Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Guliz Ozgun | -- | 1838 | 2023-02-21 23:49:55 | | | |
| 2 | Catherine Yang | Meta information modification | 1838 | 2023-02-22 01:47:58 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ozgun, G.; Nappi, L. Primary Mediastinal Germ Cell Tumors. Encyclopedia. Available online: https://encyclopedia.pub/entry/41504 (accessed on 26 July 2026).
Ozgun G, Nappi L. Primary Mediastinal Germ Cell Tumors. Encyclopedia. Available at: https://encyclopedia.pub/entry/41504. Accessed July 26, 2026.
Ozgun, Guliz, Lucia Nappi. "Primary Mediastinal Germ Cell Tumors" Encyclopedia, https://encyclopedia.pub/entry/41504 (accessed July 26, 2026).
Ozgun, G., & Nappi, L. (2023, February 21). Primary Mediastinal Germ Cell Tumors. In Encyclopedia. https://encyclopedia.pub/entry/41504
Ozgun, Guliz and Lucia Nappi. "Primary Mediastinal Germ Cell Tumors." Encyclopedia. Web. 21 February, 2023.
Copy Citation
Primary mediastinal germ cell tumors (PMGCTs) are a rare type of cancer affecting young adults. They have different molecular and clinical features compared to testicular germ cell tumors. Non-seminoma PMGCTs have the shortest 5-year overall survival and the poorest prognosis among all of the germ cell tumor presentations, while seminomas share the same survival and prognosis as their testicular counterparts.
mediastinal germ cell tumors (MGCTs)
clinical features
poor prognosis
treatment
molecular features
1. Clinical Features
PMGCTs arise more frequently from the anterior mediastinum and rarely from the posterior, middle, or superior mediastinum. PMGCTs account for 15–20% of mediastinal tumors in young men aged between 25 and 35 [1]. Dyspnea, cough, chest pain, and weight loss are typical symptoms at the time of diagnosis. The other accompanying symptoms include hemoptysis, superior vena cava (SVC) syndrome, recurrent laryngeal nerve palsy, fever, night sweats, nausea, and gynecomastia. PMGCTs primarily metastasize to the mediastinal lymph nodes. However, they can also metastasize to the lungs, liver, bone, retroperitoneum, central nervous system, and heart [2]. As extra-mediastinal metastasis is related to poorer prognosis, patients should be staged with appropriate diagnostic tests that include whole-body CT scans.
In differential diagnosis, thymic diseases, thyroid goiter, NUT carcinoma (a rare SALL4- and AFP-expressing poorly differentiated squamous cell cancer), metastatic melanoma, sarcomas (the SMARCA4-deficient subtype often expresses SOX2, SALL4, and vimentin), lymphomas (which can be identified with CD45a and B/T cell markers), and metastatic carcinoma to the mediastinum should be considered. Moreover, dissemination following the course of the thoracic duct can be associated with a testicular or a retroperitoneal primary. Therefore, additional workup should be carried out to evaluate testicular primary.
Although the histological and pathological features of PMGCTs are not different from gonadal GCTs, the clinical presentation, symptomatology, and prognosis may differ from their gonadal counterparts. Non-seminoma PMGCTs have a poor prognosis, with a 5-year overall survival (OS) of 40%–50% despite chemotherapy and surgery (Table 1). Independent prognostic factors associated with shorter survival in non-seminoma PMGCTs are non-pulmonary visceral metastases and elevated β-HCG. Conversely, seminoma PMGCTs have an excellent prognosis with an OS exceeding 90% using the current curative treatment modalities [2][3][4][5].
Table 1. Survival updates on poor-risk GCTs.
| Years | Number of Patients | 5-Year PFS (95% CI) |
5-Year OS (95% CI) |
Ref. | |
|---|---|---|---|---|---|
| IGCCCG | 1975–1990 | 832 | 41% (35 to 47) | 48% (42 to 54) | [6] |
| Gillessen et al. (IGCCCG Update) |
1990–2013 | 2514 | 54% (52 to 56) | 67% (65 to 69) | [7] |
| Adra et al. | 1990–2014 | 273 | 58% (51% to 63%) | 73% (67% to 78%) | [8] |
IGCCCG, International Germ Cell Cancer Collaborative Group; PFS, progression-free survival; OS, overall survival; CI, confidence interval.
2. Histopathology and Embryogenesis
Interrupted migration of progenitor germ cells during embryogenesis, burnt-out primary (healed testis primary at extragonadal GCT diagnosis), and reverse migration of transformed germ cells from testes are the proposed mechanisms so far for developing extragonadal GCTs [9][10][11].
PMGCTs are classified as non-seminoma, including teratoma (mature, immature, and teratoma with somatic malignancies), yolk sac tumors, choriocarcinoma, embryonal carcinoma, mixed germ cell tumors, germ cell tumors with associated hematological malignancy (WHO 2021 classification), and seminoma [12]. While teratoma (58%) and yolk sac tumors (42%) make up a substantial part of prepubertal extra-gonadal GCTs, the most common histological subtype in adults is mature teratoma [13]. GCTs are also classified into five groups regardless of their primary site, defined by chromosomal changes and developmental potential. Type I GCTs comprise infantile teratomas and yolk sac tumors with the loss of chromosomes 1p, 4, and 6q and the gain of chromosomes 1q, 12(p13), and 20q. Type II GCTs include seminomas and non-seminomas in adolescent/adult men and typically have a gain of chromosomes 7, 8, 12p, 21, and X and a loss of chromosomes 1p, 11, 13, and 18 [14].
Primary mediastinal choriocarcinoma is rare and most patients have hematogenous dissemination at diagnosis. Therefore, it has a poorer prognosis when compared to other histologic subtypes [15]. Embryonal carcinoma cells are considered malignant variants of embryonal stem cells and they share biochemical and morphologic similarities. A yolk sac tumor usually includes malignant endodermal and extraembryonic mesenchymal cells, which are more commonly observed in children. Yolk sac tumors and embryonal carcinoma are associated with poor prognosis.
Teratomas arise from the three germinal layers and have the potential to differentiate into any tissue in the body. Immaturity and quantification of the neuroepithelial component are used for teratoma grading. Grade 1 is defined as tumors with some degree of immaturity, but neuroepithelium presence is limited to a maximum of one focus. Grade 3 is defined as the existence of a significant immaturity and neuroepithelium, with neuroepithelial components in ≥4 fields within individual parts. Grade 2 stands between grades 1 and 3 [16]. Unlike testicular GCTs (TGCTs), mature and immature teratoma differentiation is critical for MGCT patients’ management since immature teratomas have the potential for aggressive behavior. Mature teratoma is the most common form of teratoma in PMGCTs (63%), while immature teratoma is diagnosed in about 4% of patients. Teratoma with sarcoma, other malignant germ cell elements, or carcinoma is observed in about 33% of cases [17].
SALL4, PLAP (placental alkaline phosphatase), organic cation transporter (OCT) 3–4, NANOG, c-kit (CD117), CD30, EMA (epithelial membrane antigen), cytokeratins, FP, -HCG, and glycipan-3 are the validated immunohistochemical markers for the pathological diagnosis confirmation of GCTs. Since there are often variable and focal stainings depending on the tumor phenotype, immunohistochemical antibodies should be used to detect the proteins.
Seminomas are positive for PLAP, OCT-4, c-kit, and SALL-4 and negative for CD30 and cytokeratins. Conversely, embryonal carcinoma is almost always positive for cytokeratins. EMA, CD30, OCT-4, and SALL-4 can also be positive while PLAP is positive in about 50% of cases. Likewise, yolk sac tumors are positive for cytokeratins and SALL-4 but negative for CD30 and c-kit (Table 2). Leucocyte common antigen (LCA; CD45) and desmin/vimentin are used for the differential diagnosis of lymphomas and sarcomas, respectively. Non-germ cell malignant transformation occurs more frequently in teratoma PMGCTs than in gonadal or primary retroperitoneal GCTs and consists of sarcoma and adenocarcinoma differentiation [18].
Table 2. Immunohistochemistry in germ cell tumors.
| Seminoma | Immature Teratoma | Yolk Sac Tumor | Embryonal Carcinoma | Choriocarcinoma | |
|---|---|---|---|---|---|
| Positive Markers | SALL4 OCT 3–4 NANOG c-kit PLAP |
SALL4 SOX2 Cytokeratins EMA |
SALL4 Glypican-3 Cytokeratins AFP |
SALL4 OCT 3–4 CD30 SOX2 Cytokeratins NANOG |
HCG Cytokeratins Glypican-3 SALL4 EMA |
| Negative Markers | CD30 Glypican-3 SOX2 |
OCT 3–4 CD30 c-kit NANOG SOX2 |
CD30 c-kit SOX2 NANOG OCT 3–4 |
c-kit Glypican-3 |
OCT 3–4 c-kit CD30 SOX2 NANOG |
Post-chemotherapy residual disease is characterized by 40–50% fibrosis and necrosis and mixed inflammatory aggregates, 10–20% viable GCTs, and 30–40% teratoma [19][20]. A pathology report should contain the ratio of viable non-teratoma GCTs since treatment response is one of the most critical factors in predicting long-term outcomes. Viable tumor cells below 10% represents a good prognostic factor [21]. Overall, sampling should be extensive to better appreciate the residual tumor, if present.
3. Genomic Features
The i(12p) is a cytogenetic aberration identified in approximately 80% PMGCTs regardless of the histological subtype [22]. In suspicious cases, which have lost their typical GCT appearance, documenting i(12p) persistence is a helpful tool for molecular diagnosis. Therefore, identification of i(12p) in a mediastinal mass specimen (usually with fluorescence in situ hybridization) could confirm a diagnosis of PMGCTs [23]. Several genes including MED21, Sox5, DAD-R, BCAT1, KRAS, Cyclin D2, FGF6, ATF7IP, GDF3, LRP6, Wnt5B, FGF23, Nanog, and Dppa3 are located on the short arm of chromosome 12 and could play a role in GCT development [24]. However, the exact molecular mechanisms leading to GCT initiation and progression remain unclear. Other chromosomal abnormalities involve chromosomes 1p, 1q, and 6q and the sex chromosomes. Compared to testicular GCTs, PMGCTs have a higher tumor mutational burden and specific pathogenic oncogene alterations. The most common mutations described in PMGCTs are: TP53 (46%), c-KIT (18%), KRAS (18%), PTEN (11%), NRAS (4%), and PIK3CA (4%) (Figure 1). These alterations are more common in non-seminoma PMGCTs than in seminomas and non-seminoma TGCTs. Few studies have correlated TP53 mutations and MDM2 alterations to cisplatin resistance in GCTs. While TP53 mutations are primarily found in mediastinal GCTs, MDM2 amplifications are mainly found in the testis [25]. In a retrospective analysis, TP53 alterations were detected in 16.3% (17/104) of patients with cisplatin-resistant GCTs. Of note, TP53 mutations were observed in 72.2% (13/18) of the non-seminoma PMGCTs samples analyzed in that study [26]. In a recent multi-institutional study focused on PMGCTs, TP53 genomic alterations were described in 56% of non-seminoma tumors and were associated with significantly shorter OS than in patients with wild-type TP53 PMGCTs, suggesting a distinct genomic background in patients with PMGCTs which could explain the poor prognosis of this patient population [27]. TP53 alterations are even more frequent in PMGCTs associated with hematologic malignancies where they have been observed in 91% of the patients [28].
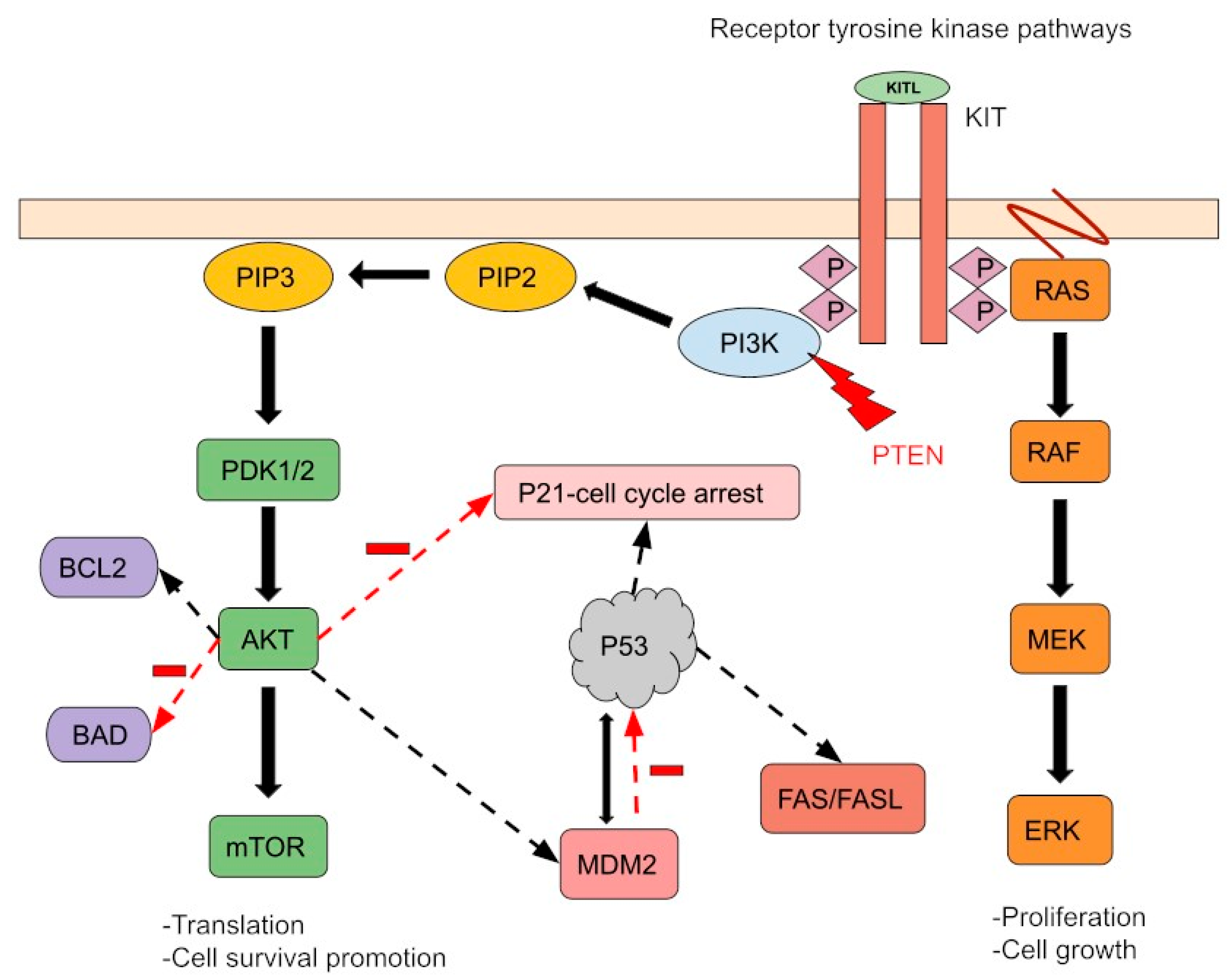
Figure 1. A simple schema of molecular aberrations in PMGTs. The most common mutations described in PMGCTs are TP53 (46%), c-KIT (18%), KRAS (18%), PTEN (11%), NRAS (4%), and PIK3CA (4%). K-RAS and N-RAS mutations cause Ras-Raf-MEK-ERK pathway activation regardless of receptor activity. The PTEN tumor suppressor gene controls cell growth and migration. Its mutation activates the PI3K pathway, leading to impaired mitotic arrest and cell survival promotion. MDM2 and P53 regulate each other through feedback mechanisms. P53 mutation and/or MDM2-induced ubiquitination and disruption of P53 inhibit apoptosis. Finally, the c-KIT gain-of-function mutation and/or KIT overexpression activates downstream signaling by increasing receptor tyrosine kinase activity. Furthermore, these pathways can interact and co-regulate downstream processes, promoting cell survival and proliferation.
4. Association with Other Diseases and Malignancies
Non-seminoma PMGCTs can be associated with chromosomopathies and hematologic malignancies (HMs). Klinefelter’s syndrome (KS; 47, XXY) is the most common chromosomal disease associated with non-seminoma PMGCTs. According to the Children’s Oncology Group, approximately one-third of patients with PMGCT have KS [29]. Therefore, PMGCT patients should be screened for KS. There are few case reports describing PMGCT in neurofibromatosis type 1 patients [30].
HMs are seen in 2–3% of non-seminoma PMGCT patients, either simultaneously (31%) or after initial diagnosis (46%) and are associated with a very poor prognosis and OS of less than 2 years. The most common HM in PMGCTs patients is acute megakaryoblastic leukemia (AML-M7). Other HMs include other AMLs, chronic myeloid leukemia, acute lymphoblastic leukemia, myelodysplasia, malignant mastocytosis, essential thrombocytopenia, and malignant histiocytosis [31][32][33]. i(12p) is detected in about 47% of secondary HM cells. Other genomic alterations including TP53, KRAS, and PTEN can also be present, suggesting that these cells are derived from a mutual descent [31][34]. Therefore, hematologic malignancies secondary to PMGCTs genetically resemble PMGCTs rather than primary HMs. There are no guidelines for non-seminoma PMGCT-associated hematologic malignancy treatment. The case reports/series show a limited response rate to current hematologic chemotherapy protocols. Therefore, treating this disease as AML with poor prognostic features is reasonable. They should also be promptly considered for allogeneic hematopoietic stem cell transplantation, even if definitive GCT treatment is delayed [24].
Etoposide-related HMs should be considered when a PMGCT patient is diagnosed with hematologic malignancy. Therapy-related diseases would emerge later, approximately 25–60 months after chemotherapy treatment. Furthermore, they possibly have etoposide-related translocations such as 11q23 and usually are negative for i(12p).
Somatic transformation occurs in 1–2% of male GCTs of which 25–30% are PMGCTs. The most common somatic transformations include carcinoma (adenocarcinoma, adenoid cystic carcinoma, and high-grade neuroendocrine carcinoma), sarcoma (angiosarcoma, neurogenic sarcoma, rhabdomyosarcoma, and high-grade sarcoma, not otherwise specified), primitive neuroectodermal tumors, and Wilms tumors, which are related to poor prognosis [35]. Conversely, somatic transformation in a metastatic rather than primary TGCT lesion is associated with an increased risk of mortality [36]. The primary treatment of these aggressive variants of PMGCTs is surgery, as most of them are not sensitive to cisplatin-based chemotherapies. Inoperable cases may be treated with chemotherapy regimens chosen according to the somatic transformation type [37][38].
References
- Rosti, G.; Secondino, S.; Necchi, A.; Fornarini, G.; Pedrazzoli, P. Primary mediastinal germ cell tumors. Semin. Oncol. 2019, 46, 107–111.
- Bokemeyer, C.; Nichols, C.R.; Droz, J.P.; Schmoll, H.J.; Horwich, A.; Gerl, A.; Fossa, S.D.; Beyer, J.; Pont, J.; Kanz, L.; et al. Extragonadal germ cell tumors of the mediastinum and retroperitoneum: Results from an international analysis. J. Clin. Oncol. 2002, 20, 1864–1873.
- Kesler, K.A.; Rieger, K.M.; Hammoud, Z.T.; Kruter, L.E.; Perkins, S.M.; Turrentine, M.W.; Schneider, B.P.; Einhorn, L.H.; Brown, J.W. A 25-year single institution experience with surgery for primary mediastinal nonseminomatous germ cell tumors. Ann. Thorac. Surg. 2008, 85, 371–378.
- Rivera, C.; Arame, A.; Jougon, J.; Velly, J.F.; Begueret, H.; Dahan, M.; Riquet, M. Prognostic factors in patients with primary mediastinal germ cell tumors, a surgical multicenter retrospective study. Interact. Cardiovasc. Thorac. Surg. 2010, 11, 585–589.
- Hartmann, J.T.; Nichols, C.R.; Droz, J.P.; Horwich, A.; Gerl, A.; Fossa, S.D.; Beyer, J.; Pont, J.; Kanz, L.; Einhorn, L.; et al. Prognostic variables for response and outcome in patients with extragonadal germ-cell tumors. Ann. Oncol. 2002, 13, 1017–1028.
- Group, I.G.C.C.C. International Germ Cell Consensus Classification: A prognostic factor-based staging system for metastatic germ cell cancers. J. Clin. Oncol. 1997, 15, 594–603.
- Gillessen, S.; Sauvé, N.; Collette, L.; Daugaard, G.; de Wit, R.; Albany, C.; Tryakin, A.; Fizazi, K.; Stahl, O.; Gietema, J.A.; et al. Predicting Outcomes in Men with Metastatic Nonseminomatous Germ Cell Tumors (NSGCT): Results from the IGCCCG Update Consortium. J. Clin. Oncol. 2021, 39, 1563–1574.
- Adra, N.; Althouse, S.K.; Liu, H.; Brames, M.J.; Hanna, N.H.; Einhorn, L.H.; Albany, C. Prognostic factors in patients with poor-risk germ-cell tumors: A retrospective analysis of the Indiana University experience from 1990 to 2014. Ann. Oncol. 2016, 27, 875–879.
- Hailemariam, S.; Engeler, D.S.; Bannwart, F.; Amin, M.B. Primary mediastinal germ cell tumor with intratubular germ cell neoplasia of the testis--further support for germ cell origin of these tumors: A case report. Cancer 1997, 79, 1031–1036.
- Ariel-Glenn, O.; Barkovich, A.J. Intracranial Germ Cell Tumors: A Comprehensive Review of Proposed Embryologic Derivation. Pediatr. Neurosurg. 1996, 24, 242–251.
- Chaganti, R.S.; Rodriguez, E.; Mathew, S. Origin of adult male mediastinal germ-cell tumours. Lancet 1994, 343, 1130–1132.
- Marx, A.; Chan, J.K.C.; Chalabreysse, L.; Dacic, S.; Detterbeck, F.; French, C.A.; Hornick, J.L.; Inagaki, H.; Jain, D.; Lazar, A.J.; et al. The 2021 WHO Classification of Tumors of the Thymus and Mediastinum: What Is New in Thymic Epithelial, Germ Cell, and Mesenchymal Tumors? J. Thorac. Oncol. 2022, 17, 200–213.
- Williamson, S.R.; Ulbright, T.M. Germ cell tumors of the mediastinum. In Pathology of the Mediastinum; Marchevsky, A.M., Wick, M.R., Eds.; Cambridge University Press: Cambridge, UK, 2000; pp. 146–168.
- Oosterhuis, J.W.; Looijenga, L.H.J. Testicular germ-cell tumours in a broader perspective. Nat. Rev. Cancer 2005, 5, 210–222.
- Moran, C.A.; Suster, S. Primary mediastinal choriocarcinomas: A clinicopathologic and immunohistochemical study of eight cases. Am. J. Surg. Pathol. 1997, 21, 1007–1012.
- Heerema-McKenney, A.; Bowen, J.; Hill, D.A.; Suster, S.; Qualman, S.J. Protocol for the examination of specimens from pediatric and adult patients with extragonadal germ cell tumors. Arch. Pathol. Lab. Med. 2011, 135, 630–639.
- Moran, C.A.; Suster, S. Primary germ cell tumors of the mediastinum: I. Analysis of 322 cases with special emphasis on teratomatous lesions and a proposal for histopathologic classification and clinical staging. Cancer 1997, 80, 681–690.
- Gonzalez-Vela, J.L.; Savage, P.D.; Manivel, J.C.; Torkelson, J.L.; Kennedy, B.J. Poor prognosis of mediastinal germ cell cancers containing sarcomatous components. Cancer 1990, 66, 1114–1116.
- Carver, B.S.; Bianco, F.J., Jr.; Shayegan, B.; Vickers, A.; Motzer, R.J.; Bosl, G.J.; Sheinfeld, J. Predicting teratoma in the retroperitoneum in men undergoing post-chemotherapy retroperitoneal lymph node dissection. J. Urol. 2006, 176, 100–103.
- Steyerberg, E.W.; Keizer, H.J.; Fosså, S.D.; Sleijfer, D.T.; Toner, G.C.; Schraffordt Koops, H.; Mulders, P.F.; Messemer, J.E.; Ney, K.; Donohue, J.P.; et al. Prediction of residual retroperitoneal mass histology after chemotherapy for metastatic nonseminomatous germ cell tumor: Multivariate analysis of individual patient data from six study groups. J. Clin. Oncol. 1995, 13, 1177–1187.
- Mead, G.M.; Stenning, S.P. The International Germ Cell Consensus Classification: A new prognostic factor-based staging classification for metastatic germ cell tumours. Clin. Oncol. 1997, 9, 207–209.
- Houldsworth, J.; Korkola, J.E.; Bosl, G.J.; Chaganti, R.S. Biology and genetics of adult male germ cell tumors. J. Clin. Oncol. 2006, 24, 5512–5518.
- Bosl, G.J.; Ilson, D.H.; Rodriguez, E.; Motzer, R.J.; Reuter, V.E.; Chaganti, R.S. Clinical relevance of the i(12p) marker chromosome in germ cell tumors. J. Natl. Cancer Inst. 1994, 86, 349–355.
- Zhao, G.-Q.; Dowell, J.E. Hematologic malignancies associated with germ cell tumors. Expert Rev. Hematol. 2012, 5, 427–437.
- Timmerman, D.M.; Eleveld, T.F.; Gillis, A.J.M.; Friedrichs, C.C.; Hillenius, S.; Remmers, T.L.; Sriram, S.; Looijenga, L.H.J. The Role of TP53 in Cisplatin Resistance in Mediastinal and Testicular Germ Cell Tumors. Int. J. Mol. Sci. 2021, 22, 11774.
- Bagrodia, A.; Lee, B.H.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic Determinants of Cisplatin Resistance in Patients with Advanced Germ Cell Tumors. J. Clin. Oncol. 2016, 34, 4000–4007.
- Bacon, J.V.W.; Giannatempo, P.; Cataldo, G.; Fazli, L.; Saxena, N.; Ozgun, G.; Soleimani, M.; Chi, K.; Nichols, C.; Necchi, A.; et al. TP53 Alterations Are Associated with Poor Survival in Patients with Primary Mediastinal Nonseminoma Germ Cell Tumors. Oncologist 2022, 27, e912–e915.
- Taylor, J.; Donoghue, M.T.; Ho, C.; Petrova-Drus, K.; Al-Ahmadie, H.A.; Funt, S.A.; Zhang, Y.; Aypar, U.; Rao, P.; Chavan, S.S.; et al. Germ cell tumors and associated hematologic malignancies evolve from a common shared precursor. J. Clin. Investig. 2020, 130, 6668–6676.
- Williams, L.A.; Pankratz, N.; Lane, J.; Krailo, M.; Roesler, M.; Richardson, M.; Frazier, A.L.; Amatruda, J.F.; Poynter, J.N. Klinefelter syndrome in males with germ cell tumors: A report from the Children’s Oncology Group. Cancer 2018, 124, 3900–3908.
- Kashima, S.; Saito, M.; Tsuchiya, N.; Saito, H.; Nanjo, H.; Numakura, K.; Tsuruta, H.; Akihama, S.; Inoue, T.; Narita, S.; et al. Primary Mediastinal Germ Cell Tumor Arising in A Patient with Neurofibromatosis Type 1. Nihon Hinyokika Gakkai Zasshi 2015, 106, 178–184.
- Nichols, C.R.; Roth, B.J.; Heerema, N.; Griep, J.; Tricot, G. Hematologic neoplasia associated with primary mediastinal germ-cell tumors. N. Engl. J. Med. 1990, 322, 1425–1429.
- Hartmann, J.T.; Nichols, C.R.; Droz, J.P.; Horwich, A.; Gerl, A.; Fossa, S.D.; Beyer, J.; Pont, J.; Fizazi, K.; Einhorn, L.; et al. Hematologic disorders associated with primary mediastinal nonseminomatous germ cell tumors. J. Natl. Cancer Inst. 2000, 92, 54–61.
- Mukherjee, S.; Ibrahimi, S.; John, S.; Adnan, M.M.; Scordino, T.; Khalil, M.O.; Cherry, M. Non-seminomatous mediastinal germ cell tumor and acute megakaryoblastic leukemia. Ann. Hematol. 2017, 96, 1435–1439.
- Akizuki, K.; Sekine, M.; Kogure, Y.; Kameda, T.; Shide, K.; Koya, J.; Kamiunten, A.; Kubuki, Y.; Tahira, Y.; Hidaka, T.; et al. TP53 and PTEN mutations were shared in concurrent germ cell tumor and acute megakaryoblastic leukemia. BMC Cancer 2020, 20, 5.
- Nichols, C.R. Mediastinal germ cell tumors. Semin. Thorac. Cardiovasc. Surg. 1992, 4, 45–50.
- Guo, C.C.; Punar, M.; Contreras, A.L.; Tu, S.M.; Pisters, L.; Tamboli, P.; Czerniak, B. Testicular germ cell tumors with sarcomatous components: An analysis of 33 cases. Am. J. Surg. Pathol. 2009, 33, 1173–1178.
- Ehrlich, Y.; Beck, S.D.W.; Ulbright, T.M.; Cheng, L.; Brames, M.J.; Andreoiu, M.; Foster, R.S.; Einhorn, L.H. Outcome analysis of patients with transformed teratoma to primitive neuroectodermal tumor. Ann. Oncol. 2010, 21, 1846–1850.
- Al-Hader, A.A.; Jain, A.; Al-Nasrallah, N.; Einhorn, L.H. Metastatic malignant transformation of teratoma to primitive neuroectodermal tumor (PNET): Results with PNET-based chemotherapy. Am. J. Clin. Oncol. 2015, 38, 364–366.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
885
Revisions:
2 times
(View History)
Update Date:
22 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No