Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vladimir Ya. LEE | -- | 9384 | 2023-02-24 08:06:49 | | | |
| 2 | Jason Zhu | Meta information modification | 9366 | 2023-03-01 04:03:30 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lee, V.Y. Organogermanium Analogues. Encyclopedia. Available online: https://encyclopedia.pub/entry/41616 (accessed on 23 July 2026).
Lee VY. Organogermanium Analogues. Encyclopedia. Available at: https://encyclopedia.pub/entry/41616. Accessed July 23, 2026.
Lee, Vladimir Ya.. "Organogermanium Analogues" Encyclopedia, https://encyclopedia.pub/entry/41616 (accessed July 23, 2026).
Lee, V.Y. (2023, February 24). Organogermanium Analogues. In Encyclopedia. https://encyclopedia.pub/entry/41616
Lee, Vladimir Ya.. "Organogermanium Analogues." Encyclopedia. Web. 24 February, 2023.
Copy Citation
One of the most fundamental topics in modern organogermanium chemistry is the study of low-coordinate species, and within this realm, the field of multiply bonded compounds is now one of the mainstreams. It therefore comes as no surprise that the literature covering the latter field is vast. Heteronuclear multiply bonded combinations of germanium with the Main Group elements of groups 13, 15, and 16, >Ge=E13–, >Ge=E15–, and >Ge=E16, respectively, are excluded from the consideration. Moreover, numerous compounds, in which the low-coordinate Ge center is intramolecularly (by n-donor substituents) or intermolecularly (through external donor ligands) coordinated, thus experiencing remarkable electronic perturbation, are also not considered, except for silagermenylidenes >Si=Ge(NHC): and digermanium(0) complexes :Ge0(NHC/or NHSi)=Ge0(NHC/or NHSi):, which otherwise cannot be stabilized for their isolation.
bond length
double bond
germanium
main group element
multiple bonding
1. Introduction
One of the most fundamental topics in modern organogermanium chemistry is the study of low-coordinate species, and within this realm, the field of multiply bonded compounds is now one of the mainstreams. It therefore comes as no surprise that the literature covering the latter field is vast. Heteronuclear multiply bonded combinations of germanium with the Main Group elements of groups 13, 15, and 16, >Ge=E13–, >Ge=E15–, and >Ge=E16, respectively, are excluded from the consideration. Moreover, numerous compounds, in which the low-coordinate Ge center is intramolecularly (by n-donor substituents) or intermolecularly (through external donor ligands) coordinated, thus experiencing remarkable electronic perturbation, are also not considered, except for silagermenylidenes >Si=Ge(NHC): and digermanium(0) complexes :Ge0(NHC/or NHSi)=Ge0(NHC/or NHSi):, which otherwise cannot be stabilized for their isolation.
2. Heavy Analogues of Alkenes
2.1. Homonuclear Derivatives
2.1.1. Digermenes >Ge=Ge<
Acyclic Digermenes
The very first isolable digermene, namely tetra(alkyl)digermene Dis2Ge=GeDis2 [Dis = CH(SiMe3)2] 1, reported by Lappert and coworkers in 1976, was synthesized by the reaction of bis(amino)germylene [(Me3Si)2N]2Ge: with DisLi [1], and its crystal structure was reported ten years later (Scheme 1) [2]. Dimeric was in the solid state (as was confirmed by X-ray diffraction study), and digermene 1 dissociated in solution into a pair of monomeric germylenes Dis2Ge:, thus implying the easy breaking of the weak Ge=Ge double bond (Scheme 1). Remarkably, crystallographic studies revealed that the Ge=Ge bond in 1 was notably short (rGe=Ge = 2.347(2) Å) and not twisted, the geometry around both Ge centers was pyramidal (ΣGe = 348.5°), and the Dis-substituents at the Ge=Ge double bond were arranged in a trans-bent fashion (θ = 32°).
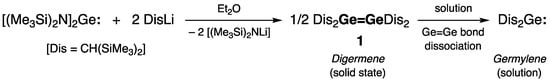
Scheme 1. Synthesis of the first isolable digermene Dis2Ge=GeDis2 1.
Since 1976, numerous digermenes have been isolated and, in the majority of cases, structurally characterized, with most of these cases being reported after 2000. The synthetic strategies toward stable digermenes can be categorized into the four main approaches: (1) photolysis of cyclotrigermanes (route A); (2) reduction of dihalo- or monohalogermylenes with Grignard or organolithium reagents (route B); (3) reductive dehalogenation of 1,1-dihalogermanes (route C); and (4) 1,2-addition or cycloaddition to digermynes (route D) (Scheme 2).
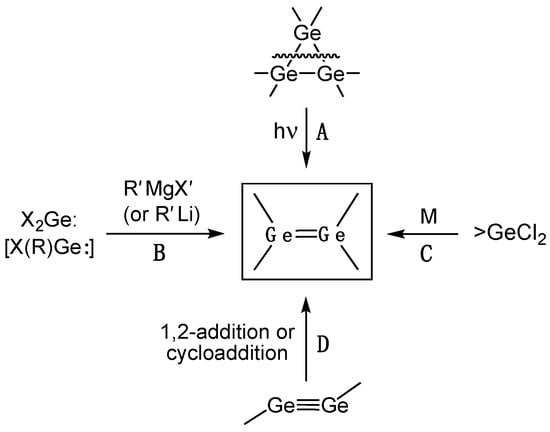
Scheme 2. General methods for the synthesis of isolable digermenes.
Route A (photolysis of hexaarylcyclotrigermanes) is mostly of a historical importance as the method employed by Masamune and coworkers for preparation of the first stable tetra(aryl)digermenes Ar2Ge=GeAr2 [3]. However, because of the synthetic limitations of this approach, which requires cyclotrogermane precursors that are not readily available, currently this method is not commonly used. Since 2000, there was only one report from the Baines group on an improved synthetic procedure for the tetra(mesityl)digermene Mes2Ge=GeMes2 2 that was generated by photolysis of hexa(mesityl)cyclotrigermane precursor in THF at −70 °C [4].
According to approach B, the reduction of isolable di(halo)germylenes X2Ge:/(X2Ge:–dioxane complex) or mono(halo)germylenes X(R)Ge: with Grignard RMgX or organolithium RLi reagents generates at first transient germylenes R2Ge: that dimerize forming digermenes R2Ge=GeR2, and this method was used for the preparation of Lappert’s germylene Dis2Ge=GeDis2 1 [1][2]. Since 2000, a few other isolable digermenes were prepared by method B: Ar(R)Ge=Ge(Ar)R [R = Me, Et, Ph; Ar = 2,6-(2,4,6-iPr3-C6H2)2-C6H3] 3a–c [5], [Me3Si–C≡C](Ar′)Ge=Ge[C≡C–SiMe3]Ar′ [Ar′ = 2,6-(2,6-iPr2-C6H3)2-C6H3] 4 [6], Ar*Ge=GeAr* [Ar* = 2,5-tBu2-C6H3] 5 [7], Bbt(Br)Ge=Ge(Bbt)Br [Bbt = 2,6-[(Me3Si)2CH]2-4-[(Me3Si)3C]-C6H2] 6 (equilibrating in solution with the monomeric germylene Bbt(Br)Ge:) [8], and Tbb(Br)Ge=Ge(Tbb)Br [Tbb = 2,6-[(Me3Si)2CH]2-4-tBu-C6H2] 7 [9]. A donor-acceptor Lewis base–Lewis acid digermene complex {[(OC)5W]←:GeH2–H2Ge←[:IPr]} [IPr = 1,3-bis(2,6-diisopropylphenyl)-2H-imidazol-2-ylidene] 8 was also prepared, featuring however single (instead of double) germanium–germanium bond, being therefore not classified as a true digermene [10].
Reductive dehalogenation of 1,1-di(halo)germanes (method C) is by far the most popular protocol for synthesis of stable digermenes, due to the ready availability of the starting R2GeX2. Using this approach, the synthesis and crystal structure of the following digermenes were reported: (R3Si)2Ge=Ge(SiR3)2 [R3Si = SiMe2tBu] 9 [11][12], Tbt(Mes)Ge=Ge(Tbt)Mes [Tbt = 2,4,6-[(Me3Si)2CH]3-C6H2] 10 [13], Fc(Tip)Ge=Ge(Fc)Tip [Tip = 2,4,6-iPr3-C6H2, Fc = ferrocenyl] 11 [10]. In tetra(aryl)digermene 10, the strong repulsive interaction of the bulky aryl substituents caused the room-temperature dissociation of the Ge=Ge bond into the corresponding germylenes Tbt(Mes)Ge:, as was monitored by UV–Vis spectroscopy (ΔH = 14.7 ± 0.2 kcal/mol, ΔS = 42.4 ± 0.8 cal/mol·deg) [14].
Method D, namely 1,2-addition or cycloaddition across the Ge≡Ge triple bond of digermynes, is the latest approach toward digermenes that was enabled by the recent availability of the stable digermynes. This approach is exemplified by the preparation of Ar′(H)Ge=Ge(Ar′)H [Ar′ = 2,6-(2,6-iPr2-C6H3)2-C6H3] 12 [15] (which is also available by method B [16]) and Ar′(H)Ge=Ge(Ar′)R [Ar′ = 2,6-(2,6-iPr2-C6H3)2-C6H3, R = cyclopentyl] 13 [17]. Using method D, several compounds with a Ge–Ge bond were classified as digermenes [18][19][20]. However, given their remarkably long (even longer than many Ge–Ge single bonds) and accordingly quite weak Ge=Ge bonds, this classification is somewhat doubtful.
Below, some recently published representative examples of the stable digermenes are described.
Thus, employing method C, Lee, Sekiguchi and coworkers reported the tetra(silyl)digermene (tBu2MeSi)2Ge=Ge(SiMetBu2)2 14, featuring very bulky substituents, that was readily available in large-scale by the reductive dehalogenation of the (tBu2MeSi)2GeCl2 precursor with potassium graphite [21][22][23]. In the solid state, digermene 14 manifested a quite unusual combination of the structural features, namely a very long [rGe=Ge = 2.346(2) Å] and exceptionally twisted (τ = 52.8°) Ge=Ge double bond, nevertheless featuring practically planar geometry at its sp2-Ge centers (ΣGe = 358.8 and 359.2°) [21][22]. In solution, digermene 14 has a very distinct deep sapphire-blue color, in contrast to all other isolable digermenes that are yellow, orange, or red. Accordingly, its longest wavelength UV band was observed at 618 nm [π(HOMO)–π*(LUMO)], a value that was extraordinarily red-shifted compared to those of other stable digermenes [21][22]. This was caused by the extreme twisting of the Ge=Ge double bond due to the severe steric repulsion of bulky silyl substituents, resulting in rather poor 4pπ(Ge)–4pπ(Ge) orbital overlap, destabilization of the HOMO, and accordingly to the overall decrease in the HOMO–LUMO energy gap. Moreover, exceptional twisting of the Ge=Ge bond in 14 results in its partial breaking and progressively increasing biradical contribution. Digermene 14 does not dissociate in solution into the germylenes (tBu2MeSi)2Ge:, maintaining its structural integrity up to 80 °C, as confirmed by Raman and UV–Vis spectroscopy measurements, as well as trapping reactions study [21][22][23]. Nevertheless, 14 can behave as a germylene source when strongly nucleophilic Lewis bases (isocyanide or ortho-benzoquinone) are applied, forming germylene reactivity products although not involving “free” germylenes into the reaction process [21][22]. CV measurement of digermene 14 in ortho-dichlorobenzene in the presence of [nBu4N][B(C6F5)4] inert electrolyte afforded two reversible redox couples, for both oxidation and reduction processes, with E1/2(ox) = 0.38 V and E1/2(red) = −1.50 V [22]. This implies that both cation-radical [4]+∙ and anion-radical [4]−∙ are persistent under the CV measurement conditions.
Scheschkewitz and coworkers synthesized tetra(silyl)digermene 15 by dimerization of a cyclic germylene–NHC complex (Scheme 3) [24]. The exocyclic Ge=Ge bond in 15 revealed structural features that are typical for digermenes: short bond [rGe=Ge = 2.2944(4) Å], strongly pyramidalized Ge centers [ΣGe = 334.5°], and trans-bending of the silyl-substituents [θ = 37.7°]. Accordingly, digermene 15 was stable both in the solid state and in solution.
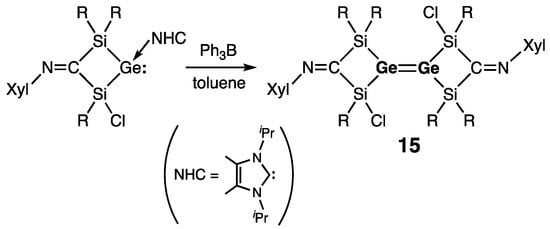
Scheme 3. Synthesis of the tetra(silyl)digermene 15.
The same group also recently reported the first isolable digermenide Tip2Ge=Ge(Tip)Li 16 made by the reduction of Tip2GeCl2 with metallic lithium [25]. In this anionic digermene, the Ge=Ge double bond is short [rGe=Ge = 2.284(6) Å], unremarkably trans-bent (θ = 7.1/12.8°), and twisted (τ = 19.9°). Digermenide 16 can be further functionalized at the anionic Ge site forming novel silyl-substituted digermenes Tip2Ge=Ge(Tip)R (R = SiMe3 [25], SiPhMe2 [25], SiPh3 [26], SiMe2Cl [26], SiMePhCl [26], and SiPh2Cl [26]) and even persistent (acyl)digermenes Tip2Ge=Ge(Tip)[C(O)R] (R = tBu [26], 2-methylbutan-2-yl [26], 1-adamantyl [26]).
Matsuo, Sasamori and coworkers prepared 1,2-di(halo)digermenes Eind(X)Ge=Ge(X)Eind (X = Cl, Br) 17a,b (a: X = Cl, b: X = Br) by the redistribution of stable di(aryl)germylene Eind2Ge: and GeX2∙dioxane complex [27]. Bearing electronegative substituents, 17a,b expectedly revealed strong structural deformation at the Ge=Ge double bond: trans-bending of substituents (θ = 44.3/43.3°) and pyramidalization at the Ge centers (ΣGe = 335.9 and 337.1°). In line with this, the Ge=Ge bond in 17a,b is rather long [rGe=Ge = 2.4119(5) and 2.4145(3) Å], as a manifestation of the weak bonding between Ge atoms, which was translated into the ready dissociation of this bond. Accordingly, in solution, di(halo)digermenes Eind(X)Ge=Ge(X)Eind dissociate to (halo)germylenes Eind(X)Ge:.
Dicationic derivative 18, which can be viewed as a digermene with cationic imidazolium substituents, as reported by Aldridge and coworkers, was synthesized by the reaction of a (chloro)germylene–NHC complex with either Na{B[3,5-(CF3)2-C6H3]4} or Li{Al[OC(CF3)3]4} (Scheme 4) [28]. In 18, the Ge=Ge bond is short [rGe=Ge = 2.300(2) Å], and Ge centers are only insignificantly pyramidalized (ΣGe = 353.1 and 353.6°). Exchanging in 18 NHC substituents for Me4-NHC and boryl substituents for Ar-groups [Ar = 2,6-Mes2-C6H3 (Mes = 2,4,6-Me3-C6H2)], the same group synthesized another dicationic digermene 19 manifesting longer Ge=Ge distance of 2.380(1) Å [29].
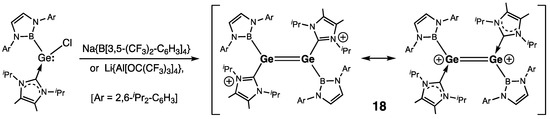
Scheme 4. Synthesis of the dicationic digermene 18.
Very recently, Scheschkewitz and coworkers found that the thermolysis (benzene, 65 °C, 18 h) of the unsymmetrically substituted digermene Tip2Ge=Ge(Tip)[SiR2Dma] (R = Me, Ph; Dma = 2-Me2N-C6H4) produced a mixture of redistribution products, Tip2Ge=GeTip2 and trans-[DmaR2Si](Tip)Ge=Ge(SiR2Dma)Tip 20 [Ge=Ge bond (for R = Me): rGe=Ge = 2.2576(5) Å, θ = 21.5°, τ = 0°] [30]. This approach was then applied toward the development of ADMET (acyclic diene metathesis) polymerization of digermenes. Accordingly, thermolysis (benzene, 65 °C, 48 h) of a diene with terminal digermene fragments linked by a p-phenylene spacer, Tip2Ge=Ge(Tip)–SiMe2–1,4-[2,5-(Me2N)2-C6H2]–SiMe2–Ge(Tip)=GeTip2 21 [Ge=Ge bonds: rGe=Ge = 2.3038(4) Å, θ = 24.9/31.9°, τ = 18.0°], formed ADMET-polyene Tip2Ge{=Ge(Tip)–SiMe2–1,4-[2,5-(Me2N)2-C6H2]–SiMe2–Ge(Tip)=}nGeTip2, with the number-average degree of polymerization being 23, mass-average degree of polymerization being 45, and dispersity index being 1.95.
In comparison to organic alkenes >C=C<, structural deformations of the double bond in digermenes >Ge=Ge< (stretching, trans-bending, and twisting) are even more pronounced compared to those of the corresponding disilenes >Si=Si<. The extent of these structural distortions in digermenes follows some general tendencies: electronegative substituents provoke notable elongation and weakening of the Ge=Ge bond, as well as trans-bending at the doubly bonded Ge centers, whereas electropositive substituents cause shortening and strengthening of the Ge=Ge bond and planarization at the doubly bonded Ge centers. Accordingly, the shortest Ge=Ge bond [rGe=Ge = 2.2576(5) Å] was found in the di(aryl)di(silyl)digermene 20, whereas the longest one [rGe=Ge = 2.5087(7) Å] was detected in the di(aryl)di(bromo)digermene 6, with the exceptionally bulky Bbt substituents. Moreover, in line with what was mentioned above, tetra(silyl)digermene 9 has planar (the least trans-bent) geometry at its sp2-Ge centers (θ = 0.3°), whereas the greatest trans-bending was observed in the di(aryl)digermene 12 (θ = 45.0°). Twisting in digermenes is controlled by the substituent effect, to range from non-twisted Ge=Ge double bonds (τ = 0.0° in tetra(alkyl)digermene 1 and di(aryl)di(silyl)digermene 20) to extraordinarily twisted (τ = 52.8° in tetra(silyl)digermene 14 with very bulky silyl substituents).
Cyclic Digermenes
To date, 22 neutral organogermanium compounds featuring an endocyclic Ge=Ge double bond, 14 three-membered rings, 4 four-membered rings, 3 five-membered rings, and 1 six-membered ring, are reported.
Three-Membered Ring Compounds
There are currently 14 heavy cyclopropene analogues incorporating a Ge=Ge double bond into the three-membered ring: 12 homonuclear cyclotrigermenes cyclo-[Ge3] and 2 heteronuclear 1H-siladigermirenes cyclo-[Si–Ge=Ge]. Unlike their acyclic congeners, all of these cyclic digermenes are synthesized by special methods which are not outlined in Scheme 2.
The first isolable cyclotrigermenes 22a,b were synthesized by Sekiguchi and coworkers by the reaction of tBu3EM (a: E = Si, M = Na; b: E = Ge, M = Li) with GeCl2•diox (Scheme 5) [31]. Remarkably, one-electron oxidation of 22a with [Ph3C]+[BPh4]− produced tris(tri-tert-butylsilyl)cyclotrigermenylium tetraphenylborate [(tBu3Si)3Ge3]+[BPh4]−, as the germanium analogue of aromatic 2π-electron cyclopropenylium ion [32].
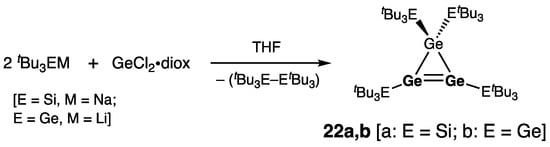
Scheme 5. Synthesis of the first cyclotrigermenes 22a,b.
Unsymmetrically substituted cyclotrigermenes 23a–e (a: R = SitBu3; b: R = GetBu3; c: R = Si(SiMe3)3; d: R = Ge(SiMe3)3; e: R = Mes) were later reported by the same authors prepared by the reaction of tris(tri-tert-butylsilyl)cyclotrigermenylium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate with the alkali metal salts RM (R = silyl, germyl, aryl; M = alkali metal) (Scheme 6) [33]. Likewise, treatment of (tri-tert-butylsilyl)cyclotrigermenylium tetrakis(2,3,5,6-tetrafluorophenyl)borate with potassium halides KX (X = Cl, Br, I) formed halogen-substituted cyclotrigermenes 24a–c (a: X = Cl, b: X = Br; c: X = I) (Scheme 7) [34].
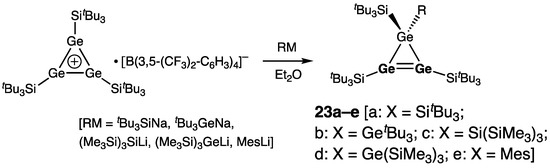
Scheme 6. Synthesis of the unsymmetrically substituted cyclotrigermenes 23a–e.
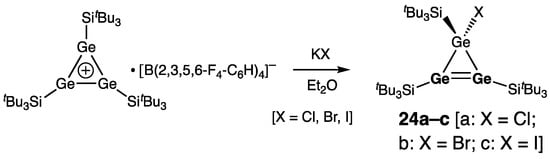
Scheme 7. Synthesis of the halogen-substituted cyclotrigermenes 24a–c.
Lee, Sekiguchi, and coworkers reported novel three-membered ring cyclic digermenes, 1H-siladigermirene 25a and 1H-trigermirene (cyclotrigermene) 25b, synthesized by the reaction of 1,1,2,2-tetra(chloro)digermane R–GeCl2–GeCl2–R (R = SiMetBu2) with 1,1-di(lithio)silane R2SiLi2 or 1,1-di(lithio)germane R2GeLi2, respectively (Scheme 8) [35][36].
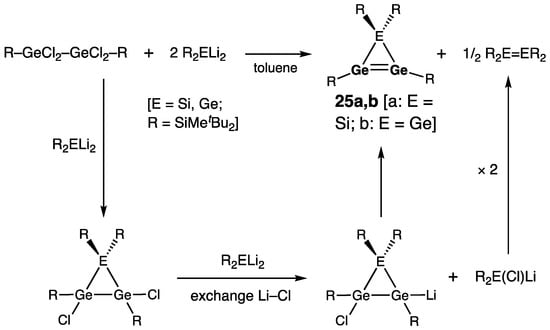
Scheme 8. Synthesis of 1H-siladigermirene 25a and 1H-trigermirene 25b.
In due course, the solid-state thermolysis of the tetra(silyl)digermene (tBu2MeSi)2Ge=Ge(SiMetBu2)2 1427 (170 °C, 1 h, evacuated sealed tube) was found to be an attractive alternative to the above-described preparation of 1H-trigermirene 25b, improving the isolated yield of the latter up to 48% [37].
The alkyl-substituted heavy cyclopropene analogues 26a,b (a: E = Si; b: E = Ge), representing the nearest homologues of the above-described 1H-siladigermirene 25a and 1H-trigermirene 25b, being distinguished from them by only one CH2-unit, were prepared by Lee, Sekiguchi, and coworkers by the reductive dehalogenation of 1,3-di(chloro)cyclobutane derivatives (Scheme 9) [38]. The overall process was proposed to proceed via the transient bicyclo[1.1.0]butane derivatives, rapidly isomerizing at room temperature to the more stable heavy cyclopropenes 26a,b.
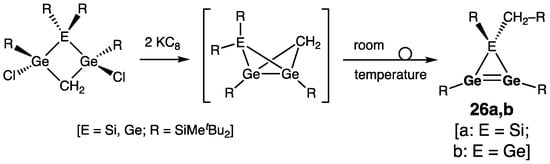
Scheme 9. Synthesis of the alkyl-substituted heavy cyclopropene analogues 26a,b.
In cyclotrigermenes, the Ge=Ge double bonds [rGe=Ge = 2.239(4)–2.2743(8) Å] are typically shorter than those of acyclic digermenes [rGe=Ge = 2.2576(5)–2.5087(7) Å]. The longest Ge=Ge bonds were detected in halogen-substituted cyclotrigermenes 24a–c [rGe=Ge = 2.2721(6)–2.2743(8) Å], and they were realized in terms of πGe=Ge–σ*Ge–X orbitals mixing facilitated by the electronegative halogen atoms X at the sp3-Ge [34]. Usually, cyclotrigermenes exhibited significant twisting about their Ge=Ge double bonds: τ = 8.1–51.0°. Cyclotrigermene 25b with very bulky tBu2MeSi substituents showed a record twisting for the endocyclic Ge=Ge bond in heavy cyclopropenes (τ = 60.5°) [36].
-
Four-Membered Ring Compounds
Four neutral cyclobutene derivatives containing heavy group 14 elements and incorporating Ge=Ge double bond are currently known: one homonuclear (tetragermetene cyclo-[Ge4]) and three heteronuclear (disiladigermetene cyclo-[Si2Ge2], trigermetene cyclo-[Ge3C], and digermetene cyclo-[Ge2C2]).
The very first compound of this type, namely disiladigermetene 27, was reported by Lee, Sekiguchi, and coworkers, formed by the unexpected ring expansion of either 3H- or 1H-disilagermirenes [39] with GeCl2•diox (Scheme 10) [40]. The Ge=Ge double bond in 27 [rGe=Ge = 2.2911(4) Å] is one of the longest reported for the cyclic digermenes, whereas the endocyclic Si–Ge bonds are sizably shortened, and the exocyclic Si–Cl bonds are elongated. This was explained by the important extent of πGe=Ge–σ*Si–Cl negative hyperconjugation promoted by the presence of electronegative chlorine atoms and folding of the Si2Ge2-ring (folding angle = 28.3°). The geometry around the sp2-Ge atoms in 27 is only slightly pyramidal: ΣGe = 357.2°/358.6°.
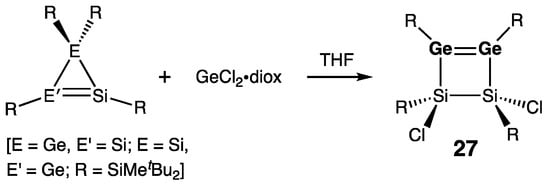
Scheme 10. Synthesis of disiladigermetene 27.
Likewise, Lee, Sekiguchi, and coworkers found that the similar reaction of 1H-trigermirene 25b with the GeCl2•diox yielded the first (and still the only known) homonuclear heavy cyclobutene analogue, tetragermetene 28 (Scheme 11) [41]. Structurally, tetragermetene 28 is similar to the above-described disiladigermetene 27: rGe=Ge = 2.2993(5) Å (28) vs. 2.2911(4) (27), and ΣGe = 354.4°/356.4° (28) vs. 357.2°/358.6° (27).
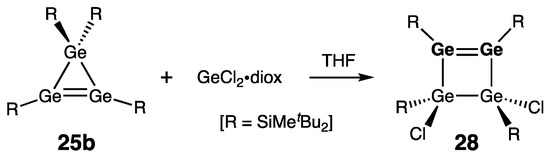
Scheme 11. Synthesis of tetragermetene 28.
Weidenbruch and coworkers developed an alternative approach toward heavy cyclobutene analogues by reacting tetra(germa)buta-1,3-diene Tip2Ge=Ge(Tip)–Ge(Tip)=GeTip2 with 2-methoxyphenyl isocyanide to produce 1,2,3-trigermet-1-ene 29, featuring a germanium–germanium double bond within the Ge3C-skeleton (Scheme 12) [42]. The Ge=Ge bond in 29 was marginally shorter that those in disiladigermetene 27 and tetragermetene 28: 2.2808(7) Å vs. 2.2911(4) and 2.2993(5) Å, respectively.
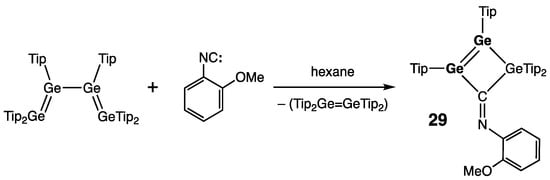
Scheme 12. Synthesis of 1,2,3-trigermet-1-ene 29.
The four-membered ring 1,2-digermet-1-ene 30 with an endocyclic Ge=Ge double bond was prepared by Sasamori, Tokitoh, and coworkers by [2 + 2] cycloaddition of the stable digermyne BbtGe≡GeBbt (Bbt = 2,6-[(Me3Si)2CH]2-4-[(Me3Si)3C]-C6H2) and ethylene, (Scheme 13) [43]. In 30, its remarkable ring strain and trans-bending of the substituents (θ = 39.5 and 39.7°) caused elongation and weakening of the Ge=Ge double bond [rGe=Ge = 2.4132(5) Å].
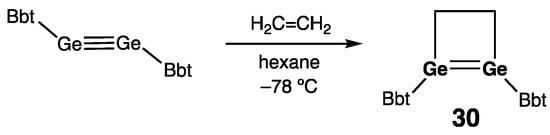
Scheme 13. Synthesis of 1,2-digermet-1-ene 30.
Except for the above-described neutral heavy cyclobutene analogues 27–30, there are several four-membered ring compounds where the Ge=Ge double bond is a part of the tri(germa)allylic system (cationic, radical, or anionic). Thus, Weidenbruch and coworkers reported the tetra(germa)cyclobutenyl anion 31 unexpectedly produced by exhaustive reduction of Tip2Ge=GeTip2 with excess of lithium (Scheme 14) [44]. In 31, the Ge4-ring is practically planar (sum of the internal bond angles = 360°), and within the tri(germa)allylic system, the Ge–Ge bonds length of 2.3679(6) Å is just in-between those of the typical Ge–Ge single and Ge=Ge double bonds.
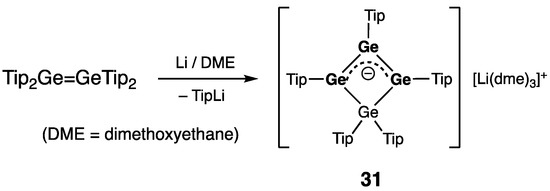
Scheme 14. Synthesis of tetra(germa)cyclobutenyl anion 31.
Cyclic compound 31 can be directly compared with acyclic tri(germa)allyl anion derivative 32, reported by Power and coworkers and prepared by the reductive ring opening of a cyclotrigermenyl radical 33 with KC8 (Scheme 15) [45]. The germanium–germanium bond length [2.422(2) Å] in 32 is also intermediate between those of the Ge–Ge single and Ge=Ge double bonds, and the Ge3-array is characterized by a wide Ge–Ge–Ge bond angle [159.19(10)°].
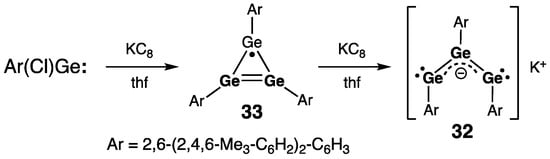
Scheme 15. Synthesis of tri(germa)allyl anion 32.
Most recently, Lee, Sekiguchi, and coworkers synthesized silatri(germa)cyclobutenylium ion derivative 34 by the oxidative demethylation of the cyclotrigermene 25b [35] with [Et3Si]+∙[B(C6F5)4]− (Scheme 16) [46]. In 34, the SiGe3 four-membered ring is strongly folded (folding angle 40.4°), thus enabling Ge2∙∙∙Ge3 through-space orbital interaction manifested in the short transannular Ge2–Ge3 distance of only 2.9346(3) Å. Overall, the structural peculiarities of 34 (ring folding and short transannular distance) testify to the important extent of its homoaromaticity. Accordingly, 34 is to be classified as a germanium analogue of the cyclobutenylium ion, i.e., a homo-tri(germa)cyclopropenylium ion. The homoaromaticity of 34 was further confirmed by the calculation of the nucleus-independent chemical shift (NICS) at 1 Å above the Ge3-ring center, which was diagnostically negative (−17.3) as a manifestation of the diatropic ring current. The “homoaromatization energy” of 34, calculated as the barrier to inversion of the Ge3Si-ring (through the planar allylic-type cationic transition state lacking homoaromatic stabilization), was exceedingly low, i.e., only 3.7 kcal/mol [46]. In accordance with its homoaromaticity, 34 showed practically no alteration in the Ge–Ge bond lengths of its Ge3-fragment [rGe=Ge = 2.3327(3) Å (Ge1–Ge2) and 2.3400(3) Å (Ge1–Ge3)], and it showed essentially planar geometry at all skeletal Ge atoms [ΣGe = 360.0° (Ge1), 358.7° (Ge2), and 357.5° (Ge3)].
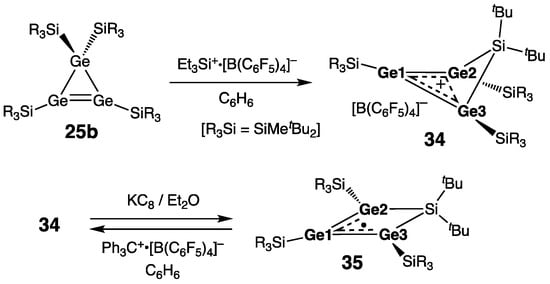
Scheme 16. Synthesis of silatri(germa)cyclobutenylium ion derivative 34.
Upon the one-electron reduction of 34 with KC8, a free-radical species, namely silatri(germa)cyclobutenyl radical 35, was cleanly formed (Scheme 16) [46]. The homoaromaticity of 34 is completely lost upon its reduction, which was seen in the remarkable flattening of the SiGe3-ring in 35 (folding angle was reduced from 40.4° in the starting 34 to only 6.9° in the resulting 35) and great elongation of the Ge2∙∙∙Ge3 transannular distance [3.3315(4) Å in 35, or 14% elongation compared with 34]. Thus, 35 is to be classified as the allylic free radical, featuring the unpaired electron delocalized over the two terminal Ge atoms, [∙Ge2–Ge1=Ge3 ↔ Ge2=Ge1–Ge3∙]. Accordingly, the lengths of both Ge–Ge bonds in 35 are intermediate between those for the single and double bonds [2.3458(3) Å (Ge1–Ge2) and 2.3206(3) Å (Ge1–Ge3)], and all Ge atoms maintain essential planarity of their geometry upon reduction [ΣGe = 358.6° (Ge1), 355.2° (Ge2), and 359.9° (Ge3)]. The EPR resonance g-value of 35 (2.0227) is in the range typical for the silyl-substituted Ge-centered free radicals. In the Ge3-unit of 35, the hyperfine coupling constants (hfcc) for the terminal Ge nuclei are markedly greater than that for the central Ge nucleus: a(73Ge2) [or a(73Ge3)] = 1.54 mT [or 1.44 mT] vs. a(73Ge1) = 0.59 mT. This observation agrees well with the allylic radical formulation of 35, in which the odd electron is mostly localized at the Ge2 and Ge3 termini. Given that the small values of the 73Ge hfcc in 35 imply the location of its unpaired electron in the orbital of π-symmetry, 35 should be categorized as a π-radical. The allylic free radical 35 can be compared with the cyclotrigermenyl radical 33 prepared by Power and coworkers by the stoichiometric reduction of the aryl(chloro)germylene Ar(Cl)Ge: [Ar = 2,6-(2,4,6-Me3-C6H2)2-C6H3] with KC8 (Scheme 15) [45]. In the cyclotrigermenyl radical 33, the average Ge–Ge bond distance within the Ge3-ring of 2.35(7) Å is comparable with those of the silatri(germa)cyclobutenyl radical 35 [2.3458(3) Å and 2.3206(3) Å]. The EPR characteristics of 33 are also comparable to those of 35: g = 2.0069 [2.0227 in 35], and a(73Ge) = 1.6 mT [a(73Ge2) or a(73Ge3) = 1.54 or 1.44 mT in 35].
-
Five-Membered Ring Compounds
Three compounds of this type with the Ge=Ge double bond within the five-membered ring skeleton have been reported to date. Weidenbruch and coworkers synthesized the first two representatives by [1 + 4] cycloaddition of sulfur/or selenium to the tetra(germa)buta-1,3-diene Tip2Ge=Ge(Tip)–Ge(Tip)=GeTip2, forming thia- or selena-tetra(germa)cyclopentenes 36a,b (Scheme 17) [42][47]. The structural features of heavy cyclopentenes 36a,b are typical for the cyclic digermenes: rGe=Ge = 2.2841(5) Å (36a) and 2.2975(5) Å (36b), and ΣGe = 348.4°/351.4° (36a) and 349.2°/349.9° (36b); and Ge4E rings are practically planar (sums of the internal bond angles are 539.5° (36a) and 537.7° (36b)).
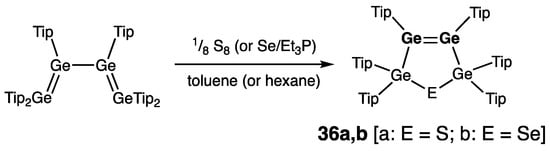
Scheme 17. Synthesis of heavy cyclopentene analogues 36a,b with endocyclic Ge=Ge double bonds.
The latest example of the five-membered ring cyclic digermene, namely bicyclic digermene 37 with a bridging Ge=Ge double bond, was prepared by Marschner and coworkers; it was unexpectedly formed by the reaction of 1,3-di(potassio)trisilane with GeBr2∙dioxane and PEt3 (Scheme 18) [48]. In 37, the Ge=Ge double bond revealed the structural parameters that are characteristic for the cyclic digermenes: shortened bond [rGe=Ge = 2.2663(9) Å], negligible trans-bending of substituents [θ = 2.5°], and moderate twisting [τ = 16.2°]. Upon its electrochemical (CV) reduction, digermene 37 showed two reversible reduction waves corresponding to generation of anion-radical and dianion.
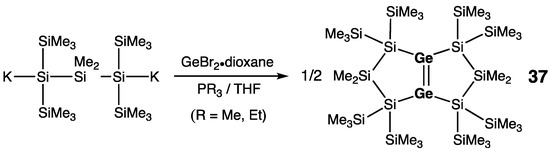
Scheme 18. Synthesis of bicyclic digermene 37.
-
Six-Membered Ring Compounds
Only one “heavy cyclohexene” derivative has been reported to date. Marschner and coworkers prepared this tetracyclic digermene 38 with an endocyclic Ge=Ge double bond bridging polycyclic scaffold (Scheme 19) [48]. Digermene 38 was available via the synthetic strategy applied for the preparation of the above-described bicyclic digermene 37, namely by the reaction of 1,4-di(potassio)cyclohexasilane with GeBr2∙dioxane in the presence of PEt3. The metric parameters of 38 are comparable to those of 37: rGe=Ge = 2.2896(6) Å, θ = 2.1/8.3°, and τ = 5.2°.
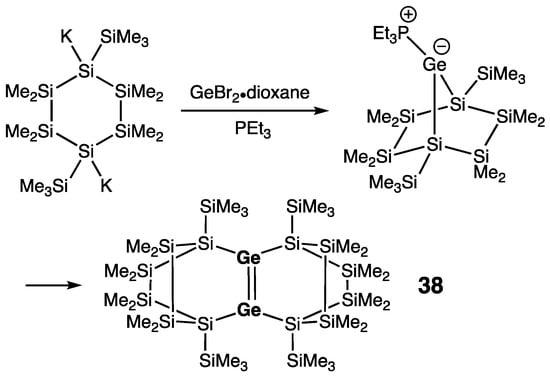
Scheme 19. Synthesis of tetracyclic digermene 38.
2.2. Heteronuclear Derivatives
2.2.1. Germenes >Ge=C<
The first isolable germenes were independently synthesized in 1987 by the groups of Berndt and Escudié. Berndt and coworkers prepared germenes 39a,b by the coupling of electrophilic “cryptocarbene” and bis(amino)germylenes (Scheme 20) [49]. The formulation of 39a,b as germenes Ge=C was supported by the observation of low-field resonance for their doubly bonded C atoms [δ(13C) = 115.0 ppm (39a) and 93.2 ppm (39b)] and rather short Ge=C bond [rGe=C = 1.827(4) Å (39a)]. The Ge=C double bond in 39a was significantly twisted [τ = 36°], although the sp2-Ge center featured a planar geometry [ΣGe = 359.9°]. The spectroscopic and crystallographic data of 39 are indicative of the substantial contribution of the ylide resonance structure with a positive charge on the Ge atom and a negative charge delocalized over the B–C–B allylic fragment of the B2C2 four-membered ring (Scheme 20) [49]. This was exemplified by the shielding of the B atoms [δ(11B) = 66.0 ppm (39a) and 65.0 ppm (39b)] and shortening of the cyclic sp2-C–B bonds in 39a [rC–B = 1.534(7)/1.523(6) Å].
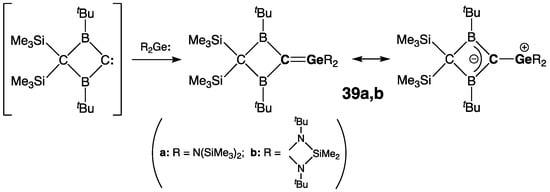
Scheme 20. Synthesis of stable germenes 39a,b.
Escudié and coworkers applied another approach toward their stable dimesityl(fluorenylidene)germene 40: dehydrofluorination of the (fluorenyl)fluorogermane percursor by its lithiation with tBuLi with subsequent elimination of LiF from the intermediate lithium salt (Scheme 21, R = R′ = Mes) [50]. In 40, the Ge=C double bond of 1.803(4) Å (mean value for the two crystallographically independent molecules) was notably shorter than the typical Ge–C single bonds and even shorter than that in 39a, being practically undistorted [ΣGe = ΣC = 360.0°, τ = 6°] [51]. Except for the steric protection of the Ge=C bond by the bulky Mes groups, extra stabilization in 40 results from the contribution of the charge-separated Geδ+=Cδ− resonance form enabled by the effective aromatic delocalization of the negative charge over the cyclopentadiene fragment of the fluorenylidene group.
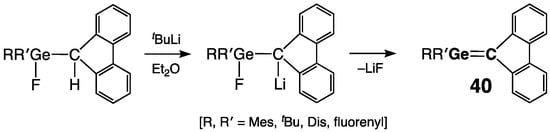
Scheme 21. Synthesis of stable germene 40.
Applying the same synthetic protocol, Escudié and coworkers prepared other stable germenes 40, as outlined in Scheme 21: R = R′ =Dis [52]; R = Dis, R′ = Mes [52]; R = R′ = fluorenyl [53]; and R = t-Bu, R′ = fluorenyl [53]. They also reported generation of the halogen-substituted germenes (Me5C5)(X)Ge=CR2 [CR2 = fluorenylidene] 40 (Scheme 21: R = C5Me5; R′ = F, Cl) by β-elimination of Me3SiF from the (Me5C5)(X)FGe–(Me3Si)CR2 precursors [54]. Lacking resonance stabilization, dimesityl(neopentyl)germene Mes2Ge=CH–CH2tBu 41 was synthesized by Couret and coworkers via the addition of tBuLi to Mes2(F)Ge–CH=CH2, followed by β-elimination of LiF from the intermediate lithium salt Mes2(F)Ge–CH(Li)–CH2tBu [55].
A series of stable germenes 42–44, prepared from a germylene Ar2Ge: (Ar = 2-tBu-4,5,6-Me3-C6H), were reported by Weidenbruch and coworkers (Scheme 22) [7][56][57][58][59]. The germene 42, similar to Berndt’s germene 39a, disclosed a short [rGe=C = 1.845(10) Å] and remarkably twisted [τ = 33°] Ge=C bond, and planar geometry at both sp2-Ge and sp2-C atoms [ΣGe = ΣC = 359.9°] [56]. The reaction of two equivalents of germylene Ar2Ge: with two equivalents of phosphaalkyne tBu–C≡P unexpectedly produced a germene 43 with an exocyclic Ge=C double bond [rGe=C = 1.833(4) Å] (Scheme 22) [57]. A series of bis(germenes) connected via acetylene spacer, 44a–c, were synthesized by the double [1 + 2] cycloaddition of germylenes Ar2Ge: to the C≡C bonds of bis(alkynes), followed by the ring-opening isomerization of the transient bis(germirenes) (Scheme 22) [7][58][59]. The length of the Ge=C double bond in 44a–c was in the range expected for germenes: 1.819(6) Å in 44a [7], 1.819(2) Å in 44b [58], and 1. 840(4) Å in 44c [59]. The observable conjugation between the terminal Ge=C bonds and the central C≡C bond in 44a–c was seen in the diagnostic bathochromic shift of their longest wavelength UV absorptions [500 nm (44a) [7], 518 nm (44b) [58], and 595 nm (44c) [59], as well as in the shortening of the central C–C bonds in the Ge=C–C≡C fragments.
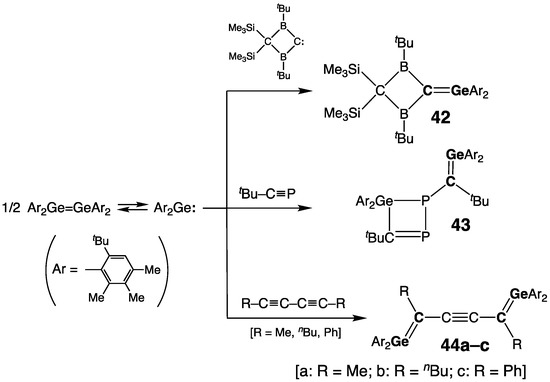
Scheme 22. Synthesis of germenes 42–44.
Two stable germenes, exemplifying the shortest and the longest extremes for the range of Ge=C bond lengths, are of particular interest. The germene with the shortest Ge=C double bond, 45, was prepared by Okazaki and coworkers by the reaction of di(aryl)germylene Tbt(Tip)Ge: (generated by the reduction of di(bromo)germane Tbt(Tip)GeBr2 with lithium naphthalenide) with CS2 (Scheme 23) [60]. The length of the Ge=C double bond in 45 was only 1.771(16) Å, a value which was substantially smaller than those of Berndt’s germene 39a (1.827(4) Å) [49] and Escudié’s germene 40 (1.803(4) Å) [51]. Furthermore, the Ge=C bond in 45 showed no signs of structural distortion, exhibiting a trigonal-planar geometry at both sp2-Ge and sp2-C centers [ΣGe = 359.7°, ΣC = 360.0°] and a negligible twisting [τ = 4°].
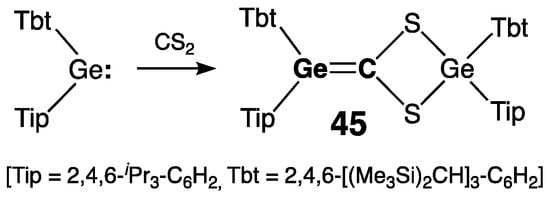
Scheme 23. Synthesis of the germene 45 with the shortest Ge=C double bond.
The germene 46 with the longest ever reported Ge=C bond [rGe=C = 1.895(3) Å] was reported by Lee, Sekiguchi, and coworkers in 2002 [61]. This endocyclic Ge=C bond was embedded into the norbornene bicyclic skeleton of 46, which was unexpectedly formed by the reaction of 2,4-disila-1-germatricyclo[2.1.0.02,5]pentane cage 47 with benzaldehyde (Scheme 24). The major factor responsible for such extraordinary stretching of the germanium–carbon bond is most likely a steric repulsion between the bulky substituents. Nevertheless, despite this exceptional steric crowding around the double bond, the Ge=C bond in 46 revealed practically no structural deformations, neither pyramidalization at the doubly bonded Ge and C atoms nor twisting of the Ge=C bond: ΣGe = 359.8°/ΣC = 358.9°, τ = 7.1°.
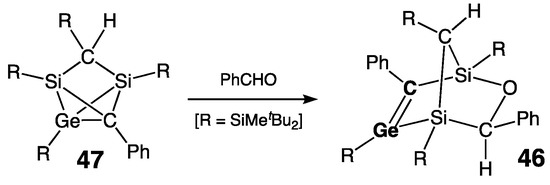
Scheme 24. Synthesis of the germene 46 with the longest Ge=C double bond.
The latest representative of isolable germenes, namely the first stable Brook-type germene 48 with an exocyclic Ge=C double bond, was prepared by Haas and coworkers [62]. This O-silylated germene 48 was prepared by the reaction of Me3SiCl with the stable germenolate 49, predominantly exhibiting acyl germyl anion character with a negatively charged Ge, Ge–C single bond, and C=O double bond (Scheme 25). Spectroscopic and structural characteristics of 48 are typical for the isolable germenes: strongly deshielded sp2-C atom [δ(13C) = 210.0 ppm]; short [rGe=C = 1.835(2) Å], pyramidalized at Ge [ΣGe = 351.7° (cf.: ΣC = 360.0°)], and twisted (torsional angles: O–C=Ge–Si = 10.0° and CMes–C=Ge–Si = 18.1°) Ge=C double bond.
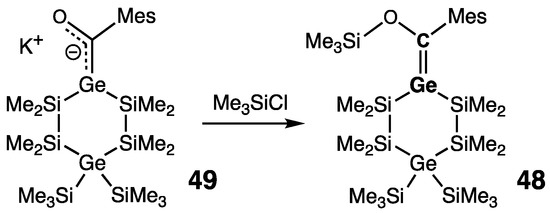
Scheme 25. Synthesis of the Brook-type germene 48.
2.2.2. Silagermenes >Si=Ge<
The first ever reported silagermene, metastable Mes2Si=GeMes2, was thermally or photochemically generated from hexa(mesityl)siladigermirane precursor, cyclo-(Mes2Si–GeMes2–GeMes2), by Baines and coworker [63]. Stable in solution only below −70 °C, Mes2Si=GeMes2 was identified by low-temperature NMR and UV measurements (δ(29Si) = 80.6 ppm and 414 nm (πSi=Ge–π*Si=Ge), respectively), as well as by the quenching with methanol forming 1,2-addition product Mes2Ge(H)–Si(OMe)Mes2. Above −70 °C, Mes2Si=GeMes2 underwent irreversible 1,2-migration of Mes-group from Ge to Si generating transient mesityl(silyl)germylene Mes(Mes3Si)Ge:.
The first isolable silagermene, 1H-disilagermirene 50, with a cyclic Si=Ge double bond within the three-membered ring GeSi2-skeleton, was reported by Lee, Sekiguchi, and coworkers in 2000 [39]. Moreover, 50 was quantitatively available from 3H-disilagermirene precursor 51 by either thermal or photochemical isomerization, driven by the thermodynamic preference for the silagermene >Si=Ge< over the disilene >Si=Si< (Scheme 26). In 50, the sp2-Si center was expectedly deshielded [δ(29Si) = 100.7 ppm], and the Si=Ge double bond was notably twisted (torsional angle R–Si=Ge–R = 40.3°).
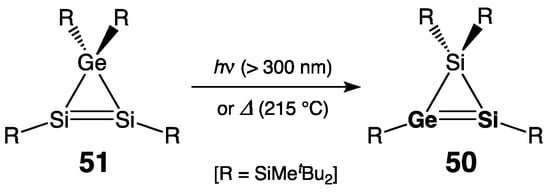
Scheme 26. Synthesis of the first isolable silagermene 50.
Lee, Sekiguchi, and coworkers later developed another approach toward isolable silagermenes by the thermal isomerization of disilagermabicyclo[1.1.0]butane 52, forming novel 1H-disilagermirene 53, as the nearest homologue of 50, distinguished from the latter by only a CH2 unit (Scheme 27) [38]. Compared to 50, 53 revealed a more deshielded sp2-Si center: δ(29Si) = 100.7 ppm and 126.6 ppm, respectively. Bearing RCH2-substituent at the sp3-Si atom, 1H-disilagermirene 53 is the first representative of the alkyl-substituted cyclopropene analogues of the heavy group 14 elements.
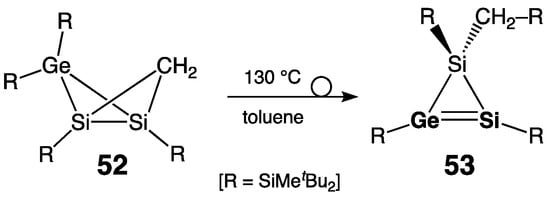
Scheme 27. Synthesis of alkyl-substituted 1H-disilagermirene 53.
The other three silagermenes, namely (tBu2MeSi)2Si=GeMes2 54 [64], Mes2Si=Ge(SiMetBu2)2 55 [65], and (tBu3Si)2Si=GeMes2 56 [66], were available by the reaction of 1,1-di(lithio)derivatives (tBu2MeSi)2SiLi2, (tBu2MeSi)2GeLi2, and (tBu3Si)2SiLi2 with di(aryl)dichlorides Mes2GeCl2, Mes2SiCl2, and Mes2GeCl2, respectively. The silagermene 55 showed a characteristic low-field signal of its sp2-Si atom [δ(29Si) = 146.9 ppm], whereas silagermenes 54 and 56 exhibited unusually shielded sp2-Si centers [δ(29Si) = 22.4 ppm and 18.7 ppm, respectively]. This striking spectroscopic distinction resulted from the differing substitution pattern in 54 and 56 (donating groups on Si and withdrawing groups on Ge), which alters the natural Siδ+=Geδ− bond polarity (like in 55) into a reversed one Siδ−=Geδ+ (like in 54 and 56). Only silagermene 56 was crystallographically identified as featuring peculiar properties caused by the exceptional steric crowding of its tBu3Si-substituents: rather long [rSi=Ge = 2.2769(8) Å] and markedly twisted [τ = 24.7°] Si=Ge double bond with planar geometry at the sp2-Si and sp2-Ge atoms [ΣSi = ΣGe = 360°]. Upon thermolysis at 100 °C, 56 quantitatively isomerized to a symmetrically substituted silagermene (E)-[tBu3Si(Mes)Si=Ge(Mes)SitBu3] [66].
Iwamoto, Kira, and coworkers reported identically substituted silagermene, (tBuMe2Si)2Si=Ge(SiMe2tBu)2 57, prepared by the reductive debromination of 1,2-di(bromo)silagermane Br(tBuMe2Si)2Si–Ge(SiMe2tBu)2Br with sodium in toluene [12]. As with other stable silagermenes, 57 manifested a low-field resonance for its sp2-Si center [δ(29Si) = 144.0 ppm], a short (compared to other silagermenes) [rSi=Ge = 2.2208(4) Å] and almost undistorted [τ = 7.5°] Si=Ge double bond, and practically planar geometry at the doubly bonded Si and Ge atoms [θ = 0.6°].
The ”inorganic ethylene” donor–acceptor push–pull complex, {[IPr:]→SiH2–H2Ge:→[W(CO)5]} 58, in which the “SiH2–H2Ge” fragment is complexed with both Lewis base [IPr (IPr = 1,3-bis(2,6-diisopropylphenyl)-2H-imidazol-2-ylidene)] and Lewis acid [W(CO)5], was reported by Rivard and coworkers [67]. Moreover, 58 was synthesized by the reaction of the di(chloro)silylene complex [IPr∙SiCl2] [68] and [Cl2Ge∙W(CO)5], giving at first [IPr∙SiCl2–Cl2Ge∙W(CO)5] a complex that was subsequently reduced with LiAlH4 to form the final complex, {[IPr:]→SiH2–H2Ge:→[W(CO)5]} 58. Given the strongly shielded Si nucleus [δ(29Si) = –71.9 ppm (triplet, 1JSi–H = 192.2 Hz)] and very long Si–Ge bond of 2.3717(14) Å, the central Si–Ge interaction in 58 is best classified as a single, rather than a double, bond. This was supported by the computations on the model compound (Me in place of the real 2,6-iPr2-C6H3 substituents on N atoms in IPr ligand; B3LYP/cc-pVDZ-pp), which yielded WBISi–Ge value of only 0.88, thus suggesting Si–Ge single-type bonding.
The latest example of the stable silagermenes was very recently reported by Scheschkewitz and coworkers. Their cyclic potassium silagermenide 59, representing a Si=Ge analogue of a vinyl anion, was synthesized by the reduction of a germylene–NHC complex with KC8 (Scheme 28) [69]. The spectroscopic and structural features of 59 conform well to those of other stable silagermenes: characteristically deshielded sp2-Si center [δ(29Si) = 142.9 ppm (in C6D6) and 138.5 ppm (in thf-d8)] and short Si=Ge bond [rSi=Ge = 2.2590(3) Å]. Moreover, the GeSi2C-ring in 59 is nearly planar, with a negligible folding of 1.9°. According to the crystallographic and UV-spectroscopic data, the π-conjugative interaction between the endocyclic Si=Ge double bond and exocyclic C=N double bond in 59 is notable with the calculated value for the πSi=Ge–π*C=N interaction energy of 23.6 kcal mol−1. Silagermenide 59 can be functionalized at the anionic Ge with the appropriate electrophiles to form novel neutral Ge-substituted silagermenes 60a,b (Scheme 28) [69]. Similar to the starting silagermene 59, both (silyl)silagermene 60a [E = SiPh3] and (phosphanyl)silagermene 60b [E = P(NiPr2)2] showed low-field resonances for their sp2-Si atoms [δ(29Si) = 136.6 ppm (singlet) and 104.5 ppm (doublet, 2JSi–P = 9.8 Hz), respectively], and short Si=Ge bonds [rSi=Ge = 2.2020(2) and 2.2252(4) Å, respectively]. The GeSi2C-ring in 60a is practically planar (folding angle = 0.2°), and the geometry around the Ge atom is slightly pyramidal (ΣGe = 357.3°).
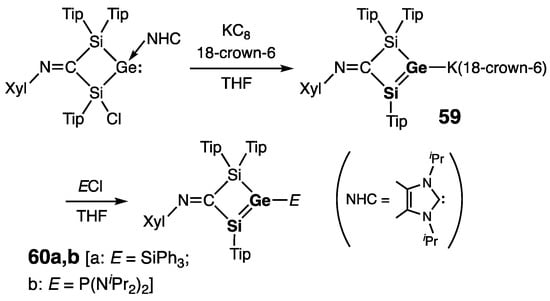
Scheme 28. Synthesis of anionic silagermenide 59 and neutral silagermenes 60a,b.
Silagermenes, in which the Si=Ge double bond is a part of the 1,3-diene (>Si=Ge–C=C<), allene (>Si=Ge=Si</>Ge=Si=Ge<), or vinylidene (>Si=Ge:) system, are discussed separately in Section 4.2.1, Section 5.1, and Section 6.2.1, respectively.
2.2.3. Germastannenes >Ge=Sn<
The first reported transient germastannene, [Mes2Ge=SnTip2], was generated via the dehydrofluorination of Mes2(H)Ge–Sn(F)Tip2 by tBuLi by Escudié and coworkers [70]. Decomposing at room temperature, this germastannene was proved to be as such on the basis of its low-field 119Sn NMR resonance [360.0 ppm, −20 °C] and trapping reactions with MeOH and PhCHO.
As for the stable germastannenes, four of them were reported in 2003/2004. The first three were synthesized by the research groups of Weidenbruch and Sekiguchi in 2003. Thus, Weidenbruch and coworkers prepared the first structurally authenticated germastannene, Tip2Ge=SnTip2 61, by a one-pot low temperature reaction of TipMgBr, GeCl2•diox and SnCl2 [71]. Expectedly, germastannene 61 revealed low-field resonance of its sp2-Sn center [δ(119Sn) = 268.0 ppm], short Ge=Sn bond [rGe=Sn = 2.5065(5) Å] that was substantially shorter than the typical Ge–Sn single bonds, and remarkable trans-bending of the substituents [θ = 30.2° (at Ge) and 43.3° (at Sn)]. Being stable in the solid form, 61 decomposed in solution via dissociation of the Ge=Sn double bond to give a cyclotristannane cyclo-(Tip2Sn)3 and a digermene Tip2Ge=GeTip2.
Another stable germastannene, unsymmetrically substituted (tBu2MeSi)2Ge=SnTip2 62, was synthesized in 2003 by Sekiguchi and coworkers by the coupling of 1,1-di(lithio)germane (tBu2MeSi)2GeLi2 and di(aryl)di(chloro)stannane Tip2SnCl2 (Scheme 29) [72]. In the absence of X-ray data, 62 was identified by its characteristic low-field tin resonance [δ(119Sn) = 525.1 ppm]. Upon thermolysis at 50 °C, 62 quantitatively isomerized to a symmetrically substituted germastannene, (E)-[Tip(tBu2MeSi)Ge=Sn(SiMetBu2)Tip] 63 (Scheme 29) [72]. A concerted isomerization pathway involving simultaneous 1,2-migration of the silyl and aryl groups in the starting germastannene 62 was proposed, based on the negative value of the activation entropy (ΔS‡ = –12.0 cal/K·mol). The doubly bonded Sn atom in isomeric 63 was more shielded than that in the starting 62, 373.4 ppm vs. 525.1 ppm, as a result of their different substitution patterns. The preliminary crystallographic data of 63 revealed the trans-bending of substituents at the Ge=Sn bond with θ angles of ca. 28.0°.
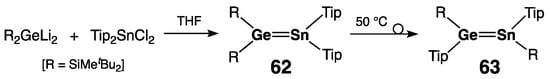
Scheme 29. Synthesis of the unsymmetrically substituted germastannene 62 and its thermal isomerization to the symmetrically substituted germastannene 63.
The only currently known cyclic germastannene, 3Δ-1,2,3,4-disilagermastannetene 64, featuring an endocyclic Ge=Sn double bond incorporated into the four-membered ring skeleton, was synthesized by Lee, Sekiguchi and coworkers in 2004 [73]. Moreover, 64 was readily available by the ring expansion of either 3H- or 1H-disilagermirenes [39] with SnCl2•diox (Scheme 30). As is typical for germastannenes, the doubly bonded Sn atom was diagnostically deshielded [δ(119Sn) = 439.3 ppm]. In a sharp contrast to the acyclic tetra(aryl)germastannenes Mes2Ge=SnTip2 [70] and Tip2Ge=SnTip2 61 [71], cyclic tetra(silyl)germastannene 64 was indefinitely stable both in the solid state and in solution, showing no signs of dissociation of its >Ge=Sn< double bond into the germylene >Ge: and stannylene >Sn:. The unexpected high thermal stability of 64 was assigned to the influence of its σ-donating silyl substituents, further enhanced by the proposed πGe=Sn→σ*Si–Cl orbital mixing lowering the πGe=Sn-orbital energy level and thus stabilizing the HOMO of the molecule [73].
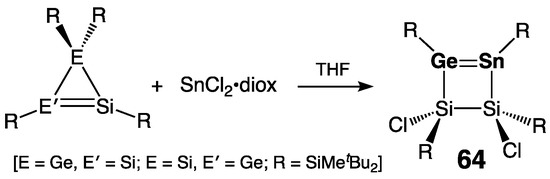
Scheme 30. Synthesis of cyclic germastannene 64.
Since 2004, no stable germastannenes >Ge=Sn< were reported.
3. Heavy Analogues of Alkynes
3.1. Homonuclear Derivatives
Digermynes –Ge≡Ge–
The first digermyne Ar′–Ge≡Ge–Ar′ [Ar′ = 2,6-(2,6-iPr2-C6H3)2-C6H3] 65 was synthesized and structurally characterized in 2002 by Power and coworkers via the reduction of the (chloro)germylene Ar′(Cl)Ge: with potassium [74]. The CAr′–Ge–Ge–CAr′ core in digermyne 65 was planar and trans-bent with the Ge–Ge–CAr′ bond angle of 128.7°. The Ge–Ge bond was rather short [rGe≡Ge = 2.2850(6) Å], which is indicative of its considerable multiply bonded character. On the other hand, accumulation of the lone pair electron density at the Ge atoms results in the trans-bending of their substituents, and consequently in the decrease (compared to the ideal triple bonding) of the Ge–Ge bond order and bond strength.
Following the report, Power’s group prepared a series of stable terphenyl-substituted digermynes, Ar*–Ge≡Ge–Ar* [Ar* = 2,6-(2,4,6-iPr3-C6H2)2-C6H3] 66 [75][76], Ar″–Ge≡Ge–Ar″ [Ar″ = 4-Cl-{2,6-(2,6-iPr2-C6H3)2}-C6H2] 67 [77], Ar‴–Ge≡Ge–Ar‴ [Ar‴ = 4-Me3Si-{2,6-(2,6-iPr2-C6H3)2}-C6H2] 68 [77], and Ar**–Ge≡Ge–Ar** [Ar** = 3,5-iPr2-{2,6-(2,4,6-iPr3-C6H2)2}-C6H] 69 [77], which were uniformly prepared by the reduction of the corresponding aryl(chloro)germylenes with K or KC8. The digermyne 66 was characterized by NMR spectroscopy, elemental analysis, and its reaction with 2,3-dimethyl-1,3-butadiene [75][76]. All structurally characterized digermynes 67–69 displayed features similar to those of the first digermyne 65, namely planar [CAr–Ge–Ge–CAr torsional angles = 180° (for 67 and 68) and 165.8° (for 69)] and trans-bent [trans-bent Ge–Ge–CAr angles = 124.2° (for 67), 128.4° (for 68), and 136.1° (for very crowded 69)] CAr–Ge–Ge–CAr core arrays [74][77]. The Ge–Ge bonds in digermynes 67–69 were short [rGe≡Ge = 2.3071(3) Å (for 67), 2.2438(8) Å (for 68), 2.2125(13) Å (for 69)], being comparable to those in digermenes >Ge=Ge<. The different Ge–Ge bond lengths in digermynes 65 and 67–69 can be understood in terms of the substituents’ electronic effects. Thus, digermynes 68 and 69 with the shortest Ge–Ge bonds feature electron donating alkyl or silyl substituents on the central aryl rings, whereas digermyne 65 (which has no such electron donating groups) and digermyne 67 (with electron withdrawing Cl substituents) have notably longer Ge–Ge bonds. Given that the electron donating groups typically reduce the extent of the trans-bending, whereas electron withdrawing groups having the opposite effect, it is reasonable that a smaller bending leads to a higher bond order and a shorter Ge–Ge bond, and vice versa.
Jones and coworkers reported bis(amido)digermyne R–Ge≡Ge–R {R = N(SiiPr3)[2,6-(CHPh2)2-4-iPr-C6H2]} 70 prepared by the reduction of the germylene R(Cl)Ge: with [(MesNacnac)Mg]2 [18]. However, given the Ge–Ge bond distance of 2.3568(3) Å in 70 that is markedly longer than those of Power’s di(aryl)digermynes [74][77], it is better formulated as a digermene (WBI = 1.75) [18], rather than a digermyne.
By contrast, the constitution of the stable di(aryl)digermyne Bbt–Ge≡Ge–Bbt (Bbt = 2,6-[(Me3Si)2CH]2-4-[(Me3Si)3C]-C6H2) 71, as the triply bonded derivative, was firmly established by Tokitoh and coworkers [78]. 71 was readily prepared by the reduction of the 1,2-di(bromo)digermene precursor trans-[Bbt(Br)Ge=Ge(Bbt)Br] with KC8. The two crystallographically independent molecules in the unit cell of 71 showed slightly twisted [CAr–Ge–Ge–CAr torsional angles = 160.2° and 168.9°] and very short [rGe≡Ge = 2.2060(7) Å and 2.2260(7) Å] Ge–Ge bonds (markedly shorter that those in Power’s digermynes 65 and 67–69).
Reducing 1,2-di(bromo)digermene (E)-Tbb(Br)Ge=Ge(Br)Tbb (Tbb = 2,6-[(Me3Si)2CH]2-4-tBu-C6H2) with KC8, Sasamori, Tokitoh and coworkers synthesized novel di(aryl)digermyne Tbb–Ge≡Ge–Tbb 72 [9]. As other digermynes, 72 displayed short Ge≡Ge bond [rGe≡Ge = 2.2410(9)/2.2221(9) Å] and trans-bent Caryl–Ge–Ge angle (130.5/130.7°) (for two independent molecules in the unit cell of 72).
3.2. Heteronuclear Derivatives
Germynes –Ge≡C–
Heteronuclear alkyne analogues of the type –Ge≡E– (E = C, Si, Sn, Pb), featuring a triple bond between germanium and different group 14 element, were always among the top challenges for experimental pursuits. However, despite numerous research efforts, such compounds (as stable derivatives) have not been isolated to date, although germynes R–Ge≡C–R′ were most closely approached, both computationally and experimentally.
Theoretical studies by Su and a coworker predicted that the germynes can be synthetically accessible, if appropriately substituted with bulky groups to prevent isomerization and oligomerization of highly reactive germynes [79].
The first chemical evidence for the transient germyne ArGe≡CSiMe3 [Ar = 2,6-(iPr2NCH2)2-C6H3], generated by the low-temperature photolysis (λ = 300 nm, −50 C°) of (diazo)germylene precursor ArGe[C(N2)SiMe3] and trapped with tBuOH forming ArGe(OtBu)2CH2SiMe3, was reported by Couret and coworkers [80]. The transient germyne ArGe≡CSiMe3 [Ar = 2,4-tBu-6-(iPr2NCH2)-C6H2] was similarly generated and trapped with water and tBuOH by Mazières and coworkers [81].
Likewise, photolyzing stable (diazo)germylene, the Kato and Baceiredo group generated metastable (stable below −30 °C) phosphine-stabilized germyne [82]. This compound, however, featured a very long Ge–C separation, [rGe–C = 1.887(5) Å], a value that was intermediate between those of typical Ge–Ge single and Ge=Ge double bonds. Accordingly, it is better described as a singly bonded bis(carbenoid) [:Ge–C:] rather than a triply bonded germyne [Ge≡C].
4. Heavy Analogues of 1,3-Dienes
4.1. Homonuclear Derivatives
Tetra(germa)buta-1,3-dienes >Ge=Ge–Ge=Ge<
The very first compound of this type, Tip2Ge=Ge(Tip)–Ge(Tip)=GeTip2 (Tip = 2,4,6-iPr3-C6H2) 73, was synthesized in 2000 by Weidenbruch and coworkers via the reduction of the digermene Tip2Ge=GeTip2 with lithium, followed by the reaction of the resulting digermenyllithium [Tip2Ge=Ge(Tip)Li] with TipBr (Scheme 31) [44]. The Ge–Ge bonds in 73 are in accord with the formulation of the 1,3-diene system >Ge1=Ge2–Ge3=Ge4<, being 2.3568(6) Å (for Ge1=Ge2), 2.4581(5) Å (for Ge2–Ge3), and 2.3439(5) Å (for Ge3=Ge4), respectively. Both Ge=Ge bonds in 73 are trans-bent [θ = 35.4°/31.1° (for Ge1=Ge2) and 33.3°/31.1° (for Ge3=Ge4)] and twisted [τ = 22.4° (Ge1=Ge2) and 21.3° (for Ge3=Ge4)], as is typical for the digermenes. The longest wavelength absorption of 73 in hexane is 560 nm, which remarkably exceeded those of yellow or orange digermenes by 140 nm [and even those of tetra(silyl)digermenes by ca. 100 nm], thus testifying to the presence of conjugation between the two Ge=Ge double bonds.

Scheme 31. Synthesis of the first isolable tetragermabuta-1,3-diene 73.
The same authors later reported improved synthesis of 73 by the one-pot reaction of GeCl2∙dioxane complex, TipMgBr, and Mg in THF, allowing for an increase of the isolated yield of 73 to 31% (cf.: 11% [40]) [47]. In o-dichlorobenzene, tetra(germa)buta-1,3-diene 73 revealed a reversible reduction [E1/2(red) = –0.46 V] and irreversible oxidation [Ep(ox) = 0.15 V], whereas in THF both reduction and oxidation processes were irreversible [Ep(red) = –1.75/–1.3 V and Ep(ox) = 0.6 V] [83].
Apart from 73, there is only one tetra(germa)butadiene derivative reported by Matsuo and coworkers, tetra(germa)cyclobutadiene Ge4[EMind]4 (EMind = 1,1,7,7-tetraethyl-3,3,5,5-tetramethyl-s-hydrindacen-4-yl) 74 (as the germanium analogue of the isostructural tetra(sila)cyclobutadiene [84]) [85]. Moreover, 74 was available by the reduction of di(chloro)digermene [EMind](Cl)Ge=Ge(Cl)[EMind] with lithium naphthalenide (Scheme 32). In contrast to tetra(sila)cyclobutadiene, tetra(germa)cyclobutadiene 74 was thermally stable, with the UV-absorptions observed at 458 (ε = 15000), 510 (ε = 7400), and 836 nm (ε = 150). The Ge4-ring is rhombic-planar [Σinternal bond angles = 360°] with the cyclic Ge–Ge bonds of 2.430 Å (av.). Two germanium atoms feature sp2-like trigonal-planar geometry (ΣGe = 360.0°), whereas the other two germanium atoms have sp3-like pyramidal configuration (ΣGe = 334.0 and 327.7°) with the trans-bending of substituents (θ = 37.9 and 40.6°). Theoretical studies revealed that the Ge–Ge bond orders in 74 = 1.08–1.09 (WBI). Within the Ge4-ring, there is a clear charge separation: if the two diagonal Ge atoms are slightly positively charged (+0.146 and +0.151, NPA charges), the other two Ge atoms are strongly positively charged (+0.602 and +0.573, NPA charges). All of these data are indicative of the crucial contribution of the charge-separated resonance form cyclo-[Ge+–Ge−–Ge+–Ge−], which was also the case for the previously reported tetra(sila)cyclobutadiene. The polar Jahn–Teller distortion results in the relaxation of the inherent 4π-antiaromaticity of 74, thus forming a charge-separated rhombic–planar singlet structure, which is nonaromatic (based on the NICS values).
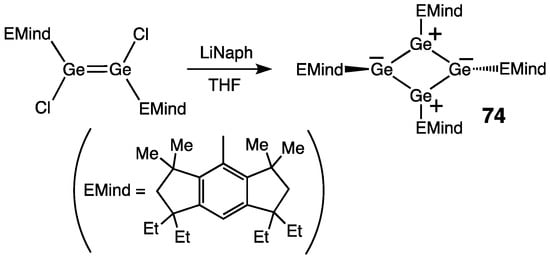
Scheme 32. Synthesis of tetra(germa)cyclobutadiene 74.
4.2. Heteronuclear Derivatives
4.2.1. 1-Sila-2-germabuta-1,3-dienes >Si=Ge–C=C<
Lee, Sekiguchi, and coworkers developed a different approach toward 1,3-dienes containing Ge atoms. They synthesized five-membered ring derivatives including the >Si=Ge–C=C< 1,3-diene fragment, starting from the [2 + 2] cycloaddition of the heavy cyclopropene (1H-disilagermirene 50 [39]) with terminal alkynes to yield bicyclo[2.1.0]pentenes 75, which underwent valence isomerization, finally forming 1,2-disila-3-germacyclopenta-2,4-dienes 76 [29Si NMR (sp2-Si): +124.2 ppm (76a), +123.6 ppm (76b), and +127.3 ppm (76c)] (Scheme 33) [86][87][88]. Such heavy cyclopentadienes 76 constitute rather unusual cyclic systems with two formally conjugated double bonds, Si=Ge and C=C, which are actually isolated, despite the planarity of the five-membered ring. This conclusion was based on the crystallographic and UV spectroscopic data. Thus, in the Si=Ge–C=C fragment, all bonds are within the standard ranges (for example, for 76a, 2.250(1) Å (Si=Ge), 1.972(3) Å (Ge–C), and 1.343(5) Å (C=C)); that is, no elongation of the double bonds and shortening of the single bonds expected for the conjugated system were observed. Likewise, there was no notable red-shift in the longest wavelength UV absorption (π–π*) of 76a compared to that of the starting 50 with isolated Si=Ge bond, 472 vs. 467 nm, respectively. Most likely, the lack of 1,3-conjugation between the Si=Ge and C=C bonds in 1,2-disila-3-germacyclopenta-2,4-dienes 76 results from the substantial spatial and energetic mismatch of the Si=Ge and C=C bonds molecular orbitals, because effective overlap of the latter is a prerequisite for the π-electrons delocalization.
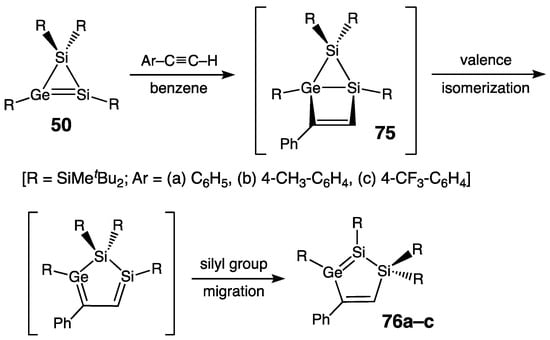
Scheme 33. Synthesis of 1,2-disila-3-germacyclopenta-2,4-dienes 76a–c containing Si=Ge–C=C system.
4.2.2. 1,2-di(germa)cyclobuta-1,3-diene >Ge=Ge–C=C<
This synthetic strategy toward Ge=Ge–C=C conjugated systems was pioneered by Power’s group and then developed by Sasamori and coworkers, who prepared and isolated stable 1,2-di(germa)cyclobutadienes 77a,b via the reaction of their respective digermynes with diphenylacetylene (Scheme 34) [89][90][91]. In both 77a and 77b, the Ge2C2-ring is practically planar (Σinternal bond angles = 359.8° (77a) [89] and 359.9° (77b) [91]), albeit the Ge centers are strongly pyramidalized [ΣGe = 318.8° and 317.3° (77a [89])]. Within the Ge=Ge–C=C fragment, the bonds germanium–germanium/germanium–carbon/carbon–carbon are classified as double/single/double bonds, respectively: 2.4708(9)/[2.022(5) and 2.027(5)]/1.365(7) Å (77a) [89], and 2.4160(5)/2.022(2)/1.362(5) Å (77b) [91]. Accordingly, the structure of 77a,b is best described by the resonance form cyclo-[Ge=Ge–C=C] with the weak Ge=Ge double bond, as depicted in Scheme 34.
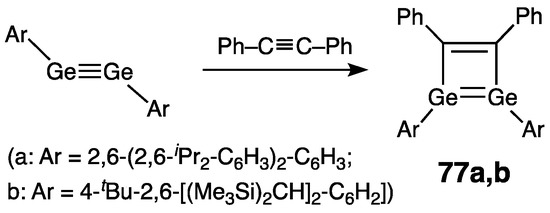
Scheme 34. Synthesis of stable 1,2-di(germa)cyclobutadienes 77a,b.
By reacting 1,2-di(germa)cyclobutadiene 77b [91] with (Me2N)3P=Se, Sasamori and coworkers synthesized 2,5-di(germa)selenophene 78, formed through the transient “housene”-type bicyclic selenadigermirane with its subsequent isomerization to 78 (Scheme 35) [92]. The selenium nucleus in 78 resonated in the low-field region at 481.8 ppm (77Se NMR). On the basis of its structural data [rGe–C = 1.921(3)/1.922(3) Å, rC–C = 1.375(4) Å, θGe = 7.8°(trans), nonplanar SeGe2C2-ring] and computational studies, 78 is better formulated as a singlet (digerma)biradicaloid 78B, while classical Lewis representation 78A is a minor contributor (Scheme 35). Nevertheless, 78 exhibited some degree of aromaticity, as its NICS(1) of –8.0 was comparable with that of the organic selenophene.
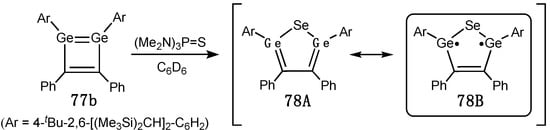
Scheme 35. Synthesis of 2,5-di(germa)selenophene 78.
5. Heavy Analogues of Allene
5.1. Tri(germa)allene >Ge=Ge=Ge<, Germadi(sila)allene >Si=Ge=Si<, and Di(germa)silaallene >Ge=Si=Ge<
Following their original report on the synthesis of the stable tri(sila)allene >Si=Si=Si< [93], Kira and coworkers subsequently prepared tri(germa)allene 79a [94], 1,3-di(germa)silaallene 79b [94], and 2-germadi(sila)allene 79c [95]. Of them, 79a and 79b were prepared by the co-reduction of tetra(chloro)digermane 80a and chloro(trichlorosilyl)germane 80b with KC8, whereas 79c was synthesized by the co-reduction of di(alkyl)silylene 81 and GeCl2∙diox with KC8 (Scheme 36). All sp2-Si atoms were diagnostically deshielded, with the central Si atom in 79b being more strongly deshielded than the peripheral Si atoms in 79c: +236.6 ppm vs. +219.4 ppm. The longest wavelength UV absorptions in 79a, 79b, and 79c were found at 630, 612, and 599 nm, respectively, thus pointing to the significant conjugation between the cumulated double bonds. The allenic bonds in 79 were all in the range of the typical double bonds: rGe=Ge = 2.321(2) Å and 2.330(3) Å in 79a, rGe=Si = 2.2697(8) Å in 79b, and rSi=Ge = 2.2366(7) Å and 2.2373(7) Å in 79c. The allenic E=E′=E framework in 79 was notably bent [bending angle = 122.61(6)° (in 79a), 125.71(7)° (in 79b), and 132.38(2)° (in 79c)], and the terminal atoms E were strongly pyramidalized [θE = 348.5°/348.6° (in 79a), 349.3° (in 79b), and 353.9°/354.0° (in 79c)].
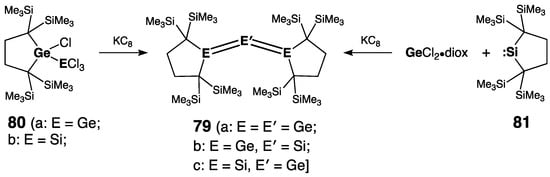
Scheme 36. Synthesis of tri(germa)allene 79a, 1,3-di(germa)silaallene 79b, and 2-germadi(sila)allene 79c.
Apart from Kira’s heavy allenes described above, there was only one recent report on the title compounds: Sasamori, Tokitoh, and coworkers prepared cyclic 1,3-di(germa)-2-silaallene 82 via the reductive dechlorination of tetra(chloro)-2,5-digerma-1-silacyclopentane with KC8 (Scheme 37) [96]. In 82, the SiGe2C2-ring is planar, and the germanium–silicon bonds are typical double bonds [rSi=Ge = 2.2681(18) and 2.2900(18) Å]. However, the central allenic Si in 82 was extraordinarily shielded [δ(29Si) = –16.5 ppm] [96] compared to that of Kira’s 1,3-di(germa)-2-silallene (236.6 ppm) [94]. This was explained by the contribution of the “silylone” resonance structure [>Ge:→Si0←:Ge<] of 82, resulting from the acute Ge–Si–Ge bond angle (80.1°) due to incorporation of the Ge–Si–Ge unit into the five-membered ring skeleton. This was supported by calculations which showed that the germanium–silicon bonds in 82 are highly polarized as Geδ+–Siδ−, –0.27 for Si and +0.90/+0.91 for Ge (NPA charges), and this is indicative of an important Ge-to-Si σ-donation.
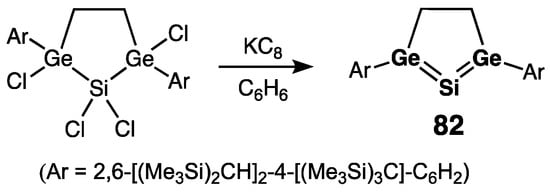
Scheme 37. Synthesis of cyclic 1,3-di(germa)-2-silaallene 82.
5.2. Germaallenes >Ge=C=C<
Only a couple of isolable 1-germaallenes >Ge=C=C< are currently known. The first one, Tip2Ge=C=C(tBu)Ph 83, was reported by West and coworkers synthesized by the reaction of Tip2Ge(F)–C≡C–Ph with tBuLi via the transient [Tip2Ge(F)–C(Li)=C(tBu)Ph], which eliminated LiF to finally yield 5 [97]. In solution, 83 decomposed at room temperature within 15 h. In 83, as in other heteroallenes, the Ge=C and C=C bonds were short [rGe=C = 1.783(2) Å and rC=C = 1.314(2) Å], the Ge=C=C fragment was bent (159.2°), the geometry at the Ge atom was pyramidal (ΣGe = 348.4°), and the central C atom was greatly deshielded [δ(13C) = +235.1 ppm].
The second 1-germaallene, Tbt(Mes)Ge=C=CR2 (CR2 = fluorenylidene) 84, was prepared by the reductive dechlorination of Tbt(Mes)ClGe–C(Cl)=CR2 with tBuLi [98]. Like in 83, the central allenic carbon of 84 resonated in the diagnostic low-field at +243.6 ppm. Without trapping reagents, 84 underwent slow intramolecular cyclization via C–H activation of one of the (Me3Si)2CH groups by the Ge=C bond to form a benzogermacyclobutene derivative.
6. Heavy Analogues of Vinylidenes
Vinylidenes R2C=C:, with substituent-free terminal carbon atoms, are the valence isomers of alkynes RC≡CR. They are exceptionally reactive, and for their stabilization, external bases, such as NHC, are particularly effective. Accordingly, all—except for Aldridge’s di(germa)vinylidene (vide infra)—isolable heavy group 14 analogues of vinylidenes R2E=E′: (E, E′ = heavy group 14 elements) were stabilized by NHC-coordination.
6.1. Homonuclear Derivatives
Di(germa)vinylidene >Ge=Ge:
The first di(germa)vinylidene 85, synthesized by Aldridge and coworkers, was free from external NHC-coordination, in contrast to the NHC-supported silagermenylidenes (vide infra) (Scheme 38) [99].
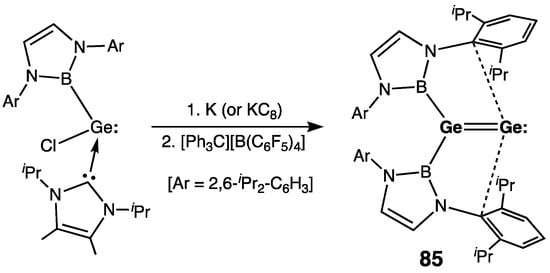
Scheme 38. Synthesis of the first digermavinylidene 85.
85 was prepared by the reduction of the boryl(chloro)germylene–NHC complex with K (or KC8), followed by the oxidation of the intermediate dianion by [Ph3C]+[B(C6F5)4]− (Scheme 38). The germanium–germanium bond in 85 is planar and short [rGe=Ge = 2.312(1) Å] in order to formulate it as a Ge=Ge double bond. In the crystal, weakly stabilizing interactions were found between one of the aryl π-systems of each of the boryl substituents and the vacant p-orbital at the monocoordinated Ge atom. The HOMO of 85 is the Ge=Ge π-bond, whereas the HOMO–1 is mostly the naked Ge lone pair. The longest wavelength UV-absorption in 85, observed at 460 nm, is due to the Ge=Ge bond π–π* electronic transition.
Another stable di(germa)vinylidene 86 was reported by Wesemann and coworkers, synthesized by the reduction of intramolecularly phosphine-stabilized chloro(germyl)germylene with [(MesNacnac)Mg]2 (MesNacnac = {[(2,4,6-Me3-C6H2)N–(Me)C]2CH}−) or metallic sodium (Scheme 39) [100]. The germanium–germanium bond in 86 [rGe=Ge = 2.3060(2) Å] is within the range of the Ge=Ge double bonds, but it is slightly shorter than that in 85 [rGe=Ge = 2.312(1) Å]; the bicyclic framework is planar, and the tricoordinate sp2-Ge disclosed nearly planar geometry (ΣGe = 358.8°). As the manifestation of the Ge=Ge double bonding in 86, πGe=Ge and π*Ge=Ge orbitals represent the HOMO and LUMO, respectively. The bonding situation in 86 can be alternatively viewed as a double Lewis base-stabilized Ge atom in the formal oxidation state 0. Thus, di(germa)vinylidene 86 can potentially serve as a source for Ge0-single atom transfer by the germanium abstraction reactions.
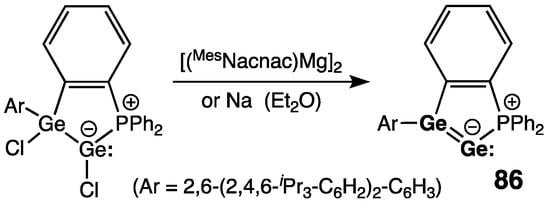
Scheme 39. Synthesis of digermavinylidene 86.
6.2. Heteronuclear Derivatives
6.2.1. Silagermenylidene >Si=Ge:
The first isolable heavy Group 14 elements analogue of the vinylidene, NHC-stabilized silagermenylidene Tip2Si=Ge:(⟵NHC) (NHC = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene) 87, was prepared by the co-reduction of Tip2SiCl2 and [NHC→GeCl2] complex with lithium naphthalenide, as reported by Scheschkewitz and coworkers [101]. As is typical for the Si=Ge doubly bonded system, the Si atom in 87 is markedly deshielded [δ(29Si) = 158.9 ppm] and has a planar configuration (ΣSi = 359.8°), and the Si=Ge bond is short [rSi=Ge = 2.2521(5) Å]. Moreover, the Si=Ge bond in 87 is practically untwisted (τ = 3.1°), which favors an effective pπ–pπ orbital overlap, and the NHC-ligand coordinates to the Ge nearly orthogonally (CNHC–Ge–Si bond angle = 98.9°), thus maximizing the [nNHC lone pair → pGe] orbital interaction. Subsequently, the same group prepared another NHC-stabilized silagermenylidene, [Tip2(Cl)Si](Tip)Si=Ge:(⟵NHC) (NHC = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene) 88, whose spectral and structural characteristics are similar to those of 87: δ (29Si) = 162.5 ppm, rSi=Ge = 2.2757(10) Å, CNHC–Ge–Si bond angle = 101.9° [102].
References
- Davidson, P.J.; Harris, D.H.; Lappert, M.F. Subvalent group 4B metal alkyl and amides. Part I. The synthesis and physical properties of kinetically stable bis-germanium(II), -tin(II), and -lead(II). J. Chem. Soc., Dalton Trans. 1976, 2268–2274.
- Goldberg, D.E.; Hitchcock, P.B.; Lappert, M.F.; Thomas, K.M.; Thorne, A.J.; Fjeldberg, T.; Haaland, A.; Schilling, B.E.R. Subvalent group 4B metal alkyl and amides. Part 9. Germanium and tin alkene analogues, the dimetallenes M2R4 : X-ray structures, molecular orbital calculations for M2H4, and trends in the series M2R′4 . J. Chem. Soc. Dalton Trans. 1986, 2387–2394.
- Snow, J.T.; Murakami, S.; Masamune, S.; Williams, D.J. Synthesis and characterization of tetrakis(2,6-diethylphenyl)digermene. Tetrahedron Lett. 1984, 25, 4191–4194.
- Hurni, K.L.; Rupar, P.A.; Payne, N.C.; Baines, K.M. On the synthesis, structure, and reactivity of tetramesityldigermene. Organometallics 2007, 26, 5569–5575.
- Stender, M.; Pu, L.; Power, P.P. Stabilized terphenyl-substituted digermene derivatives of simple organic groups and their halide precursors: Preference for symmetrically bonded structures. Organometallics 2001, 20, 1820–1824.
- Lei, H.; Fettinger, J.C.; Power, P.P. Synthesis and structures of low-valent alkynyl tin and germanium complexes supported by terphenyl ligands: Heavier group 14 element enediyne analogues. Organometallics 2010, 29, 5585–5590.
- Pampuch, B.; Saak, W.; Weidenbruch, M. New compounds with Ge/Ge and Ge/C multiple bonds. J. Organomet. Chem. 2006, 691, 3540–3544.
- Sasamori, T.; Sugiyama, Y.; Takeda, N.; Tokitoh, N. Structure and properties of an overcrowded 1,2-dibromodigermene. Organometallics 2005, 24, 3309–3314.
- Sasamori, T.; Sugahara, T.; Agou, T.; Guo, J.-D.; Nagase, S.; Streubel, R.; Tokitoh, N. Synthesis and characterization of a 1,2-digermabenzene. Organometallics 2015, 34, 2106–2109.
- Al-Rafia, S.M.I.; Momeni, M.R.; Ferguson, M.J.; McDonald, R.; Brown, A.; Rivard, E. Stable complexes of parent digermene: An inorganic analogue of ethylene. Organometallics 2013, 32, 6658–6665.
- Kira, M.; Iwamoto, T.; Maruyama, T.; Kabuto, C.; Sakurai, H. Tetrakis(trialkylsilyl)digermenes. Salient effects of trialkylsilyl substituents on planarity around the Ge=Ge Bond and remarkable thermochromism. Organometallics 1996, 15, 3767–3769.
- Iwamoto, T.; Okita, J.; Yoshida, N.; Kira, M. Structure and reactions of an isolable Ge=Si doubly bonded compound, tetra(t-butyldimethylsilyl)germasilene. Silicon 2010, 2, 209–216.
- Tokitoh, N.; Kishikawa, K.; Okazaki, R.; Sasamori, T.; Nakata, N.; Takeda, N. Synthesis and characterization of an extremely hindered tetraaryl-substituted digermene and its unique properties in the solid state and in solution. Polyhedron 2002, 21, 563–577.
- Sasamori, T.; Miyamoto, H.; Sakai, H.; Furukawa, Y.; Tokitoh, N. 1,2-Bis(ferrocenyl)digermene: A d–π electron system containing a Ge=Ge unit. Organometallics 2012, 31, 3904–3910.
- Spikes, G.H.; Fettinger, J.C.; Power, P.P. Facile activation of dihydrogen by an unsaturated heavier main group compound. J. Am. Chem. Soc. 2005, 127, 12232–12233.
- Richards, A.F.; Phillips, A.D.; Olmstead, M.M.; Power, P.P. Isomeric forms of divalent heavier group 14 element hydrides: Characterization of Ar′(H)GeGe(H)Ar′ and Ar′(H)2GeGeAr′•PMe3 (Ar′ = C6H3-2,6-Dipp2; Dipp = C6H3-2,6-Pri2). J. Am. Chem. Soc. 2003, 125, 3204–3205.
- Summerscales, O.T.; Caputo, C.A.; Knapp, C.E.; Fettinger, J.C.; Power, P.P. The role of group 14 element hydrides in the activation of C–H bonds in cyclic olefins. J. Am. Chem. Soc. 2012, 134, 14595–14603.
- Hadlington, T.J.; Hermann, M.; Li, J.; Frenking, G.; Jones, C. Activation of H2 by a multiply bonded amido-digermyne: Evidence for the formation of a hydrido-germylene. Angew. Chem. Int. Ed. 2013, 52, 10199–10203.
- Hadlington, T.J.; Li, J.; Hermann, M.; Davey, A.; Frenking, G.; Jones, C. Reactivity of amido-digermynes, LGeGeL (L = bulky amide), toward olefins and related molecules: Facile reduction, C–H activation, and reversible cycloaddition of unsaturated substrates. Organometallics 2015, 34, 3175–3185.
- Kelly, J.A.; Juckel, M.; Hadlington, T.J.; Fernández, I.; Frenking, G.; Jones, C. Synthesis and reactivity studies of amido-substituted germanium(I)/tin(I) dimers and clusters. Chem. Eur. J. 2019, 25, 2773–2785.
- Lee, V.Y.; McNiece, K.; Ito, Y.; Sekiguchi, A. A blue digermene (t-Bu2MeSi)2Ge=Ge(SiMet-Bu2)2. Chem. Commun. 2011, 47, 3272–3274.
- Lee, V.Y.; McNiece, K.; Ito, Y.; Sekiguchi, A.; Geinik, N.; Becker, J.Y. Tetrakis(di-tert-butylmethylsilyl)digermene: Synthesis, structure, electrochemical properties, and reactivity. Heteroatom Chem. 2014, 25, 313–319.
- Aysin, R.R.; Bukalov, S.S.; Leites, L.A.; Lee, V.Y.; Sekiguchi, A. Electronic structure and conformational isomerism of the digermene (tBu2MeSi)2Ge=Ge(SiMet-Bu2)2 as studied by temperature-dependent Raman and UV–vis spectra and quantum-chemistry calculations. J. Organomet. Chem. 2019, 892, 18–23.
- Jana, A.; Huch, V.; Rzepa, H.S.; Scheschkewitz, D. A Multiply functionalized base-coordinated GeII compound and its reversible dimerization to the digermene. Angew. Chem. Int. Ed. 2015, 54, 289–292.
- Nieder, D.; Klemmer, L.; Kaiser, Y.; Huch, V.; Scheschkewitz, D. Isolation and reactivity of a digerma analogue of vinyllithiums: A lithium digermenide. Organometallics 2018, 37, 632–635.
- Klemmer, L.; Kaiser, Y.; Huch, V.; Zimmer, M.; Scheschkewitz, D. Persistent digermenes with acyl and α-chlorosilyl functionalities. Chem. Eur. J. 2019, 25, 12187–12195.
- Hayakawa, N.; Sugahara, T.; Numata, Y.; Kawaai, H.; Yamatani, K.; Nishimura, S.; Goda, S.; Suzuki, Y.; Tanikawa, T.; Nakai, H.; et al. 1,2-Dihalodigermenes bearing bulky Eind groups: Synthesis, characterization, and conversion to halogermylenoids. Dalton Trans. 2018, 47, 814–822.
- Mangan, R.J.; Rit, A.; Sindlinger, C.P.; Tirfoin, R.; Campos, J.; Hicks, J.; Christensen, K.E.; Niu, H.; Aldridge, S. Activation of protic, hydridic and apolar E–H bonds by a boryl-substituted GeII cation. Chem. Eur. J. 2020, 26, 306–315.
- Mangan, R.J.; Davies, A.R.; Hicks, J.; Sindlinger, C.P.; Thompson, A.L.; Aldridge, S. Synthesis, structure and reactivity of terphenyl-substituted germylium-ylidene cations. Polyhedron 2021, 196, 115006.
- Klemmer, L.; Thömmes, A.-L.; Zimmer, M.; Huch, V.; Morgenstern, B.; Scheschkewitz, D. Metathesis of Ge=Ge double bonds. Nature Chem. 2021, 13, 373–377.
- Sekiguchi, A.; Yamazaki, H.; Kabuto, C.; Sakurai, H.; Nagase, S. Cyclotrigermenes: A new unsaturated ring system. J. Am. Chem. Soc. 1995, 117, 8025–8026.
- Sekiguchi, A.; Tsukamoto, M.; Ichinohe, M. A Free Cyclotrigermenium cation with a 2π-electron system. Science 1997, 275, 60–61.
- Sekiguchi, A.; Fukaya, N.; Ichinohe, M.; Takagi, N.; Nagase, S. Synthesis of unsymmetrically substituted cyclotrigermenes and the first example of cis configuration around the Ge=Ge double Bond. J. Am. Chem. Soc. 1999, 121, 11587–11588.
- Sekiguchi, A.; Ishida, Y.; Fukaya, N.; Ichinohe, M.; Takagi, N.; Nagase, S. The first halogen-substituted cyclotrigermenes: A unique halogen walk over the three-membered ring skeleton and facial stereoselectivity in the Diels-Alder reaction. J. Am. Chem. Soc. 2002, 124, 1158–1159.
- Lee, V.Y.; Yasuda, H.; Ichinohe, M.; Sekiguchi, A. SiGe2 and Ge3: Cyclic digermenes that undergo unexpected ring-expansion reactions. Angew. Chem. Int. Ed. 2005, 44, 6378–6381.
- Lee, V.Y.; Yasuda, H.; Ichinohe, M.; Sekiguchi, A. Heavy cyclopropene analogues R4SiGe2 and R4Ge3 (R = SiMetBu2)—New members of the cyclic digermenes family. J. Organomet. Chem. 2007, 692, 10–19.
- McNeice, K.; Lee, V.Y.; Sekiguchi, A. Making a cyclotrigermene from a digermene. Organometallics 2011, 30, 4796–4797.
- Lee, V.Y.; Yasuda, H.; Sekiguchi, A. Interplay of EnE′3–nC valence isomers (E, E′ = Si, Ge): Bicyclobutanes with very short bridging bonds and their isomerization to alkyl-substituted cyclopropenes. J. Am. Chem. Soc. 2007, 129, 2436–2437.
- Lee, V.Y.; Ichinohe, M.; Sekiguchi, A.; Takagi, N.; Nagase, S. The first three-membered unsaturated rings consisting of different heavier group 14 elements: 1-disilagermirene with a Si=Si double bond and its isomerization to a 2-disilagermirene with a Si=Ge double bond. J. Am. Chem. Soc. 2000, 122, 9034–9035.
- Lee, V.Y.; Takanashi, K.; Ichinohe, M.; Sekiguchi, A. A chemical trick: How to make a digermene from a disilene, formation of 3Δ-1,2,3,4-disiladigermetene. J. Am. Chem. Soc. 2003, 125, 6012–6013.
- Lee, V.Y.; Ito, Y.; Yasuda, H.; Takanashi, K.; Sekiguchi, A. From tetragermacyclobutene to tetragermacyclobutadiene dianion to tetragermacyclobutadiene transition metal complexes. J. Am. Chem. Soc. 2011, 133, 5103–5108.
- Ramaker, G.; Saak, W.; Haase, D.; Weidenbruch, M. Reaction modes of a tetragermabutadiene: Cycloadditions versus Ge–Ge bond cleavages. Organometallics 2003, 22, 5212–5216.
- Sasamori, T.; Sugahara, T.; Agou, T.; Sugamata, K.; Guo, J.-D.; Nagase, S.; Tokitoh, N. Reaction of a diaryldigermyne with ethylene. Chem. Sci. 2015, 6, 5526–5530.
- Schäfer, H.; Saak, W.; Weidenbruch, M. Hexaaryltetragermabuta-1,3-diene: A molecule with conjugated Ge–Ge double bonds. Angew. Chem. Int. Ed. 2000, 39, 3703–3705.
- Olmstead, M.M.; Pu, L.; Simons, R.S.; Power, P.P. Reduction of Ge(Cl)C6H3mes2-2,6 to give the cyclotrigermenyl radical (GeC6H3mes2-2,6)3∙ and the trigermenyl anion salt K(GeC6H3mes2-2,6)3. Chem. Commun. 1997, 1595–1596.
- Lee, V.Y.; Ito, Y.; Gapurenko, O.A.; Minyaev, R.M.; Gornitzka, H.; Sekiguchi, A. From a (silatrigerma)cyclobutenylium ion to a (silatrigerma)cyclobutenyl radical and back. J. Am. Chem. Soc. 2020, 142, 16455–16460.
- Ramaker, G.; Schäfer, A.; Saak, W.; Weidenbruch, M. One-pot synthesis of a tetragermabutadiene and its reactions with oxygen and sulfur. Organometallics 2003, 22, 1302–1304.
- Hlina, J.; Baumgartner, J.; Marschner, C.; Albers, L.; Müller, T.; Jouikov, V.V. Formation and properties of a bicyclic silylated digermene. Chem. Eur. J. 2014, 20, 9357–9366.
- Meyer, H.; Baum, G.; Massa, W.; Berndt, A. Stable germaethenes. Angew. Chem., Int. Ed. Engl. 1987, 26, 798–799.
- Couret, C.; Escudié, J.; Satgé, J.; Lazraq, M. The first stable germene: A compound with a germanium–carbon double bond. J. Am. Chem. Soc. 1987, 109, 4411–4412.
- Lazraq, M.; Escudié, J.; Couret, C.; Satgé, J.; Dräger, M.; Dammel, R. (Mesityl)2Ge(fluorenylidene)–stabilization of a Ge–C double bond by charge transfer into an aromatic system. Angew. Chem. Int. Ed. Engl. 1988, 27, 828–829.
- Lazraq, M.; Couret, C.; Escudié, J.; Satgé, J.; Soufiaoui, M. New stable germenes. Polyhedron 1991, 10, 1153–1161.
- Anselme, G.; Escudié, J.; Couret, C.; Satgé, J. Difluorenyl- and tert-butylfluorenyl(fluorenylidene)germenes: Synthesis, stabilization and first aspects of their reactivity. J. Organomet. Chem. 1991, 403, 93–100.
- Chaubon, M.-A.; Escudié, J.; Ranaivonjatovo, H.; Satgé, J. Halogenogermenes: Evidence for the formation of the chloro- or fluoro-germenes (Me5C5)(X)Ge–CR2 (CR2 = fluorenylidene). J. Chem. Soc. Dalton Trans. 1996, 893–897.
- Couret, C.; Escudié, J.; Delpon-Lacaze, G.; Satgé, J. Dimesitylneopentylgermene, a new stable germene. Organometallics 1992, 11, 3176–3177.
- Stürmann, M.; Saak, W.; Weidenbruch, M.; Berndt, A.; Scheschkewitz, D. Molecular structures of new compounds with Ge=C and Sn=C double bonds. Heteroatom Chem. 1999, 10, 554–558.
- Meiners, F.; Saak, W.; Weidenbruch, M. Reaction of a diarylgermylene with a phosphaalkyne: Formation of a germadiphosphacyclobutene with an exocyclic C=Ge double bond. Chem. Commun. 2001, 215–216.
- Meiners, F.; Saak, W.; Weidenbruch, M. Acetylene-linked bis(germaethenes): The first molecules with conjugated germanium–carbon double bonds. Organometallics 2000, 19, 2835–2836.
- Meiners, F.; Haase, D.; Koch, R.; Saak, W.; Weidenbruch, M. Cycloaddition reactions of an acetylene-linked bis(germaethene). Organometallics 2002, 21, 3990–3995.
- Tokitoh, N.; Kishikawa, K.; Okazaki, R. Synthesis and structure of the first germaketenedithioacetal. J. Chem. Soc. Chem. Commun. 1995, 1425–1426.
- Lee, V.Y.; Ichinohe, M.; Sekiguchi, A. 2,4-Disila-1-germatricyclopentane: A new type of cage compound of group 14 elements with an extremely long Ge–C bridge bond and an “umbrella”-type configuration of a Ge atom. J. Am. Chem. Soc. 2002, 124, 9962–9963.
- Haas, M.; Leypold, M.; Schnalzer, D.; Torvisco, A.; Stueger, H. Stable germenolates and germenes with exocyclic structures. Organometallics 2015, 34, 5291–5297.
- Baines, K.M.; Cooke, J.A. Tetramesitylgermasilene: The first relatively stable germasilene and its rearrangement to a silylgermylene. Organometallics 1992, 11, 3487–3488.
- Ichinohe, M.; Arai, Y.; Sekiguchi, A.; Takagi, N.; Nagase, S. A new approach to the synthesis of unsymmetrical disilenes and germasilene: Unusual 29Si NMR chemical shifts and regiospecific methanol addition. Organometallics 2001, 20, 4141–4143.
- Sekiguchi, A.; Izumi, R.; Ihara, S.; Ichinohe, M.; Lee, V.Y. The first isolable 1,1-dilithiogermane and its unusual dimeric structure—An effective reagent for the preparation of double-bonded derivatives of group 14 elements. Angew. Chem. Int. Ed. 2002, 41, 1598–1600.
- Igarashi, M.; Ichinohe, M.; Sekiguchi, A. A stable silagermene (tBu3Si)2Si=Ge(Mes)2 (Mes = 2,4,6-trimethylphenyl): Synthesis, X-ray crystal structure, and thermal isomerization. Heteroatom Chem. 2008, 19, 649–653.
- Al-Rafia, S.M.I.; Malcolm, A.C.; McDonald, R.; Ferguson, M.J.; Rivard, E. Trapping the parent inorganic ethylenes H2SiGeH2 and H2SiSnH2 in the form of stable adducts at ambient temperature. Angew. Chem. Int. Ed. 2011, 50, 8354–8357.
- Ghadwal, R.S.; Roesky, H.W.; Merkel, S.; Henn, J.; Stalke, D. Lewis base stabilized dichlorosilylene. Angew. Chem. Int. Ed. 2009, 48, 5683–5686.
- Majhi, P.K.; Huch, V.; Scheschkewitz, D. A mixed heavier Si=Ge analogue of a Vinyl Anion. Angew. Chem. Int. Ed. 2021, 60, 242–246.
- Chaubon, M.-A.; Escudié, J.; Ranaivonjatovo, H.; Satgé, J. First characterization of a compound with a tin-germanium double bond: The dimesityl(diisitylstanna)germene (Is)2Sn=Ge(Mes)2. Chem. Commun. 1996, 2621–2622.
- Schäfer, A.; Saak, W.; Weidenbruch, M. Tetraarylstannagermene: A molecule with a Ge=Sn double bond. Organometallics 2003, 22, 215–217.
- Sekiguchi, A.; Izumi, R.; Lee, V.Y.; Ichinohe, M. R2Ge=SnR′2 and RR′Ge=SnRR′ (R = SiMetBu2, R′ = 2,4,6-iPr3C6H2): The new stable germastannenes. Organometallics 2003, 22, 1483–1486.
- Lee, V.Y.; Takanashi, K.; Nakamoto, M.; Sekiguchi, A. 3Δ-1,2,3,4-Disilagermastannetene: The first cyclic germastannene. Russ. Chem. Bull. Int. Ed. 2004, 53, 1102–1104.
- Stender, M.; Phillips, A.D.; Wright, R.J.; Power, P.P. Synthesis and characterization of a digermanium analogue of an alkyne. Angew. Chem. Int. Ed. 2002, 41, 1785–1787.
- Stender, M.; Phillips, A.D.; Power, P.P. Formation of 2 (Ar* = C6H3-2,6-Trip2; Trip = C6H2-2,4,6-i-Pr3) via reaction of Ar*GeGeAr* with 2,3-dimethyl-1,3-butadiene: Evidence for the existence of a germanium analogue of an alkyne. Chem. Commun. 2002, 1312–1313.
- Pu, L.; Phillips, A.D.; Richards, A.F.; Stender, M.; Simons, R.S.; Olmstead, M.M.; Power, P.P. Germanium and tin analogues of alkynes and their reduction products. J. Am. Chem. Soc. 2003, 125, 11626–11636.
- Peng, Y.; Fischer, R.C.; Merrill, W.A.; Fischer, J.; Pu, L.; Ellis, B.D.; Fettinger, J.C.; Herber, R.H.; Power, P.P. Substituent effects in ditetrel alkyne analogues: Multiple vs. single bonded isomers. Chem. Sci. 2010, 1, 461–468.
- Sugiyama, Y.; Sasamori, T.; Hosoi, Y.; Furukawa, Y.; Takagi, N.; Nagase, S.; Tokitoh, N. Synthesis and properties of a new kinetically stabilized digermyne: New insights for a germanium analogue of an alkyne. J. Am. Chem. Soc. 2006, 128, 1023–1031.
- Wu, P.-C.; Su, M.-D. Theoretical design for germaacetylene (RC≡GeR′): A new target for synthesis. Dalton Trans. 2011, 40, 4253–4259.
- Bibal, C.; Mazières, S.; Gornitzka, H.; Couret, C. A route to a germanium–carbon triple bond: First chemical evidence for a germyne. Angew. Chem. Int. Ed. 2001, 40, 952–954.
- Bonnefille, E.; Mazières, S.; Saffon, N.; Couret, C. Reactivity of a germa-alkyne: Evidence for a germanone intermediate in the hydrolysis and alcoholysis processes. J. Organomet. Chem. 2009, 694, 2246–2251.
- Berthe, J.; Garcia, J.M.; Ocando, E.; Kato, T.; Saffon-Merceron, N.; De Cózar, A.; Cossío, F.P.; Baceiredo, A. Synthesis and reactivity of a phosphine-stabilized monogermanium analogue of alkynes. J. Am. Chem. Soc. 2011, 133, 15930–15933.
- Schäfer, A.; Weidenbruch, M.; Müller, T.; Pravinkumar, K.; Becker, J.Y. Electrochemical properties of a disiliene, a tetrasila-1,3-butadiene, and their germanium analogues. Chem. Eur. J. 2009, 15, 8424–8428.
- Suzuki, K.; Matsuo, T.; Hashizume, D.; Fueno, H.; Tanaka, K.; Tamao, K. A planar rhombic charge-separated tetrasilacyclobutadiene. Science 2011, 331, 1306–1309.
- Suzuki, K.; Numata, Y.; Fujita, N.; Hayakawa, N.; Tanikawa, T.; Hashizume, D.; Tamao, K.; Fueno, H.; Tanaka, K.; Matsuo, T. A stable free tetragermacyclobutadiene incorporating fused-ring bulky EMind groups. Chem. Commun. 2018, 54, 2200–2203.
- Lee, V.Y.; Ichinohe, M.; Sekiguchi, A. The first metalladiene of group 14 elements with a silole-type structure with Si=Ge and C=C double bonds. J. Am. Chem. Soc. 2000, 122, 12604–12605.
- Lee, V.Y.; Ichinohe, M.; Sekiguchi, A. Reaction of disilagermirenes with phenylacetylene: From a germasilene –Ge=Si– to a metalladiene of the type –Si=Ge–C=C–. J. Organomet. Chem. 2001, 636, 41–48.
- Lee, V.Y.; Kato, R.; Aoki, S.; Sekiguchi, A. 1,3-Disila-3-germacyclopentadienes: Cyclopentadiene analogs based on heavier group 14 elements. Russ. Chem. Bull., Int. Ed. 2011, 60, 2434–2435.
- Cui, C.; Olmstead, M.M.; Power, P.P. Reaction of Ar′GeGeAr′ (Ar′ = C6H3-2,6-Dipp2, Dipp = C6H3-2,6-iPr2) toward alkynes: Isolation of a stable digermacyclobutadiene. J. Am. Chem. Soc. 2004, 126, 5062–5063.
- Cui, C.; Olmstead, M.M.; Fettinger, J.C.; Spikes, G.H.; Power, P.P. Reactions of the heavier alkyne analogues Ar′GeGeAr′ (Ar′ = C6H3-2,6-(C6H3-2,6-Pri2)2; E = Ge, Sn) with unsaturated molecules: Probing the character of the EE multiple bonds. J. Am. Chem. Soc. 2005, 127, 17530–17541.
- Sugahara, T.; Guo, J.-D.; Sasamori, T.; Karatsu, Y.; Furukawa, Y.; Ferao, A.E.; Nagase, S.; Tokitoh, N. Reaction of a stable digermyne with acetylenes: Synthesis of a 1,2-digermabenzene and a 1,4-digermabarrelene. Bull. Chem. Soc. Jpn. 2016, 89, 1375–1384.
- Sugahara, T.; Sasamori, T.; Tokitoh, N. 2,5-Digermaselenophenes: Germanium analogues of selenophenes. J. Am. Chem. Soc. 2018, 140, 11206–11209.
- Ishida, S.; Iwamoto, T.; Kabuto, C.; Kira, M. A stable silicon-based allene analogue with a formally sp-hybridized silicon atom. Nature 2003, 421, 725–727.
- Iwamoto, T.; Masuda, H.; Kabuto, C.; Kira, M. Trigermaallene and 1,3-digermasilaallene. Organometallics 2005, 24, 197–199.
- Iwamoto, T.; Abe, T.; Kabuto, C.; Kira, M. A missing allene of heavy group 14 elements: 2-germadisilaallene. Chem. Commun. 2005, 5190–5192.
- Sugahara, T.; Sasamori, T.; Tokitoh, N. Highly bent 1,3-digerma-2-silaallene. Angew. Chem. Int. Ed. 2017, 56, 9920–9923.
- Eichler, B.E.; Powell, D.R.; West, R. Synthesis and structure of a 1-germapropadiene. Organometallics 1998, 17, 2147–2148.
- Tokitoh, N.; Kishikawa, K.; Okazaki, R. Synthesis and reactions of the first stable 1-germaallene. Chem. Lett. 1998, 27, 811–812.
- Rit, A.; Campos, J.; Niu, H.; Aldridge, S. A stable heavier group 14 analogue of vinylidene. Nature Chem. 2016, 8, 1022–1026.
- Krebs, K.M.; Hanselmann, D.; Schubert, H.; Wurst, K.; Scheele, M.; Wesemann, L. Phosphine-stabilized digermavinylidene. J. Am. Chem. Soc. 2019, 141, 3424–3429.
- Jana, A.; Huch, V.; Scheschkewitz, D. NHC-stabilized silagermenylidene: A heavier analogue of vinylidene. Angew. Chem. Int. Ed. 2013, 52, 12179–12182.
- Jana, A.; Majumdar, M.; Huch, V.; Zimmer, M.; Scheschkewitz, D. NHC-coordinated silagermenylidene functionalized in allylic position and its behaviour as a ligand. Dalton Trans. 2014, 43, 5175–5181.
More
Information
Subjects:
Chemistry, Inorganic & Nuclear
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
01 Mar 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No