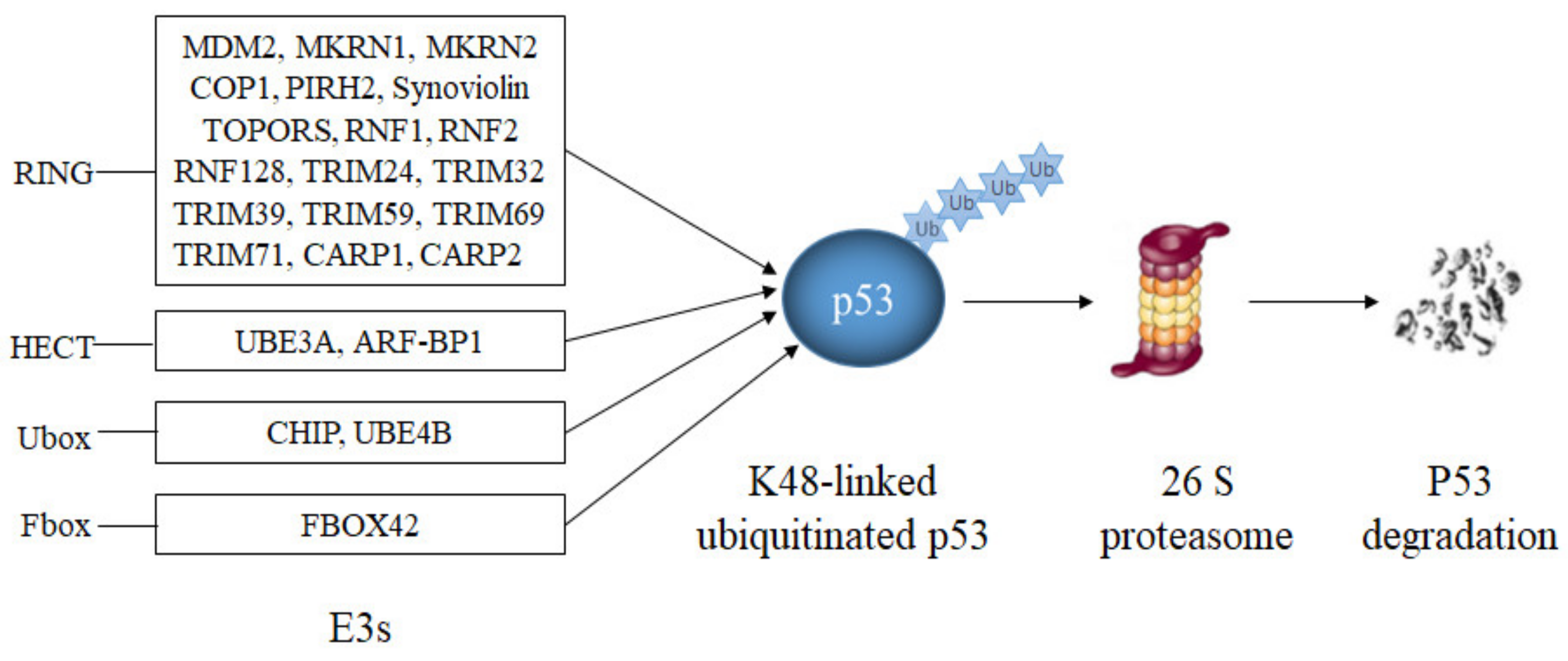
Figure 1. E3s that target p53 for degradation. RING-domain E3s (MDM2, Synoviolin, MKRN1, MKRN2, COP1, PIRH2, TOPORS, CARP1, CARP2, RNF1, RNF2, RNF128, TRIM24, TRIM32, TRIM39, TRIM59 and TRIM71), a HECT-domain E3 (ARF-BP1), two U-Box-domain E3s (CHIP, UBE4B) and a SCF-complex with the F-Box-domain protein FBXO42 modify p53 with lysine K48-linked polyubiquitin chains resulting in p53 degradation.
Table 1. E3s that target p53 for proteasomal degradation.
| E3 | Alias | Type | References |
|---|---|---|---|
| MDM2 | Hdm2 | RING | [1][2][3][4][5] |
| MKRN1 | RNF61 | RING | [6] |
| MKRN2 | RNF62 | RING | [7] |
| COP1 | RNF200 | RING | [8] |
| PIRH2 | ZN363 | RING | [9][10][11] |
| Synoviolin | HRD1 | RING | [12][13] |
| TOPORS | P53BP3 | RING | [14][15][16] |
| RNF1 | RING1 | RING | [17] |
| RNF2 | RING1B | RING | [18][19] |
| RNF128 | Grail | RING | [20] |
| TRIM24 | TIF1A | RING | [21] |
| TRIM32 | HT2A | RING | [22][23] |
| TRIM39 | RNF23 | RING | [24] |
| TRIM59 | RNF104 | RING | [25] |
| TRIM 69 | RNF36 | RING | [26][27] |
| TRIM71 | Lin41 | RING | [28] |
| CARP1/2 | RNF34/RNF189 | RING | [29] |
| UBE3A | E6-AP | HECT | [30][31][32][33] |
| ARF-BP1 | HUWE1 | HECT | [34][35] |
| CHIP | UBOX1 | U-box | [36][37][38] |
| UBE4B | UBOX3 | U-box | [39][40] |
1.1. RING-Domain E3s
RING-domain E3s are characterized by the presence of a RING (Really Interesting New Gene) domain. The amino acid sequence of the RING normally follows the pattern C-X2-C-X(9-39)-C-X(1-3)-H-X(2-3)-C-X2-C-X(4-48)-C-X2-C, also abbreviated as C3HC4 or RING-HC. Two zinc atoms coordinate the conserved seven cysteine residues and the histidine residue and stabilize the structure. Variations of the RING pattern, where a cysteine residue is replaced by a histidine residue as in the RING-H2 variant or where cysteine residues and histidine residues are swapped or replaced by another amino acid that can coordinate zinc, have also been observed [41]. RING-domains are sites of protein-protein interactions, in particular with E2s. Although some E3s act as single proteins, they have a preference to form homo and heterodimers [41].
The most studied RING-type E3 is MDM2. MDM2 binds to the transactivation domain of p53, inhibits its transcriptional activity and mediates p53 ubiquitination and degradation via 26S proteasomes [1][2][3]. Since MDM2 is also a transcriptional target of p53, the two proteins form a negative feedback loop. This feedback loop restrains p53 abundance since as soon as p53 levels rise, MDM2 levels rise as well and foster p53 degradation [4]. MDM2 mediates ubiquitination of p53 with K48-linked polyubiquitin chains at six key lysine residues in the C-terminus (K370, K372, K373, K381, K382 and K386) resulting in p53 degradation [5]. Genetic deletion of MDM2 leads to embryonic death that is rescued by simultaneous deletion of p53, demonstrating that MDM2 is a key regulator of p53 [42]. After DNA damage, p53 is released from MDM2, allowing its accumulation to high levels and transcriptional activation of its target genes [43]. Thirty-seven different isoforms can be generated from the MDM2 gene, although it is unclear whether they are all translated. Most of them lack a p53-binding site. MDM2 isoforms are more frequently expressed in poorly differentiated and late-stage tumors [44][45]. Apart from full-length MDM2, only the smallest isoform (MDM2-E) co-precipitates with p53 [44]. Although the isoform MDM2-A does not bind to p53, it causes p53-dependent perinatal lethality and senescence [45]. As MDM2 forms dimers, the full-length protein can heterodimerize with MDM2 isoforms, allowing the isoforms to control p53 abundance and activity in trans. Some heterodimers consisting of full-length MDM2 and one of its isoforms are even more potent in ubiquitinating p53 than homodimers of the full-length protein [46]. Of note, despite the increase in p53 ubiquitination, heterodimers of MDM2-B and full-length MDM2 even increased p53 activity [47][48]. More recently, it was found that MDM2-C also has E3 activity and ubiquitinates wild-type and mutated (R273H) p53 [49]. However, it was not determined whether this ubiquitination of p53 by MDM-C leads to p53 degradation.
Other RING-domain E3s that mediate p53 degradation are MKRN1 and MKRN2. MKRN1 and MKRN2 are members of the MKRN (Makorin Ring) protein family, a highly conserved but not very well-investigated family of RING-domain containing proteins. MKRN1 mediated polyubiquitination of p53 at lysine residues K291 and K292 resulted in p53 degradation and downregulation of MKRN1 induced p53-dependent cell cycle arrest [6]. Interestingly, MKRN1 is also an E3 for p21 and promotes p21 poly-ubiquitination and degradation. Upon DNA damage, the interaction of p53 and MKRN1 is strongly reduced resulting in elevated p53 levels while degradation of p21 is still ongoing. Therefore, under normal growth conditions, MKRN1 keeps cells alive by suppressing p53 and p21. However, when cells experience cellular stress, MKRN1 primarily induces the degradation of p21 leading to increased cell death [6]. MKRN2 also binds and ubiquitinates p53 resulting in downregulation of the tumor suppressor protein. MKRN2 is highly expressed in human malignant melanoma cell lines and downregulation of MKRN2 inhibited melanoma growth in a p53-dependent manner [7].
COP1 (constitutive photomorphogenic protein 1) is another RING-domain E3 that mediates p53 degradation. COP1 was identified by mass spectrometric analysis as a binding partner of p53. COP1 increased p53 turnover by targeting it for ubiquitin-mediated degradation and deletion of COP1 stabilized p53, inhibited p53-dependent transcription and arrested cells in the G1 phase of the cell cycle. Although COP1 was able to target p53 for degradation by itself, depletion of both, COP1 and MDM2, by siRNA showed that these two E3s can cooperate and sensitize U2OS cells to ionizing-radiation-induced cell death. COP1 is, furthermore, a target gene of p53, which connects the two proteins by a negative feed-back loop [8]. Like Mdm2, COP1 also possesses splice variants. A total of eleven transcripts have been generated from the human COP1 pre-mRNA and shown to exist in human cell lines. Together with the full-length protein, the COP1D variant is the most abundantly expressed isoform and the only one that has been investigated in human cells and tissues. COP1 and COP1D are nuclear proteins. Their expression varies between different cell lines and tissues. Both isoforms are induced by UV-light albeit with different ratios. Full length COP1 and COP1D form homo- and heterodimers. While COP1D reduced the ability of the full-length protein to target c-Jun for degradation, both isoforms decreased p53 levels under steady-state conditions and after UV-exposure [50].
A further RING-E3 for p53 is PIRH2 (p53-induced protein with a RING-H2 domain). PIRH2 was identified as a p53 target with a RING-domain by differential display [9]. Further analysis showed that PIRH2 binds to p53 and that overexpression of PIRH2 reduces p53 levels because of PIRH2-mediated polyubiquitination and degradation. Accordingly, transcriptional and growth inhibitory activities of p53 were also reduced after overexpression of PIRH2 [9]. The overall ligase activity of PIRH2 towards p53 was lower than that of MDM2 and PIRH2 modified a different set of lysine residues [10]. Like MDM2, PIRH2 is a transcriptional target of p53 and was induced after ionizing irradiation in murine embryonic fibroblasts, yet not to the same extent as MDM2. Of note, activation of p53 in MCF-7 and A549 cells by DNA damage did not lead to the accumulation of PIRH2, pointing to a cell type-dependent regulation of PIRH2 [9][11]. PIRH2 is highly expressed in several cancer cell lines regardless of the p53 status, suggesting that it also has p53-independent oncogenic activities [11]. PIRH2 also comes in several isoforms. A total of six isoforms have been described so far (PIRH2A to PIRH2F, where PIRH2A is the full-length protein) [51][52][53]. PIRH2B and PIRH2C have been detected in different cell lines and PIRH2E and PIRH2F in HepG2 cells [51][53]. PIRH2B and PIRH2C show the same diffuse staining in the nucleus and cytoplasm as the full-length protein. While PIRH2B is able to dimerize with PIRH2A, this is not the case for PIRH2C, yet all three proteins co-precipitated with MDM2 [53]. PIRH2B associates with p53 to a similar extent as the full-length protein while PIRH2C shows only weak interactions with p53. Nevertheless, both isoforms were able to ubiquitinate p53, to target it for degradation and to reduce its transcriptional activity. However, because their RING domains are disrupted, neither PIRH2B nor PIRH2C were able to ubiquitinate p53 directly and required other proteins, eventually PIRH2A or MDM2 [53]. PIRH2D lacks the C-terminal RING domain and thus most likely also E3 activity [52].
Synoviolin is a RING-type E3 that is located in the endoplasmatic reticulum (ER). Synoviolin was identified as an E3 for p53 in cells where synoviolin had been genetically deleted and where p53 accumulated. Synoviolin binds directly to p53, sequesters p53 in the ER and mediates ER-associated polyubiquitination and degradation of p53. Knocking-down synoviolin resulted in p53 accumulation and induction of the GADD45 (growth arrest and DNA damage-inducible protein), MDM2 and p21 genes [12]. The activity of synoviolin is regulated by ER stress. In unstressed cells, synoviolin keeps the steady state levels of p53 low by ubiquitinating it for 26S proteasome-dependent degradation. After mild ER-stress, synoviolin is transcriptionally upregulated and the increased amount of synoviolin is still able to target p53 for degradation. However, under severe or prolonged ER-stress, the overwhelming amount of unfolded proteins may squelch and release p53 from synoviolin and allow its accumulation and activation [13].
Another RING-type E3 that mediates p53 degradation is TOPORS (TOP1 Binding Arginine/Serine Rich Protein, E3). TOPORS was initially found as a p53-binding protein by a yeast two hybrid system with p53 as a bait [14]. Overexpression of TOPORS resulted in a proteasome-dependent decrease in p53 protein expression in human osteosarcoma cells. TOPORS can cooperate with the E2 conjugating enzyme, UbcH5a, Ubch5c and UbcH6 for mediating p53 ubiquitination [15]. Ubiquitination and degradation of p53 by TOPORS is regulated by phosphorylation of TOPORS at serine S718 by the Polo-like kinase 1 (PLK1). Plk1-mediated phosphorylation of TOPORS enhances p53 ubiquitination and degradation. As PLK1 expression is highest in late G2 and S-phase of the cell cycle, this is probably also the time when TOPORS-mediated regulation of p53 occurs [16]. TOPORS is induced after DNA damage, however, while several E3s are target genes of p53, the DNA damage-induced induction of TOPORS is independent of p53 [54].
Two more recently discovered E3s for p53 are RNF1 and RNF2, also known as RING1 and RING1B [17][18]. RNF1 is a crucial component of the transcriptional repression complex PRC1 and RNF2, is part of the transcriptional repression complex PRC2 of the polycomb group. RNF1 directly interacts with p53 and mediates its ubiquitination and degradation by 26S proteasomes. Depletion of RNF1 resulted in p53 stabilization and p53-dependent cell cycle arrest, apoptosis and senescence of HepG2 and HCT116 cells. This effect was compromised in p53-deficient cells showing that the induction of apoptosis and senescence occured via p53 and not through other activities of RNF1 [17]. RNF1 is even capable of downregulating p53 during the DNA damage response [17]. RNF2 binds to p53 and mediates p53 ubiquitination and degradation, however, only in selected cells and cell lines including cells from germ cell tumors or ovarian cancers [18][19]. Also, RNF2 requires the Bmi1 protein of the PRC2 complex for full activity [19]. Furthermore, although RNF2 can bind directly to p53, it also binds to MDM2 and forms a ternary complex with the two proteins. Overexpression of RNF2 was not sufficient to mediate p53 ubiquitination in vitro or in HEK293T cells, suggesting that RNF2 may require MDM2, Bmi1 or a further protein to promote p53 ubiquitination [18]. Downregulation of RNF2 primarily leads to the induction of cell cycle arrest-linked target genes of p53 and of genes that contribute to cellular metabolism but not to the induction of the pro-apoptotic target genes Bax, Noxa and Puma [55]. Starvation of cells, which downregulates RNF2 expression, leads to enhanced p53-dependent apoptosis and to liver atrophy in mice [19]. Although RNF1 can form complexes with RNF2, the association of RNF1 with p53 and its ubiquitination and degradation are independent of an interaction with RNF2 [19].
Other RNF proteins that target p53 are RNF38 and RNF128. RNF38 is widely expressed in a variety of human tissues and evolutionary conserved [56]. Its interaction with p53 was first identified in a genome-wide in vitro expression pull-down screen using Drosophila p53 as a bait [57]. RNF38 is a nuclear protein and predicted to have three isoforms. At least the full-length protein interacts with the E2 enzyme UbcH5b and shows E3 activity. The interaction of RNF38 with p53 was confirmed using bacterially expressed proteins, showing that the interaction is direct. RNF38-mediated ubiquitination of p53 was observed in vitro and after overexpression in cells. Overexpression of RNF38 forced p53 into punctate structures in the nucleus, some of them colocalized with PML bodies [58]. Unfortunately, no data are available on p53 stability upon overexpression of RNF38. Thus, it is also possible that RNF38-mediated ubiquitination of p53 leads to its activation which would be consistent with the deletion of the chromosomal area, where the RNF38 gene is located, in several malignancies [58]. RNF128, also known as Grail, is a transmembrane protein that is localized in endosomes and best-known as a regulator of energy and cytokine production [59]. RNF128 usually exists as a tri-molecular complex comprising Grail, Otub1, and USP8 and possesses E3 activity [20]. The interaction of RNF128 with p53 was identified through a yeast two-hybrid SOS recruitment system for identifying p53-interacting proteins. RNF128 interacts with the N-terminal domain of p53 under normal growth conditions as well as after DNA damage. [20]. Overexpression of RNF128 reduced p53 levels and stability, and expression of its downstream target genes p21 and Bax, and p53-dependent apoptosis. RNF128 is a p53 target gene and as it is able to target p53 for degradation after treatment with actinomycin D or exposure to UV-light, it may contribute to the downregulation of p53 after DNA damage, particularly as RNF128 is induced by p53 under these conditions [20]. As RNF128 is highly abundant in heart and liver, it may regulate p53 expression especially in these tissues. Although RNF128 downregulates p53, its expression was significantly reduced in advanced urinary cancer and its low expression was correlated with high tumor stage, high histological grade, high proliferation and vascular and perineural invasion [60].
Some other RING-type E3s that mediate p53 ubiquitination and degradation belong to the TRIM-family of proteins, a protein family with more than 80 members that are characterized by an N-terminal RING-domain, one or two B-Boxes and a coiled-coiled region. Due to the presence of a RING-domain, most TRIM proteins possess E3 activity. TRIM24, TRIM32, TRIM39, TRIM59, TRIM69 and TRIM71 all mediate ubiquitination and degradation of p53 [21][23][24][27][28]. TRIM24 was first identified as a co-repressor of the retinoic acid receptor alpha [61]. Later, by a tandem-affinity-purification approach with subsequent mass spectrometry analysis, it was found to associate with p53 [21]. TRIM24 ubiquitinates p53 and promotes its degradation in 26S proteasomes. Downregulation of TRIM24 in human tumor-derived cancer cells resulted in p53-dependent apoptosis [21]. In contrast to MDM2−/− embryos, which die a few days after fertilization, TRIM24−/− mice are viable [62][63]. However, mutation of the TRIM24 homolog Bonus led in Drosophila to apoptosis that was rescued by p53 deletion [21]. Like many E3s that target p53, TRIM24 is also a target gene of p53, connecting the two proteins by a negative feedback-loop [22]. DNA damage leads to phosphorylation of TRIM24 at serine S768 by the ATM kinase, resulting in the release of p53 from TRIM24 and degradation of TRIM24 [22]. Due to the high levels of p53 after DNA damage, TRIM24 is continuously synthesized and as it can even bind to phosphorylated p53 and target it for degradation, TRIM24 contributes to the return to normal p53 levels once DNA lesions have been repaired [22]. TRIM32 is best known for its activity during neuronal and skeletal muscle cell differentiation [64]. However, the ligase is also frequently overexpressed in head and neck cancers and in skin carcinoma [65]. In line with promoting p53 degradation, TRIM32 has been shown to reduce the transcriptional activation of p53 target genes, both under non-stressed conditions and after DNA damage, and to reduce p53-mediated cell cycle arrest and apoptosis while downregulation of TRIM32 increased expression of p53-target genes in a p53-dependent manner [66]. As TRIM32 is, like MDM2 or TRIM24, a target gene of p53, both proteins are connected by a negative feedback loop [23]. Also, TRIM39, better known as a regulator of the anaphase promoting complex [67], can directly bind to p53, ubiquitinate the tumor suppressor protein and target it for degradation. Most interestingly, cells harboring wild-type p53 cannot traverse the G1/S checkpoint when TRIM39 is absent from the cell and this cell cycle arrest depends strongly on p53 [24]. Although TRIM39-mediated ubiquitination of p53 is independent of MDM2, TRIM39 synergizes with MDM2 to promote cell cycle arrest and apoptosis via promoting p53 degradation. This is possible as both proteins bind to different domains of p53 [24]. TRIM59, attracted attention when it was found to be highly overexpressed in gastric cancer. Its increased levels were, furthermore, associated with more advanced tumor stages and shorter survival times [25]. Knocking-down TRIM59 reduced proliferation and clonogenicity of gastric cancer cells and the growth of xenografts in nude mice while increased levels of TRIM59 are correlated with decreased expression of p53 target genes, probably resulting from the interaction of TRIM59 with p53 and its ubiquitination and degradation [25]. TRIM69 is a protein that is present in the cytoplasm and nucleus where it forms speckled aggregates in a RING-dependent manner [68]. It is highly expressed throughout zebrafish embryogenesis and in most adult tissues with highest expression in testis, brain, heart and ovary [26]. Knocking-down TRIM69 by morpholino-antisense-oligonucleotides induced massive cell death that was rescued by knocking-down p53 [26]. Further analysis showed that overexpression of TRIM69 reduced UVB-induced apoptosis while downregulation of TRIM69 induced it [27]. TRIM69 interacted with p53 and induced its ubiquitination [27]. TRIM69 is downregulated in cataract lenses whereas p53 levels are higher under these conditions, suggesting that the regulation of p53 by TRIM69 might play a role in this process. [27]. TRIM71 is an E3 that is especially linked to stem cell pluripotency, reprogramming and neurogenesis. It is indispensable for neural tube closure and embryonic development. TRIM71 interacts directly with p53, controls its abundance by ubiquitinating lysine residues K313-K315 within the nuclear localization signal and antagonizes p53-dependent pro-apoptotic and pro-differentiation responses [28]. TRIM71−/− cells showed upregulation of Grhl3 and Caspase-3 activation, two downstream effectors of p53 [28]. TRIM71 is particularly active after the onset of differentiation of embryonic stem cells. Overexpression of TRIM71 decreased p53 protein levels in this setting and increased cell proliferation. Most importantly, induction of neural differentiation is associated with p53-dependent accumulation of cleaved Caspase-3 and this is strongly reduced upon overexpression of TRIM71. Loss of TRIM71 resulted in aberrant p53 activation during neural tube closure leading to massive cell death and an unclosed neural tube [28].
Another protein family where members show E3 activity towards p53 are the CARPs (Caspase8/10-associated RING proteins) [29]. CARPs (CARP1 and 2) usually target Caspase 8 and 10 for degradation and are cleaved by the activated Caspases 8 and 10 once the extrinsic cell death pathway is initiated [29]. CARPs are overexpressed in tumors and their downregulation reduced cell viability. CARP1 and 2 physically interacted with p53, ubiquitinated it, promoted its degradation and reduced the expression of p53 target genes and the induction of cell cycle arrest. CARPs belong to the few E3s that also target phosphorylated p53 for degradation and thus contribute to the return to basal p53 levels once DNA lesions are repaired [69]. In addition to the direct role for p53 degradation, CARPs bind to MDM2 and stabilize MDM2 by inhibiting its self-ubiquitination. The resulting elevated levels of MDM2 also contribute to the strong decrease in p53 levels upon overexpression of CARPS [70].
1.2. HECT-Domain and U-Box-Domain E3s
Apart from RING-domain containing E3s, some U-Box and HECT-domain containing E3s target p53 for degradation. Among these E3s are the HECT-domain containing proteins UBE3A and ARF-BP1 and the U-box E3s CHIP and UBE4B.
The HECT domain of E3s roughly consists of 350 amino acids that form a larger N-terminal and a smaller C-terminal lobe that are connected by a short linker [71]. The N-terminal lobe is the landing site for the E2 while the C-terminal lobe contains the active-site cysteine that forms a thioester bond with ubiquitin resulting in an E3-ubiquitin intermediate before the ubiquitin is transferred to its substrate [72].
UBE3A was the first HECT-type E3 that was found to be involved in p53 regulation [30][31]. Over-expression of UBE3A in neuro 2a cells increased ubiquitination and degradation of p53 that could be prevented by deletion of the HECT-domain of the ligase. Partial knockdown of UBE3A increased p53 levels and p53-dependent transcription, and also promoted neuronal cell death [32]. In vitro ubiquitination assays showed that UBE3A directly interacts with and ubiquitinates p53 [33]. UBE3A comes in several isoforms. Five mRNA subtypes have been isolated that are all expressed in cells, yet to varying degree, giving rise to three isoforms. Isoform I corresponds to the previously reported open reading frame for E6-AP while isoforms II and III have additional 20 and 23 amino acids, respectively, at their N-termini and utilize different initiation codons [73]. No function has as yet been assigned to the different isoforms.
ARF-BP1 is with approximately 500 kD one of the largest proteins in the cell. It was identified as a protein that co-purified with overexpressed p14/p19ARF protein, the alternative transcript of the Ink4a/ARF tumor suppressor locus [34]. p14/p19ARF exhibits tumor suppressive activities by stabilizing p53 in response to oncogenic stimuli. Oncogenes such as Myc or Ras induce the p14/p19ARF protein resulting in nucleolar sequestration and degradation of MDM2 with the consequence of p53 stabilization and activation [74]. In wild-type cells, ARF-BP1 binds directly to p53 and ubiquitinates p53, an activity that is inhibited by the p14/p19ARF protein [34]. Inactivation of ARF-BP1 is essential for p14/p19ARF—mediated stabilization of p53 e.g., in response to oncogenic stimuli [34]. Knock-down of ARF-BP1 also elevates the levels of endogenous p53, increases expression of its target genes and induces cell death. ARF-BP1-mediated cell death is p53-dependent and not seen in p53-negative HCT116 cells. Inactivation of ARF-BP1 during embryonic development resulted in p53-activation and embryonic lethality, however, mice where ARF-BP1 was only deleted in pancreatic β-cells were viable and displayed no obvious abnormality after birth, indicating that the regulation of p53 by ARF-BP1 is particularly powerful in a subset of cells and tissues [35].
The U-box is a domain of about 70 amino acids that is present in proteins from yeast to man. The predicted structure of the U-box is similar to that of the RING domain, yet U-boxes lack the metal coordinating histidine and cysteine residues that are a hallmark of the RING domain [75]. Like RING-domain E3s, U-box E3s lack a catalytic cysteine and rather act as a scaffold to orient the E2 and the target to allow efficient ubiquitination of the target protein. In mammals, there are six U-box proteins that mediate ubiquitination of target genes in conjunction with E1s and E2s [75]. Two of them, CHIP and UBE4B are able to mediate ubiquitination and degradation of p53.
CHIP (carboxyl terminus of HSP70-interacting protein) is a protein that is highly expressed in adult striated muscle and in most cells in culture. It associates with the chaperones Hsc70 and Hsp70 and inhibits their activity [76]. Most interestingly, CHIP not only mediates poly-ubiquitination and degradation of wild-type p53 but also of mutated p53 [36]. Ubiquitination of wild-type and mutated p53 by CHIP was further enhanced by the presence of Hsc70 [37]. CHIP seems not only to degrade proteins that associate with chaperones, it also appears to be particularly required for the degradation of proteins during senescence, a permanent cell cycle arrest [77][78]. The p53 protein is also highly linked to senescence as activation of p53 can lead to cell cycle arrest, apoptosis and senescence [79]. Interestingly, p53 levels are high under pre-senescent conditions and decrease when cells reach senescence while the protein levels of CHIP show the opposite characteristic and are high in “late passage cells” [38]. This behavior is clearly distinct to many other E3s for p53 including MDM2, COP2, PIRH2 or TOPORS that are either reduced with increasing age or not altered. CHIP is not only induced with aging, it also changes from an entirely cytoplasmic localization in young cells to a partly cytoplasmic/partly nuclear localization in senescent cells [38]. These observations strongly point to a specific role of CHIP-mediated degradation of p53 in old and senescent cells, a hypothesis that is further supported by the observation that downregulation of e.g., MDM2 leads to high accumulation of p53 both in early and late passage cells while downregulation of CHIP stabilizes p53 only in late passage cells [38]
UBE4B, a mammalian homolog of the UFD2 protein of S. cerevisiae, is not only an E3 but also an E4, a protein that is in some cases required to ensure the proper formation of the ubiquitin chain [80]. UBE4B is especially expressed in neuronal tissue and its genetic deletion leads to early embryonic death due to extensive apoptosis. Heterozygote mice survive but develop a neurological disorder [81]. p53 can be ubiquitinated and degraded by UBE4B alone. However, UBE4B also interacts with MDM2 and the two proteins can form a ternary complex with p53 which boosts p53 polyubiquitination and degradation tremendously in comparison to ubiquitination and degradation of the tumor suppressor protein in the presence of the single E3 [82]. Interestingly, while p53 was observed to be primarily monoubiquitinated or multi-monoubiquitinated by MDM2 in the absence of UBE4B, this was shifted to polyubiquitination in the presence of MDM2 and UBE4B. The increased polyubiquitination of p53 under these conditions was accompanied by a decrease in p53 levels and a decrease in transcription of the p53 target genes p21 and MDM2 [82]. Similar to MDM2-mediated degradation of p53, UBE4B also enhanced p53 degradation mediated by PIRH2 and COP1 [39]. Surprisingly, a mutated form of UBE4B that was unable to mediate p53 degradation was still able to reduce p53-dependent transactivation, suggesting that UBE4B may use several ways to control p53 activity [82]. UBE4B is a target gene of p53 and this negative feed-back loop ensures low p53 levels [40]. As UBE4B also binds to p53 phosphorylated at serine 15 and promotes its degradation, it is most likely also involved in the shut-off of the DNA damage response [40].
1.3. E3s That Require Complex Formation
While most enzymes modify p53 as single enzymes or dimers, some E3s are higher order complexes. The best characterized mammalian multi-subunit E3 is the SCF complex, a complex composed of 4 subunits, Skp1, Cul1/Cdc53, Roc1/Rbx1/Hrt1 and an F-box protein. The F-box protein is responsible for substrate recognition and binding while the entire complex provides the E3 activity [83]. One of these F-box proteins is FBXO42, also known as JFK (Jinfukang). FBXO42 is expressed in most human tissues with highest expression in heart and skeletal muscle [84]. FBXO42 is part of an SCF complex and it requires this association to destabilize p53 [84]. The FBXO42-containing SCF-complex inhibits p53-dependent transcription, and FBXO42 depletion stabilizes p53, promotes apoptosis, induces cell cycle arrest and sensitizes cells to ionizing radiation-induced cell death [84]. Like most other F-box proteins, FBXO42 requires phosphorylation of its substrate for full recognition [85]. Therefore, p53 is only recognized as a substrate of FBXO42 upon prior phosphorylation in its central domain, a modification that is implemented by CSN5, a COP9 signalosome-associated kinase. Inhibition or knockdown of COP9 or CSN5 impairs FBXO42-promoted p53 degradation resulting in enhanced p53-dependent transcription, growth suppression, cell cycle arrest and apoptosis [85]. Like most other E3s for p53, FBXO42 is transcriptionally activated by p53 and thus forms an auto-regulatory negative feedback loop with p53 [85].
2. E3s That Mediate p53 Ubiquitination without Promoting Degradation
In addition to proteasome-mediated degradation, ubiquitination of p53 can also have other consequences. For instance, it has been described that ubiquitination can promote nuclear export, stabilize the p53 protein or affect its transcriptional activity even without modulating its abundance (Table 2, Figure 2). Occasionally, the same E3 can even do both, promote p53 ubiquitination and its degradation and promote ubiquitination connected to other consequences. These sometimes even opposing activities may depend on the level of the E3, on different ubiquitin-linkages, on post-translational modifications of the E3, on the presence of other enzymes and on co-factors or on the cellular context in general.
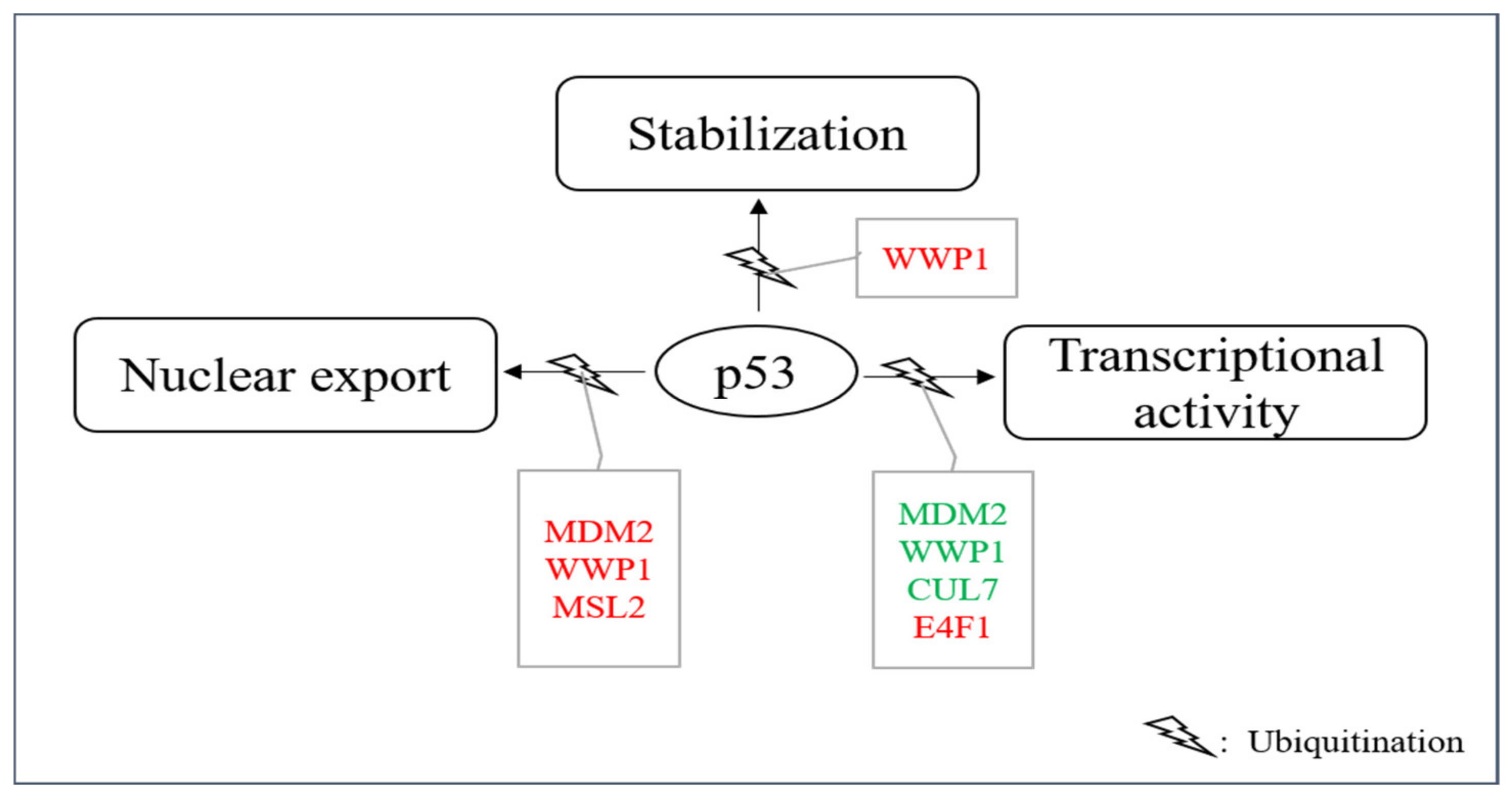
Figure 2. E3s that modify p53 without targeting it for degradation. A group of E3s including MDM2, MSL2, WWP1, CUL7, UBC13 and E4F1 decorate p53 with ubiquitin leading to nuclear export, enhanced stability and/or enhanced or reduced transcriptional stability. Ligases colored in red increase the individual activity of p53 and ligases shown in green reduce it.
Table 2. E3s that mediate p53 ubiquitination for other purposes than targeting the tumor suppressor protein for degradation.
The most prominent E3 that mediates p53 ubiquitination for other purposes than degradation is MDM2. MDM2 is able to ubiquitinates six C-terminal lysine residues of p53 [5]. While high levels of MDM2 or MDM2 together with an appropriate E4 mediate p53 polyubiquitination and subsequent degradation, low levels of MDM2 promote monoubiquitination of p53 and this monoubiquitinated p53 accumulates in the cytoplasm. p53 was also cytoplasmic when it was monoubiquitinated by genetic engineering, demonstrating that the modification of p53 is essential for its subcellular localization and not a putative shuttle function of MDM2 [86]. Ubiquitination of the six C-terminal lysine residues is essential for this process as ubiquitination at these sites leads to the exposure of a nuclear export signal of p53 in the C-terminal part of the protein [87]. According to its increased cytoplasmic localization, the transcriptional activity of p53 is reduced when it is monoubiquitinated. As a fraction of the cellular p53 protein typically localizes to the cytoplasm even under normal growth conditions, it is possible that this share of the p53 protein is normally cytoplasmic because of its monoubiquitination due to the usually low levels of MDM2 in unstressed cells [94][95]. This storage of p53 in the cytoplasm could serve as a rapid and reversable mean for downregulating p53 function. Alternatively, it could provide a mechanism for shuttling p53 to the cytoplasm where it can execute transcription-independent activities (e.g., activation of the mitochondrial cell death program) when needed.
MSL2 (male-specific lethal 2) is a nuclear, RING-containing protein of the MSL complex [96]. MSL2 was found to be associated with endogenous p53 after overexpression of the E3 in U2OS cells. MSL2 ubiquitinated p53 at lysine residues K351 and K357 in a RING-dependent manner [88]. After ubiquitination of these lysine residues, p53 accumulated in the cytoplasm (Figure 2). The cytoplasmic translocation was prevented by the drug Leptomycin B, demonstrating an involvement of the nuclear export machinery in the translocation process. Ubiquitination of p53 by MSL2 also occurred in MDM2-negative cells and is thus independent of MDM2 [88]. Although the lysine residues that are ubiquitinated by MSL2 are different from the ones that are ubiquitinated by MDM2, they might contribute to the same mechanisms which is alteration of p53 conformation and presentation of the nuclear export signal of p53 to the cellular export machinery. DNA damage leads to a rapid increase in MSL2 levels which occurs already at 30 min after ionizing irradiation and appears to be independent of p53 as there was no increase in MSL2 transcription [97]. This increase in MSL2 protein after DNA damage could contribute to an increase also of transcription-independent activities of p53 during the DNA damage response.
Also, WWP1 (WW domain-containing E3 protein 1), an E3 that is particularly found at cellular endosomes, mediates p53 nuclear export and suppresses its transcriptional activity. WWP1 is a member of the NEDD4 subfamily of E3s [98]. Like the other members of this family, WWP1 contains an N-terminal C2 domain, four tandem WW domains and a C-terminal catalytic HECT-domain. The C2 domain is a structural domain that is often found in proteins that bind phospholipids and target proteins to cell membranes. The four WW-domains mediate protein-protein interactions and bind particularly well to proline-rich polypeptides [99]. The interaction of p53 and WWP1 has been found by searching for ubiquitin ligases that associate with the proline-rich domain of p53. Later it turned out that not the proline-rich region, but the DNA binding domain, is the primary site of interaction of p53 with the E3. Nevertheless, the proline-rich domain is important for this interaction as its deletion weakens the interaction of p53 with WWP1. Although all members of the WWP family possess the proline-binding WW motif, WWP1 and to a lesser extend WWP2 are the only family members that associate with p53 [89][99]. WWP1 associates with p53 and ubiquitinates the tumor suppressor protein in a HECT-domain-dependent manner, however, without targeting it for degradation. Instead, the p53 protein becomes even more stable. Ubiquitination of p53 by WWP1 is, however, modest in comparison to ubiquitination of p53 by MDM2. WWP1-mediated stabilization of p53 is associated with an increase in cytoplasmic p53 and decreased transcription of p53 target genes (Figure 2). Knocking-down WWP1 in hepatoma carcinoma cells resulted in reduced growth and increased apoptosis in the tumor cells [89][90]. Ubiquitination of p53 by WWP1 is independent of MDM2 as it is also seen in in MDM2-knock-out cells. WWP1 is induced after DNA damage at the mRNA level, however, while p53 stimulates expression of most of the E3s that modify it, it reduces WWP1 expression and may thus not be involved in its induction after DNA damage (Chen et al.). [89][100].
Another E3 that modifies p53 without targeting it for degradation is CUL7 (Cullin7) [91]. CUL7 is a member of the Cullin protein family, a family of evolutionarily conserved proteins that bind to the RING protein ROC1 (also known as RBX1, HRT1 or Sag1), thus constituting a functional E3 [101]. The N-terminal region of the Cullin is responsible for substrate recruitment while the C-terminal part interacts with ROC1. CUL7 is localized in the cytoplasm and binds directly to p53 [91]. However, CUL7 neither affects p53 stability nor its subcellular localization, probably because it only mediates mono- or diubiquitination of p53 that is not sufficient for targeting p53 for degradation (Figure 2) [92]. Surprisingly, deletion of the ROC1-binding sequence did not alter CUL7-mediated p53 ubiquitination, implying that ROC1 is not essential for this process [92]. Whether CUL7 associates with another E3 for ubiquitinating p53 remains to be determined. Co-expression of CUL7 reduced p53-mediated transactivation and constitutive ectopic expression of CUL7 increased cell proliferation and delayed an UV-induced G2 arrest in U2OS cells but not in H1299 cells that do not express p53. Also, deletion of the N-terminal domain of CUL7 or a mutation that disrupted p53 binding abolished the ability of CUL7 to enhance U2OS cell proliferation, indicating that this effect of CUL7 depends on p53 [92]. CUL7 is induced after DNA damage at the protein and RNA level, which seems to be independent of p53 as it was also induced in p53-negative HCT116 cells and with the same kinetics as in the corresponding wild-type cells [102].
E4F1 was first identified as a cellular target of the viral oncoprotein E1A. Later it was found that it possesses E3 activity towards itself and that it binds to p53 and induces p53-dependent cell cycle arrest [93][103]. These evidences raised the intriguing possibility that E4F1 might also ubiquitinate p53 and regulate its activities. Indeed, E4F1 stimulated oligo-ubiquitylation of p53 in the hinge region of p53 that contains a cluster of three lysine residues (K319, K320, K321). Mutation of these sites rendered p53 insensitive towards E4F1-mediated ubiquitination [93]. However, despite equipping p53 with K48-linked ubiquitin chains, E4F-1-mediated ubiquitination did not increase p53 degradation. Instead, E4F1 overexpression induced a moderate, but reproducible, stabilization of p53 [93]. This E4F1-modified p53 was predominantly found at the chromatin where it stimulated transcription of growth arrest inducing genes while p53-dependent anti-apoptotic activities were suppressed (Figure 2) [93].
3. E3s That Mediate Neddylation and Sumoylation of p53
In addition to ubiquitination, p53 can also be modified with NEDD8 (neural precursor cell expressed, developmentally downregulated 8) and SUMO (small ubiquitin-like modifier) [104][105][106]. In contrast to ubiquitination and neddylation, where there is only one protein in cells that can be attached to a target protein (ubiquitin and NEDD8 respectively), four SUMO proteins are expressed in cells. SUMO1, SUMO2 and SUMO3 are widely expressed while the expression of SUMO-4 is limited to certain organs [107]. SUMO4 furthermore contains a proline residue close to the diglycine motif, which may prevent its processing [108]. Since NEDD8 or SUMO compete with ubiquitin for the same lysine, these modifications frequently lead to stabilization of the target protein. Like ubiquitination, the sumoylation and neddylation processes include three steps. Prior to neddylation of a target protein, the NEDD8 precursor protein is activated by the NEDD8-specific E1, a heterodimer consisting of APP-BP1 (amyloid precursor protein binding protein 1) and UBA3 (ubiquitin-like modifier activating protein 3) in an ATP-dependent manner. The activated NEDD8 is loaded onto the NEDD8-specific E2 UBC12. Finally, a NEDD8-specific E3 conjugates NEDD8 to the target protein. Unlike ubiquitin, NEDD8 does not form chains [109]. The process of sumoylation follows the same pattern. To start the reaction, the pro-peptide of one of the SUMO proteins is processed to expose its C-terminal diglycine motif. This is usually performed by one of several SENPs (Sentrin/SUMO-specific protease). The processed SUMO is then activated in an ATP-dependent manner by a SUMO-activating enzyme, a heterodimer of SAE1 (SUMO-activating enzyme 1) and SAE2. In the next step, the SUMO protein is loaded onto the SUMO conjugating enzyme UBC9 (ubiquitin-conjugating enzyme 9). Finally, a SUMO E3 catalyzes the covalent attachment of SUMO to the target protein. In contrast to ubiquitination, sumoylation only occurs when the target protein possesses a sumoylation consensus motif consisting of ψ-K-X-E/D where ψ is any hydrophobic amino acid, K is the lysine to be sumoylated, E is glutamic acid and D is aspartic acid [110]. Several ligases can modify p53 with NEDD8 and/or SUMO including MDM2, PIAS proteins, FBXO11, and TOPORS (Table 3, Figure 3).
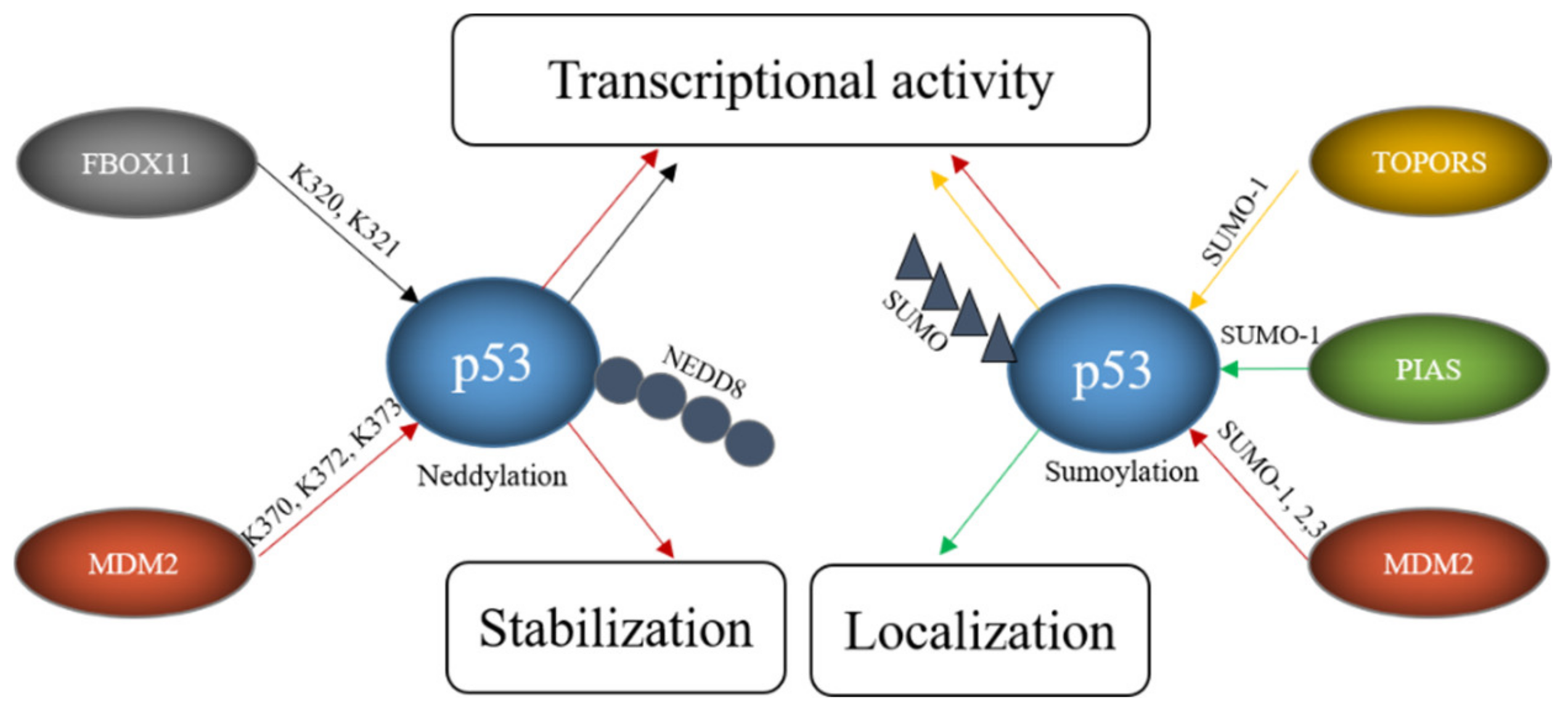
Figure 3. E3s modify p53 with SUMO and NEDD8. FBOX11 modifies p53 with NEDD8 and E3s like TOPORS or proteins of the PIAS family modify p53 with SUMO while MDM2 can modify p53 both with SUMO and NEDD8. These modifications alter p53 stability, localization and transcriptional activity.
Table 3. E3s that mediate p53 sumoylation and/or neddylation.
MDM2 can actually decorate p53 with ubiquitin, as described above, as well as with NEDD8 and SUMO. Neddylation of p53 requires a direct interaction of p53 with MDM2. It occurs at the C-terminal lysine residues K370, K372 and K373 and is accompanied by a decrease in p53 activity [106]. Sumoylation of p53 by MDM2 employs SUMO 1 and SUMO2/3 and occurs at lysine K386 within a SUMO consensus motif. Sumoylation even occurs to the conformational mutants p53R273H and p53R175H. As murine p53 lacks the consensus motif, this modification is not seen in mice [105]. While ubiquitination of p53 by MDM2 can be reconstituted in vitro, this was not possible for sumoylation, implying that a particular subcellular location or additional proteins are required [105]. Nevertheless, binding of p53 to MDM2 is required for sumoylation as shown by a p53 mutant (p5314Q, 19S) that cannot bind to MDM2 and that is poorly sumoylated [117]. The sumoylation activity of MDM2 is independent of the RING domain of MDM2 and stimulated by the presence of p14/p19ARF, the product of the alternative reading frame of the INK4a/b locus [118]. The nucleolar protein p14/p19ARF sequesters MDM2 in the nucleolus and the targeting of MDM2 and p53 to the nucleolus seems to be important for MDM2-mediated sumoylation of p53 [119]. Despite the importance of the nucleolar localization, an engineered p53 protein that was directly targeted to the nucleolus showed only modest sumoylation. Co-transfection with p14/p19ARF, however, led to a strong increase in p53 sumoylation, suggesting that nucleolar targeting, the presence of MDM2 and p14/p19ARF are all required for efficient sumoylation of p53 [117]. Mdm2-mediated sumoylation of p53 is also promoted by the ribosomal protein L11 [105]. While there is large consent that p53 is sumoylated by MDM2, the reported functional consequences are contradictory. Some researchers observed a modest increase in p53 activity after co-expression of p53, SUMO1 and UBC9 and a slightly compromised pro-apoptotic activity after mutation of the SUMO consensus motif of p53 while such an increase in p53 activity was not seen by others [111][120]. Interestingly, p53 modified by SUMO2/3 was more efficient in activating the synthetic PG13-promoter and the p21-promoter but not the BAX-promoter suggesting that sumoylation may regulate transcription of only a subset of target genes [105]. This promoter-dependent behavior of p53 modified with SUMO2/3 could eventually provide an explanation for the contradictory results that have been obtained with p53 modified with SUMO1. A similar debate is ongoing about the effect of sumoylation of p53 on its subnuclear localization. While Carter and Vousden observed at least a partial increase in nuclear export of p53 that was sumoylated with SUMO1, Kwek and co-workers found sumoylated p53 tightly bound to chromatin in the cell nucleus [120][121]. Interestingly, modification of p53 with SUMO2/3 was increased after treatment of cells with H2O2 and overexpression of SUMO2/3 promoted a premature senescence phenotype that was reduced by downregulating p53. These results suggest that modification of p53 with SUMO2/3 plays a role in the oxidative stress response and in the induction of senescence [122]. As MDM2 is also an important E3 for p53 ubiquitination and also mediates p53-neddylation, this raises the question how MDM2 can change from a ubiquitin-specific E3 to an E3 for NEDD8 or SUMO. At least part of this promiscuous behavior of MDM2 lies in the decoration of MDM2 itself with post-translational modifications. Phosphorylation of MDM2 at tyrosine Y281 and Y302 by the SRC kinase leads to the recruitment of the NEDD8-specific E2 UBC12 and by this converts MDM2 from an E3 for ubiquitin to an E3 for NEDD8 [123].
Like MDM2, TOPORS not only modifies p53 with ubiquitin, but also with SUMO1. This modification is direct as it was reproducible in an in vitro system. While the RING domain of TOPORS was obligatory for p53 ubiquitination, it was optional for p53 sumoylation [112]. Overexpression of TOPORS in Hela cells stimulated p53 sumoylation and was accompanied by an increase in p53 protein level and activity. Like in the case of MDM2, the preference of the E3 for SUMO1 or ubiquitin was regulated by post-translational modifications of the E3. While phosphorylation of TOPORS by the Polo-like kinase 1 (Plk1) enhanced p53 ubiquitination and degradation, it reduced TOPORS-mediated sumoylation of p53 [13].
A major family of E3s for p53 sumoylation is the PIAS family. All members of this family, PIAS 1, PIAS2, also known as PIASx, PIAS3 and PIAS4, also known as PIASγ, interact with p53, stimulate SUMO conjugation to p53 in a reconstituted in vitro system and increase the modification upon ectopic expression in vivo [113][114]. However, whether all family members sumoylate p53 also at physiological expression levels remains to be determined. Nevertheless, for PIAS4 it has been shown that the amount of sumoylated p53 is reduced in HEK293 cells upon its RNAi-mediated depletion [115]. As some PIAS proteins are expressed in a tissue-specific manner, it is most likely that different PIAS proteins sumoylate p53 in distinct tissues. Not all PIAS proteins have been studied in detail for activities towards p53, but for PIAS4 it has been shown that its overexpression correlates with activation of p53 target genes and induction of cellular senescence in primary human fibroblasts [115]. Since these effects depend on the ligase activity of PIAS4, it is tempting to conclude that they are a direct consequence of p53 sumoylation, although this has not been formally demonstrated. Earlier data also showed suppression of p53-mediated transcription of a p21-reporter and of endogenous p21 in the presence of PIAS4. Also, for PIAS1, it has been described that it can both, activate and repress p53-driven transcription [113][114][124]. Eventually the SUMO system may convey promoter-specific effects, an idea that is supported by the observation that PIAS4 affected p53-mediated expression of p21 but did not compromise BAX expression [125]. Thus, PIAS family members may be involved in differential regulation of selected p53 target genes.
FBXO11, a member of the F-box protein family and a component of the SCF ubiquitin ligase complex, was also found to be a NEDD8-E3 for p53. Although F-box proteins frequently mediate ubiquitination and degradation of their target proteins, this is not the case for SCF-complexes containing FBXO11 and p53. Instead, FBXO11-containing SCF-complexes attached NEDD8 to p53 involving the lysine residues K320 and K321. This modification inhibited p53 activity without altering its stability or subcellular localization [116].
4. E3s That Act. on p53 without Modifying It
While most E3s control p53 abundance and/or activity by directly decorating the tumor suppressor protein with ubiquitin or the related ubiquitin-like small modifiers, there are also some proteins with E3 activity that regulate p53 abundance and activity by a different mode. One of these proteins is TRIM25. TRIM25 is an estrogen-inducible member of the TRIM-family. As such, it has a RING domain and its ubiquitin ligase activity has been demonstrated by showing that it ubiquitinates and degrades 14-3-3σ [126]. However, although TRIM25 physically interacts with p53, it does not mediate p53 ubiquitination nor its modification with other ubiquitin-like small modifiers. Instead, TRIM25 leads to p53 stabilization by reducing the interaction of MDM2 with p300, an E4 enzyme required for the formation of polyubiquitin chains on p53 [127]. At the same time, TRIM25 inhibits the transcriptional activity of p53 by reducing the abundance of p300 and thus p53 acetylation [127].
PARC (p53-associated, Parkin-like cytoplasmic protein), also known as CUL9, an RBR-type E3, was identified during a search for cytoplasmic binding partners for p53 [128]. RBR-type E3s have more recently been categorized as a new family of E3s. They contain three highly conserved domains: Ring1, IBR (InBetweenRING) and RING2 and share common features with the RING- and the HECT- domain ligases [129]. PARC associates with p53 and the majority of cytoplasmic p53 appears to be associated with PARC. However, PARC did not modify p53 [128]. Downregulation of PARC resulted in enhanced nuclear localization of p53 and upregulation of p53-dependent apoptosis while overexpression of PARC resulted in enhanced cytoplasmic sequestration of p53 [128]. Deletion of PARC enhanced DNA damage, led to spontaneous tumor development and accelerated Eμ-Myc-induced lymphomagenesis [130][131]. In SK-N-AS neuroblastoma cells, which respond badly to the genotoxic drug etoposide, enhanced deletion of PARC the response to DNA damage [128]. As PARC retains p53 in the cytoplasm, expression of the p53 target gene p21 was significantly reduced, both under normal conditions and in response to DNA damage [131].
The PML (promyelocytic leukemia) protein, a tumor suppressor protein that is frequently implicated in leukemia, is best known for its translocation with the retinoic acid receptor alpha (RARa) resulting in the PML-RARa protein, and for the organization of PML nuclear bodies [132][133]. PML physically interacts with p53 and enhances p53′s proapoptotic and transcriptional activity [134]. Cells lacking PML showed a reduced propensity to undergo senescence or apoptosis in response to p53 activation despite the induction of several p53 target genes [135]. PML comes in several isoforms and at least one of them (PML III) targets p53 into nuclear bodies [134]. This colocalization of p53 and PML is enhanced in response to DNA damage [134]. PML is a p53 target gene and p53 stimulates PML induction in response to oncogenes and after DNA damage [135].
References
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299.
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gyuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993, 362, 857–860.
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27.
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132.
- Rodriguez, M.S.; Desterro, J.M.P.; Lain, S.; Lane, D.P.; Hay, R.T. Multiple C-Terminal Lysine Residues Target p53 for Ubiquitin-Proteasome-Mediated Degradation. Mol. Cell. Biol. 2000, 20, 8458–8467.
- Lee, E.-W.; Lee, M.-S.; Camus, S.; Ghim, J.; Yang, M.-R.; Oh, W.; Ha, N.-C.; Lane, D.P.; Song, J. Differential regulation of p53 and p21 by MKRN1 E3 ligase controls cell cycle arrest and apoptosis. EMBO J. 2009, 28, 2100–2113.
- Zhang, Y.; Cui, N.; Zheng, G. Ubiquitination of P53 by E3 ligase MKRN2 promotes melanoma cell proliferation. Oncol. Lett. 2020, 19, 1975–1984.
- Dornan, D.; Wertz, I.; Shimizu, H.; Arnott, D.; Frantz, G.D.; Dowd, P.; O’Rourke, K.; Koeppen, H.; Dixit, V.M. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 2004, 429, 86–92.
- Leng, R.P.; Lin, Y.; Ma, W.; Wu, H.; Lemmers, B.; Chung, S.; Parant, J.M.; Lozano, G.; Hakem, R.; Benchimol, S. Pirh2, a p53-Induced Ubiquitin-Protein Ligase, Promotes p53 Degradation. Cell 2003, 112, 779–791.
- Shloush, J.; Vlassov, J.E.; Engson, I.; Duan, S.; Saridakis, V.; Dhe-Paganon, S.; Raught, B.; Sheng, Y.; Arrowsmith, C.H. Structural and Functional Comparison of the RING Domains of Two p53 E3 Ligases, Mdm2 and Pirh2. J. Biol. Chem. 2011, 286, 4796–4808.
- Duan, W.; Gao, L.; Wu, X.; Zhang, Y.; Otterson, G.A.; Villalona-Calero, M.A. Differential response between the p53 ubiquitin-protein ligases Pirh2 and MdM2 following DNA damage in human cancer cells. Exp. Cell Res. 2006, 312, 3370–3378.
- Yamasaki, S.; Yagishita, N.; Sasaki, T.; Nakazawa, M.; Kato, Y.; Yamadera, T.; Bae, E.; Toriyama, S.; Ikeda, R.; Zhang, L.; et al. Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase ‘Synoviolin’. EMBO J. 2006, 26, 113–122.
- Yamasaki, S.; Nishioka, K.; Nakajima, T.; Yagishita, N. The Roles of Synoviolin in Crosstalk Between Endoplasmic Reticulum Stress-Induced Apoptosis and p53 Pathway. Cell Cycle 2007, 6, 1319–1323.
- Zhou, R.; Wen, H.; Ao, S. Identification of a novel gene encoding a p53-associated protein. Gene 1999, 235, 93–101.
- Rajendra, R.; Malegaonkar, D.; Pungaliya, P.; Henderson, M.; Rasheed, Z.; Brownell, J.; Liu, L.F.; Lutzker, S.; Saleem, A.; Rubin, E.H. Topors functions as an E3 ubiquitin ligase with specific E2 enzymes and ubiq-uitinates p53. J. Biol. Chem. 2004, 279, 36440–36444.
- Yang, X.; Li, H.; Zhou, Z.; Wang, W.-H.; Deng, A.; Andrisani, O.; Liu, X. Plk1-mediated Phosphorylation of Topors Regulates p53 Stability. J. Biol. Chem. 2009, 284, 18588–18592.
- Shen, J.; Li, P.; Shao, X.; Yang, Y.; Liu, X.-J.; Feng, M.; Pengyu, L.; Hu, R.; Wang, Z. The E3 Ligase RING1 Targets p53 for Degradation and Promotes Cancer Cell Proliferation and Survival. Cancer Res. 2018, 78, 359–371.
- Wen, W.; Peng, C.; Kim, M.O.; Jeong, C.H.; Zhu, F.; Yao, K.; Zykova, T.; Ma, W.; Carper, A.; Langfald, A.; et al. Knockdown of RNF2 induces apoptosis by regulating MDM2 and p53 stability. Oncogene 2013, 33, 421–428.
- Su, W.-J.; Fang, J.-S.; Cheng, F.; Liu, C.; Zhou, F.; Zhang, J. RNF2/Ring1b negatively regulates p53 expression in selective cancer cell types to promote tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 1720–1725.
- Chen, Y.C.; Chan, J.Y.-H.; Chiu, Y.-L.; Liu, S.-T.; Lozano, G.; Wang, S.-L.; Ho, C.-L.; Huang, S.M. Grail as a molecular determinant for the functions of the tumor suppressor p53 in tumorigenesis. Cell Death Differ. 2013, 20, 732–743.
- Allton, K.; Jain, A.K.; Herz, H.-M.; Tsai, W.-W.; Jung, S.Y.; Qin, J.; Bergmann, A.; Johnson, R.L.; Barton, M.C. Trim24 targets endogenous p53 for degradation. Proc. Natl. Acad. Sci. USA 2009, 106, 11612–11616.
- Jain, A.K.; Allton, K.L.; Duncan, A.D.; Barton, M.C. TRIM24 Is a p53-Induced E3-Ubiquitin Ligase That Undergoes ATM-Mediated Phosphorylation and Autodegradation during DNA Damage. Mol. Cell. Biol. 2014, 34, 2695–2709.
- Liu, J.; Zhang, C.; Wang, X.L.; Ly, P.; Belyi, V.; Xumonette, Z.Y.; Young, K.H.; Hu, W.; Feng, Z. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ. 2014, 21, 1792–1804.
- Zhang, L.; Huang, N.J.; Chen, C.; Tang, W.; Kornbluth, S. Ubiquitylation of p53 by the APC/C inhibitor Trim39. Proc. Natl. Acad. Sci. USA 2012, 109, 20931–20936.
- Zhou, Z.; Ji, Z.; Wang, Y.; Li, J.; Cao, H.; Zhu, H.H.; Gao, W.-Q. TRIM59 is up-regulated in gastric tumors, promoting ubiquitination and degradation of p53. Gastroenterology 2014, 147, 1043–1054.
- Han, R.; Zhao, Q.; Zong, S.; Miao, S.; Song, W.; Wang, L. A novel TRIM family member, Trim69, regulates zebrafish development through p53-mediated apoptosis. Mol. Reprod. Dev. 2016, 83, 442–454.
- Rong, X.; Rao, J.; Li, D.; Jing, Q.; Lu, Y.; Ji, Y. TRIM69 inhibits cataractogenesis by negatively regulating p53. Redox Biol. 2019, 22, 101157.
- Nguyen, D.T.T.; Richter, D.; Michel, G.; Mitsckha, S.; Kolanus, W.; Cuevas, E.; Wulczyn, F.G. The ubiquitin ligase LIN41/TRIM71 targets p53 to antagonize cell death and differ-entiation pathways during stem cell differentiation. Cell Death Differ. 2017, 24, 1063–1078.
- Yang, W.; Rozan, L.M.; McDonald, E.R., III; Navaraj, A.; Liu, J.J.; Matthew, E.M.; Wang, W.; Dicker, D.T.; El-Deiry, W.S. CARPs are ubiquitin ligases that promote MDM2-independent p53 and phos-pho-p53ser20 degradation. J. Biol. Chem. 2007, 282, 3273–3281.
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505.
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. Cloning and expression of the cDNA for E6-AP, a protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53. Mol. Cell. Biol. 1993, 13, 775–784.
- Mishra, A.; Jana, N.R. Regulation of turnover of tumor suppressor p53 and cell growth by E6-AP, a ubiquitin protein ligase mutated in Angelman mental retardation syndrome. Cell. Mol. Life Sci. 2008, 65, 656–666.
- Masuda, Y.; Saeki, Y.; Arai, N.; Kawai, H.; Kukimoto, I.; Tanaka, K.; Masutani, C. Stepwise multipolyubiquitination of p53 by the E6AP-E6 ubiquitin ligase complex. J. Biol. Chem. 2019, 294, 14860–14875.
- Chen, D.; Kon, N.; Li, M.; Zhang, W.; Qin, J.; Gu, W. ARF-BP1/Mule Is a Critical Mediator of the ARF Tumor Suppressor. Cell 2005, 121, 1071–1083.
- Kon, N.; Zhong, J.; Qiang, L.; Accili, D.; Gu, W. Inactivation of arf-bp1 Induces p53 Activation and Diabetic Phenotypes in Mice. J. Biol. Chem. 2012, 287, 5102–5111.
- Maan, M.; Pati, U. CHIP promotes autophagy-mediated degradation of aggregating mutant p53 in hypoxic conditions. FEBS J. 2018, 285, 3197–3214.
- Esser, C.; Scheffner, M.; Höhfeld, J. The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degra-dation. J. Biol. Chem. 2005, 280, 27443–27448.
- Sisoula, C.; Trachana, V.; Patterson, C.; Gonos, E.S. CHIP-dependent p53 regulation occurs specifically during cellular senescence. Free Radic. Biol. Med. 2011, 50, 157–165.
- Wu, H.; Pomeroy, S.L.; Ferreira, M.; Teider, N.; Mariani, J.; Nakayama, K.I.; Hatakeyama, S.; Tron, V.A.; Saltibus, L.F.; Spyracopoulos, L.; et al. P UBE4B promotes Hdm2-mediated degradation of the tumor suppressor p53. Nat. Med. 2011, 17, 347–355.
- Du, C.; Wu, H.; Leng, R.P. UBE4B targets phosphorylated p53 at serines 15 and 392 for degradation. Oncotarget 2015, 7, 2823–2836.
- Deshaies, R.J.; Joazeiro, C.A. RING Domain E3 Ubiquitin Ligases. Annu. Rev. Biochem. 2009, 78, 399–434.
- De Rozieres, S.; Maya, R.; Oren, M.; Lozano, G. The loss of mdm2 induces p53-mediated apoptosis. Oncogene 2000, 19, 1691–1697.
- A Boehme, K.; Blattner, C. Regulation of p53—Insights into a complex process. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 367–392.
- Sigalas, I.; Calvert, A.H.; Anderson, J.J.; Neal, D.E.; Lunec, J. Alternatively spliced mdm2 transcripts with loss of p53 binding domain sequences: Transforming ability and frequent detection in human cancer. Nat. Med. 1996, 2, 912–917.
- Bartel, F.; Taubert, H.; Harris, L.C. Alternative and aberrant splicing of MDM2 mRNA in human cancer. Cancer Cell 2002, 2, 9–15.
- Volk, E.L.; Schuster, K.; Nemeth, K.M.; Fan, L.; Harris, L.C. MDM2-A, a common Mdm2 splice variant, causes perinatal lethality, reduced longevity and enhanced senescence. Dis. Model. Mech. 2009, 2, 47–55.
- Evans, S.C.; Viswanathan, M.; Grier, J.D.; Narayana, M.; El-Naggar, A.K.; Lozano, G. An alternatively spliced HDM2 product increases p53 activity by inhibiting HDM2. Oncogene 2001, 20, 4041–4049.
- Dang, J.; Kuo, M.-L.; Eischen, C.M.; Stepanova, L.; Sherr, C.J.; Roussel, M.F. The RING domain of Mdm2 can inhibit cell proliferation. Cancer Res. 2002, 62, 1222–1230.
- Kim, J.Y.; Lee, R.; Xiao, G.; Forbes, D.; Bargonetti, J. MDM2-C Functions as an E3 Ubiquitin Ligase. Cancer Manag. Res. 2020, 12, 7715–7724.
- Savio, M.G.; Rotondo, G.; Maglie, S.; Rossetti, G.; Pardi, R. COP1D, an alternatively spliced constitutive photomorphogenic-1 (COP1) product, stabilizes UV stress-induced c-Jun through inhibition of full-length COP1. Oncogene 2008, 27, 2401–2411.
- Dong, L.; Liu, Y.; Qiu, Y.; Peng, B.; Yin, J.; Liu, W.; He, X. Identification of Pirh2E and Pirh2F, two additional novel isoforms of Pirh2 ubiquitin ligase from human hepatocellular liver carcinoma cell line. Biomed. Mater. Eng. 2012, 22, 89–95.
- Shi, J.; Huang, Y.; Sheikh, M.S. Identification of Pirh2D, an Additional Novel Isoform of Pirh2 Ubiquitin Ligase. Mol. Cell. Pharmacol. 2010, 2, 21–23.
- Corcoran, C.A.; Montalbano, J.; Sun, H.; He, Q.; Huang, Y.; Sheikh, M.S. Identification and Characterization of Two Novel Isoforms of Pirh2 Ubiquitin Ligase That Negatively Regulate p53 Independent of RING Finger Domains. J. Biol. Chem. 2009, 284, 21955–21970.
- Lin, L.; Ozaki, T.; Takada, Y.; Kageyama, H.; Nakamura, Y.; Hata, A.; Zhang, J.; Simonds, W.F.; Nakagawara, A.; Koseki, H. topors, a p53 and topoisomerase I-binding RING finger protein, is a coactivator of p53 in growth suppression induced by DNA damage. Oncogene 2005, 24, 3385–3396.
- Sen, N.; Satija, Y.K.; Das, S. PGC-1α, a Key Modulator of p53, Promotes Cell Survival upon Metabolic Stress. Mol. Cell 2011, 44, 621–634.
- Eisenberg, I.; Hochner, H.; Levi, T.; Yelin, R.; Kahan, T.; Mitrani-Rosenbaum, S. Cloning and characterization of a novel human gene RNF38 encoding a conserved pu-tative protein with a RING finger domain. Biochem. Biophys. Res. Commun. 2002, 294, 1169–1176.
- Lunardi, A.; Di Minin, G.; Provero, P.; Ferro, M.D.; Carotti, M.; Del Sal, G.; Collavin, L.L. A genome-scale protein interaction profile of Drosophila p53 uncovers additional nodes of the human p53 network. Proc. Natl. Acad. Sci. USA 2010, 107, 6322–6327.
- Sheren, J.E.; Kassenbrock, C.K. RNF38 encodes a nuclear ubiquitin protein ligase that modifies p53. Biochem. Biophys. Res. Commun. 2013, 440, 473–478.
- Seroogy, C.M.; Soares, L.; Ranheim, E.A.; Su, L.; Holness, C.; Bloom, D.; Fathman, C.G. The Gene Related to Anergy in Lymphocytes, an E3 Ubiquitin Ligase, Is Necessary for Anergy Induction in CD4 T Cells. J. Immunol. 2004, 173, 79–85.
- Lee, Y.-Y.; Wang, C.-T.; Huang, S.K.-H.; Wu, W.-J.; Huang, C.-N.; Li, C.-C.; Chan, T.-C.; Liang, P.-I.; Hsing, C.-H.; Li, C.-F. Downregulation of RNF128 Predicts Progression and Poor Prognosis in Patients with Urothelial Carcinoma of the Upper Tract and Urinary Bladder. J. Cancer 2016, 7, 2187–2196.
- Le Douarin, B.; Nielsen, A.L.; Garnier, J.M.; Ichinose, H.; Jeanmougin, F.; Losson, R.; Chambon, P. A possible involvement of TIF1 alpha and TIF1 beta in the epigenetic control of transcription by nuclear receptors. EMBO J. 1996, 15, 6701–6715.
- Khetchoumian, K.; Teletin, M.; Tisserand, J.; Mark, M.; Herquel, B.; Ignat, M.; Zucman-Rossi, J.; Cammas, F.; Lerouge, T.; Thibault, C.; et al. Loss of Trim24 (Tif1alpha) gene function confers oncogenic activity to retinoic acid receptor alpha. Nat. Genet. 2007, 39, 1500–1506.
- Jones, S.N.; Roe, A.E.; Donehower, L.A.; Bradley, A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995, 378, 206–208.
- Nicklas, S.; Otto, A.; Wu, X.; Miller, P.; Stelzer, S.; Wen, Y.; Kuang, S.; Wrogemann, K.; Patel, K.; Ding, H.; et al. TRIM32 Regulates Skeletal Muscle Stem Cell Differentiation and Is Necessary for Normal Adult Muscle Regeneration. PLoS ONE 2012, 7, e30445.
- Horn, E.J.; Albor, A.; Liu, Y.; El-Hizawi, S.; VanderBeek, G.E.; Babcock, M.; Bowden, G.T.; Hennings, H.; Lozano, G.; Weinberg, W.C.; et al. RING protein Trim32 associated with skin carcinogenesis has anti-apoptotic and E3-ubiquitin ligase properties. Carcinogenesis 2003, 25, 157–167.
- Liu, J.; Zhu, Y.; Hu, W.; Feng, Z. TRIM32 is a novel negative regulator of p53. Mol. Cell. Oncol. 2015, 2, e970951.
- Huang, N.J.; Zhang, L.; Tang, W.; Chen, C.; Yang, C.-S.; Kornbluth, S. The Trim39 ubiquitin ligase inhibits APC/CCdh1-mediated degradation of the Bax activator MOAP-1. J. Cell Biol. 2012, 197, 361–367.
- Han, Y.; Li, R.; Gao, J.; Miao, S.; Wang, L. Characterisation of human RING finger protein TRIM69, a novel testis E3 ubiquitin ligase and its subcellular localization. Biochem. Biophys. Res. Commun. 2012, 429, 6–11.
- McDonald, E.R., III; El-Deiry, W.S. Suppression of caspase-8- and -10-associated RING proteins results in sensitization to death ligands and inhibition of tumor cell growth. Proc. Natl. Acad. Sci. USA 2004, 101, 6170–6175.
- Yang, W.; Dicker, D.T.; Chen, J.; El-Deiry, W.S. CARPs enhance p53 turnover by degrading 14-3-3sigma and stabilizing MDM2. Cell Cycle 2008, 7, 670–682.
- Lorenz, S. Structural mechanisms of HECT-type ubiquitin ligases. Biol. Chem. 2018, 399, 127–145.
- Sluimer, J.; Distel, B. Regulating the human HECT E3 ligases. Cell. Mol. Life Sci. 2018, 75, 3121–3141.
- Yamamoto, Y.; Huibregtse, J.M.; Howley, P.M. The human E6-AP gene (UBE3A) encodes three potential protein isoforms generated by differential splicing. Genomics 1997, 41, 263–266.
- Saporita, A.J.; Maggi, L.B.; Apicelli, A.J.; Weber, J.D. Therapeutic Targets in the ARF Tumor Suppressor Pathway. Curr. Med. Chem. 2007, 14, 1815–1827.
- Aravind, L.; Koonin, E.V. The U box is a modified RING finger—A common domain in ubiquitination. Curr. Biol. 2000, 10, R132–R134.
- Ballinger, C.A.; Connell, P.; Wu, Y.; Hu, Z.; Thompson, L.J.; Yin, L.-Y.; Patterson, C. Identification of CHIP, a Novel Tetratricopeptide Repeat-Containing Protein That Interacts with Heat Shock Proteins and Negatively Regulates Chaperone Functions. Mol. Cell. Biol. 1999, 19, 4535–4545.
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479.
- Sisoula, C.; Gonos, E.S. CHIP E3 ligase regulates mammalian senescence by modulating the levels of oxidized proteins. Mech. Ageing Dev. 2011, 132, 269–272.
- Tonnessen-Murray, C.A.; Lozano, G.; Jackson, J.G. The Regulation of Cellular Functions by the p53 Protein: Cellular Senescence. Cold Spring Harb. Perspect. Med. 2016, 7, a026112.
- Koegl, M.; Hoppe, T.; Schlenker, S.; Ulrich, H.D.; Mayer, T.U.; Jentsch, S. A Novel Ubiquitination Factor, E4, Is Involved in Multiubiquitin Chain Assembly. Cell 1999, 96, 635–644.
- Kaneko-Oshikawa, C.; Nakagawa, T.; Yamada, M.; Yoshikawa, H.; Matsumoto, M.; Yada, M.; Hatakeyama, S.; Nakayama, K.; Nakayama, K.I. Mammalian E4 Is Required for Cardiac Development and Maintenance of the Nervous System. Mol. Cell. Biol. 2005, 25, 10953–10964.
- Wu, H.; Leng, R.P. UBE4B, a ubiquitin chain assembly factor, is required for MDM2-mediated p53 polyubiquitination and deg-radation. Cell Cycle 2011, 10, 1912–1915.
- Cardozo, P.; Pagano, M. The SCF ubiquitin ligase: Insights into a molecular machine. Nat. Rev. Mol. Cell. Biol. 2004, 5, 739–751.
- Sun, L.; Shi, L.; Li, W.; Yu, W.; Liang, J.; Zhang, H.; Yang, X.; Wang, Y.; Li, R.; Yao, X.; et al. JFK, a Kelch domain-containing F-box protein, links the SCF complex to p53 regulation. Proc. Natl. Acad. Sci. USA 2009, 106, 10195–10200.
- Sun, L.; Shi, L.; Wang, F.; Huangyang, P.; Si, W.; Yang, J.; Yao, Z.; Shang, Y. Substrate Phosphorylation and Feedback Regulation in JFK-promoted p53 Destabilization. J. Biol. Chem. 2011, 286, 4226–4235.
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono- versus polyubiquitination: Differential control of p53 fate by Mdm2. Science 2003, 302, 1972–1975.
- Carter, S.; Bischof, O.; Dejean, A.; Vousden, K.H. C-terminal modifications regulate MDM2 dissociation and nuclear export of p53. Nat. Cell. Biol. 2007, 9, 428–435.
- Kruse, J.P.; Gu, W. MSL2 promotes Mdm2-independent cytoplasmic localization of p53. J. Biol. Chem. 2009, 284, 3250–3263.
- Laine, A.; Ronai, Z. Regulation of p53 localization and transcription by the HECT domain E3 ligase WWP1. Oncogene 2007, 26, 1477–1483.
- Cheng, Q.; Cao, X.; Yuan, F.; Li, G.; Tong, T. Knockdown of WWP1 inhibits growth and induces apoptosis in hepatoma carcinoma cells through the activation of caspase3 and p53. Biochem. Biophys. Res. Commun. 2014, 448, 248–254.
- Kasper, J.S.; Arai, T.; DeCaprio, J.A. A novel p53-binding domain in CUL7. Biochem. Biophys. Res. Commun. 2006, 348, 132–138.
- Andrews, P.; He, Y.J.; Xiong, Y. Cytoplasmic localized ubiquitin ligase cullin 7 binds to p53 and promotes cell growth by antago-nizing p53 function. Oncogene 2006, 25, 4534–4548.
- Le Cam, L.; Linares, L.K.; Paul, C.; Julien, E.; Lacroix, M.; Hatchi, D.; Triboulet, R.; Bossis, G.; Shmuel, A.; Rodriguez, M.S.; et al. E4F1 is an atypical ubiquitin ligase that modulates p53 effector functions independently of degradation. Cell 2006, 127, 775–788.
- Yan, H.; Solozobova, V.; Zhang, P.; Armant, O.; Kuehl, B.; Brenner-Weiss, G.; Blattner, C. p53 is active in murine stem cells and alters the transcriptome in a manner that is remi-niscent of mutant p53. Cell Death Dis. 2015, 6, e1662.
- Maki, C.G. p53 Localization. In p53. Molecular Biology Intelligence Unit; Springer: Boston, MA, USA, 2010; Volume 1, pp. 117–126.
- Mendjan, S.; Akhtar, A. The right dose for every sex. Chromosoma 2006, 116, 95–106.
- Lai, Z.; Moravcová, S.; Canitrot, Y.; Andrzejewski, L.P.; Walshe, D.M.; Rea, S. Msl2 Is a Novel Component of the Vertebrate DNA Damage Response. PLoS ONE 2013, 8, e68549.
- Ingham, R.J.; Gish, G.; Pawson, T. The Nedd4 family of E3 ubiquitin ligases: Functional diversity within a common modular architecture. Oncogene 2004, 23, 1972–1984.
- Zhi, X.; Chen, C. WWP1: A versatile ubiquitin E3 ligase in signaling and diseases. Cell. Mol. Life Sci. 2012, 69, 1425–1434.
- Chen, J.; Shi, H.; Chen, Y.; Fan, S.; Liu, D.; Li, C. DNA damage induces expression of WWP1 to target ΔNp63α to degradation. PLoS ONE 2017, 12, e0176142.
- Sarikas, A.; Hartmann, T.; Pan, Z.-Q. The cullin protein family. Genome Biol. 2011, 12, 1–12.
- Jung, P.; Verdoodt, B.; Bailey, A.; Yates, J.R.; Menssen, A.; Hermeking, H. Induction of Cullin 7 by DNA damage attenuates p53 function. Proc. Natl. Acad. Sci. USA 2007, 104, 11388–11393.
- Sandy, P.; Gostissa, M.; Fogal, V.; De Cecco, L.; Szalay, K.; Rooney, R.J.; Schneider, C.; Del Sal, G. p53 is involved in the p120E4F-mediated growth arrest. Oncogene 2000, 19, 188–199.
- Melchior, F.; Hengst, L. SUMO-1 and p53. Cell Cycle 2002, 1, 243–247.
- Stindt, M.H.; Carter, S.; Vigneron, A.M.; Ryan, K.M.; Vousden, K.H. MDM2 promotes SUMO-2/3 modification of p53 to modulate transcriptional activity. Cell Cycle 2011, 10, 3176–3188.
- Xirodimas, D.P.; Saville, M.K.; Bourdon, J.-C.; Hay, R.T.; Lane, D.P. Mdm2-Mediated NEDD8 Conjugation of p53 Inhibits Its Transcriptional Activity. Cell 2004, 118, 83–97.
- Citro, S.; Chiocca, S. Sumo paralogs: Redundancy and divergencies. Front. Biosci. 2013, 5, 544–553.
- Owerbach, D.; McKay, E.M.; Yeh, E.T.; Gabbay, K.H.; Bohren, K.M. A proline-90 residue unique to SUMO-4 prevents maturation and sumoylation. Biochem. Biophys. Res. Commun. 2005, 337, 517–520.
- Rabut, G.; Peter, M. Function and regulation of protein neddylation. ‘Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 2008, 9, 969–976.
- Yang, Y.; He, Y.; Wang, X.; Liang, Z.; He, G.; Zhang, P.; Zhu, H.; Xu, N.; Liang, S. Protein SUMOylation modification and its associations with disease. Open Biol. 2017, 7, 7.
- Gostissa, M.; Hengstermann, A.; Fogal, V.; Sandy, P.; Schwarz, S.E.; Scheffner, M.; Del Sal, G. Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. EMBO J. 1999, 18, 6462–6471.
- Weger, S.; Hammer, E.; Heilbronn, R. Topors acts as a SUMO-1 E3 ligase for p53 in vitro and in vivo. FEBS Lett. 2005, 579, 5007–5012.
- Stehmaier, P.; Muller, S. Regulation of p53 family members by the ubiquitin-like SUMO system. DNA Repair (Amsterdam) 2009, 8, 491–498.
- Schmidt, D.; Müller, S. Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc. Natl. Acad. Sci. USA 2002, 99, 2872–2877.
- Bishof, O.; Schwamborn, K.; Martin, N.; Werner, A.; Sustmann, C.; Grosschedl, R.; Dejean, A. The E3 SUMO ligase PIASγ is a regulator of cellular senescence and apoptosis. Mol. Cell 2006, 22, 783–794.
- Abida, W.M.; Nikolaev, A.; Zhao, W.; Zhang, W.; Gu, W. FBXO11 Promotes the Neddylation of p53 and Inhibits Its Transcriptional Activity. J. Biol. Chem. 2007, 282, 1797–1804.
- Chen, L.; Chen, J. MDM2-ARF complex regulates p53 sumoylation. Oncogene 2003, 22, 5348–5357.
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.M.; Palmero, I.; Ryan, K.M.; Hara, E.; Vousden, K.H.; et al. The alternative product from the human CDKN2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998, 17, 5001–5014.
- Weber, J.D.; Taylor, L.J.; Roussel, M.F.; Sherr, C.J.; Bar-Sagi, D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat. Cell Biol. 1999, 1, 20–26.
- Kwek, S.S.; Derry, J.; Tyner, A.L.; Shen, Z.; Gudkov, A.V. Functional analysis and intracellular localization of p53 modified by SUMO-1. Oncogene 2001, 20, 2587–2599.
- Carter, S.A.; Vousden, K.H. p53-Ubl fusions as models of ubiquitination, sumoylation and neddylation of p53. Cell Cycle 2008, 7, 2519–2528.
- Li, T.; Santockyte, R.; Shen, R.F.; Tekle, E.; Wang, G.; Yang, D.C.H.; Chock, P.B. Expression of SUMO-2/3 induced senescence through p53- and pRB-mediated pathways. J. Biol. Chem. 2006, 281, 36221–36227.
- Batuello, C.N.; Hauck, P.M.; Gendron, J.M.; Lehman, J.A.; Mayo, L.D. Src phosphorylation converts Mdm2 from a ubiquitinating to a neddylating E3 ligase. Proc. Natl. Acad. Sci. USA 2015, 112, 1749–1754.
- Xu, J.H.; Megidish, T.; Xu, C.W. Activation of p53 by Protein Inhibitor of Activated Stat1 (PIAS1). J. Biol. Chem. 2002, 277, 8255–8259.
- Nelson, V.; Davis, G.E.; Maxwell, S.A. A putative protein inhibitor of activated STAT (PIASy) interacts with p53 and inhibits p53-mediated transactivation but not apoptosis. Apoptosis 2001, 6, 221–234.
- Urano, T.; Saito, T.; Tsukui, T.; Fujita, M.; Hosoi, T.; Muramatsu, M.; Ouchi, Y.; Inoue, S. Efp targets 14-3-3 sigma for proteolysis and promotes breast tumour growth. Nature 2002, 417, 871–875.
- Zhang, P.; Elabd, S.; Hammer, S.; Solozobova, V.; Yan, H.; Bartel, F.; Inoue, S.; Henrich, T.; Wittbrodt, J.; Loosli, F.; et al. TRIM25 has a dual function in the p53/Mdm2 circuit. Oncogene 2015, 34, 5729–5738.
- Nikolaev, A.Y.; Li, M.; Puskas, N.; Qin, J.; Gu, W. Parc: A cytoplasmic anchor for p53. Cell 2003, 112, 29–40.
- Spratt, D.E.; Walden, H.; Shaw, G.S. RBR E3 ubiquitin ligases: New structures, new insights, new questions. Biochem. J. 2014, 458, 421–437.
- Pei, X.-H.; Bai, F.; Li, Z.; Smith, M.D.; Whitewolf, G.; Jin, R.; Xiong, Y. Cytoplasmic CUL9/PARC Ubiquitin Ligase Is a Tumor Suppressor and Promotes p53-Dependent Apoptosis. Cancer Res. 2011, 71, 2969–2977.
- Li, Z.; Xiong, Y. Cytoplasmic E3 ubiquitin ligase CUL9 controls cell proliferation, senescence, apoptosis and genome integrity through p53. Oncogene 2017, 36, 5212–5218.
- Iaccarino, L.; Divona, M.; Ottone, T.; Cicconi, L.; Lavorgna, S.; Ciardi, C.; Alfonso, V.; Travaglini, S.; Facchini, L.; Cimino, G.; et al. Identification and monitoring of atypical PML/RARA fusion transcripts in acute pro-myelocytic leukemia. Genes Chromosomes Cancer 2019, 58, 60–65.
- Lallemand-Breitenbach, V.; de Thé, H. PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010, 2, a000661.
- Fogal, V.; Gostissa, M.; Sandy, P.; Zacchi, P.; Sternsdorf, T.; Jensen, K.; Pandolfi, P.P.; Will, H.; Schneider, C.; Del Sal, G. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 2000, 19, 6185–6195.
- de Stanchina, E.; Querido, E.; Narita, M.; Davuluri, R.V.; Pandolfi, P.P.; Ferbeyre, G.; Lowe, S.W. PML is a direct p53 target that modulates p53 effector functions. Mol. Cell 2004, 13, 523–535.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
27 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No