Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chengjun Yao | -- | 1156 | 2023-02-21 10:28:26 | | | |
| 2 | Catherine Yang | Meta information modification | 1156 | 2023-02-22 02:00:16 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yao, C.; Zhou, H.; Dong, Y.; Alhaskawi, A.; Ezzi, S.H.A.; Wang, Z.; Lai, J.; Kota, V.G.; Abdulla, M.H.A.H.; Lu, H. Mechanisms of Malignant Peripheral Nerve Sheath Tumors Pathogenesis. Encyclopedia. Available online: https://encyclopedia.pub/entry/41465 (accessed on 25 June 2026).
Yao C, Zhou H, Dong Y, Alhaskawi A, Ezzi SHA, Wang Z, et al. Mechanisms of Malignant Peripheral Nerve Sheath Tumors Pathogenesis. Encyclopedia. Available at: https://encyclopedia.pub/entry/41465. Accessed June 25, 2026.
Yao, Chengjun, Haiying Zhou, Yanzhao Dong, Ahmad Alhaskawi, Sohaib Hasan Abdullah Ezzi, Zewei Wang, Jingtian Lai, Vishnu Goutham Kota, Mohamed Hasan Abdulla Hasan Abdulla, Hui Lu. "Mechanisms of Malignant Peripheral Nerve Sheath Tumors Pathogenesis" Encyclopedia, https://encyclopedia.pub/entry/41465 (accessed June 25, 2026).
Yao, C., Zhou, H., Dong, Y., Alhaskawi, A., Ezzi, S.H.A., Wang, Z., Lai, J., Kota, V.G., Abdulla, M.H.A.H., & Lu, H. (2023, February 21). Mechanisms of Malignant Peripheral Nerve Sheath Tumors Pathogenesis. In Encyclopedia. https://encyclopedia.pub/entry/41465
Yao, Chengjun, et al. "Mechanisms of Malignant Peripheral Nerve Sheath Tumors Pathogenesis." Encyclopedia. Web. 21 February, 2023.
Copy Citation
Malignant peripheral nerve sheath tumor (MPNST) is an aggressive soft tissue sarcoma with limited therapeutic options and a poor prognosis. Although neurofibromatosis type 1 (NF1) and radiation exposure have been identified as risk factors for MPNST, the genetic and molecular mechanisms underlying MPNST pathogenesis have only lately been roughly elucidated. Plexiform neurofibroma (PN) and atypical neurofibromatous neoplasm of unknown biological potential (ANNUBP) are novel concepts of MPNST precancerous lesions, which revealed sequential mutations in MPNST development.
malignant peripheral nerve sheath tumor
neurofibromatosis type 1
plexiform neurofibroma
1. Genetic Mechanism
Significant breakthroughs have been made in understanding the genetic mechanisms of MPNST in the previous decade. Researchers now have access to more genetic data related to MPNST because of the application of genetic sequencing in clinical diagnosis. In the case of NF1-associated MPNST, sequential, multiple-hit genetic changes may eventually lead to the transformation of Schwann cells into precancerous lesions and MPNST. Knocking out the NF1 from the cell (NF1flox/flox embryos) and mouse (Nf1flox/flox; DhhCre) models has been found to cause PNs similar to those seen in humans [1]. Simultaneously, the NF1 mutation duplicates are significantly associated with the development of lesions [1]. However, only the loss of NF1 cannot result in the malignant transformation of PN to MPNST, suggesting that other genes are involved in developing MPNST [2]. Loss of CDKN2A is a common alteration of ANNUBP and MPNST, which is unrelated to NF1-associated or sporadic causes [3][4]. The transition to the premalignant state is driven by the extent of CDKN2A/B deficiency [5]. A haploid mutation can lead to atypical neurofibroma, and the homozygous deletion is defined as ANNUBP [5]. Conditional ablation of NF1 and CDKN2A in the Schwann cell lineage results in ANNUBP and can progress to MPNST in the Postn-Cre Nf1flox/flox Arfflox/flox mouse model [6]. Function loss of polycomb repressive complex 2 (PRC2) is also associated with MPNST. Lee et al. found that dysfunction of PRC2 occurs in 70% of NF1-associated MPNST, 92% of sporadic MPNST, and 90% of radiotherapy-associated MPNST [7]. EED and SUZ12 are the core components of PRC2, which work together to methylate Lys27 of histone H3 to produce H3K27Me3, which can inhibit nerve repair genes [8]. Almost 80% of MPNST exhibited EED or SUZ12 deletion, resulting in the loss of PRC2 function and leading to H3K27Me3 global hypomethylation [7]. Researchers transferred wild type SUZ12 or EED into the MPNST cell line ST88-14 (which has lost H3K27Me3) to investigate the association between PRC2 and MPNST. After SUZ12 transfer, H3K27me3 levels recovered, and cell growth was significantly reduced [7]. Ma et al. found that EED knockdown can cause epigenetic changes by reducing H3K27me3; however, this effect was inhibited by the induction of CDKN2A. It is speculated that the co-mutation of PRC2 encoding genes and CDKN2A resulted in the progression of benign neurofibroma to MPNST [9]. The present findings suggest that mutations in NF1, CDKN2A, and genes encoding PRC2 complex proteins may play a synergistic role in the development of MPNST, particularly NF1-associated MPNST. Furthermore, the mutation order may also coordinate with the biomarker changes (Figure 1).
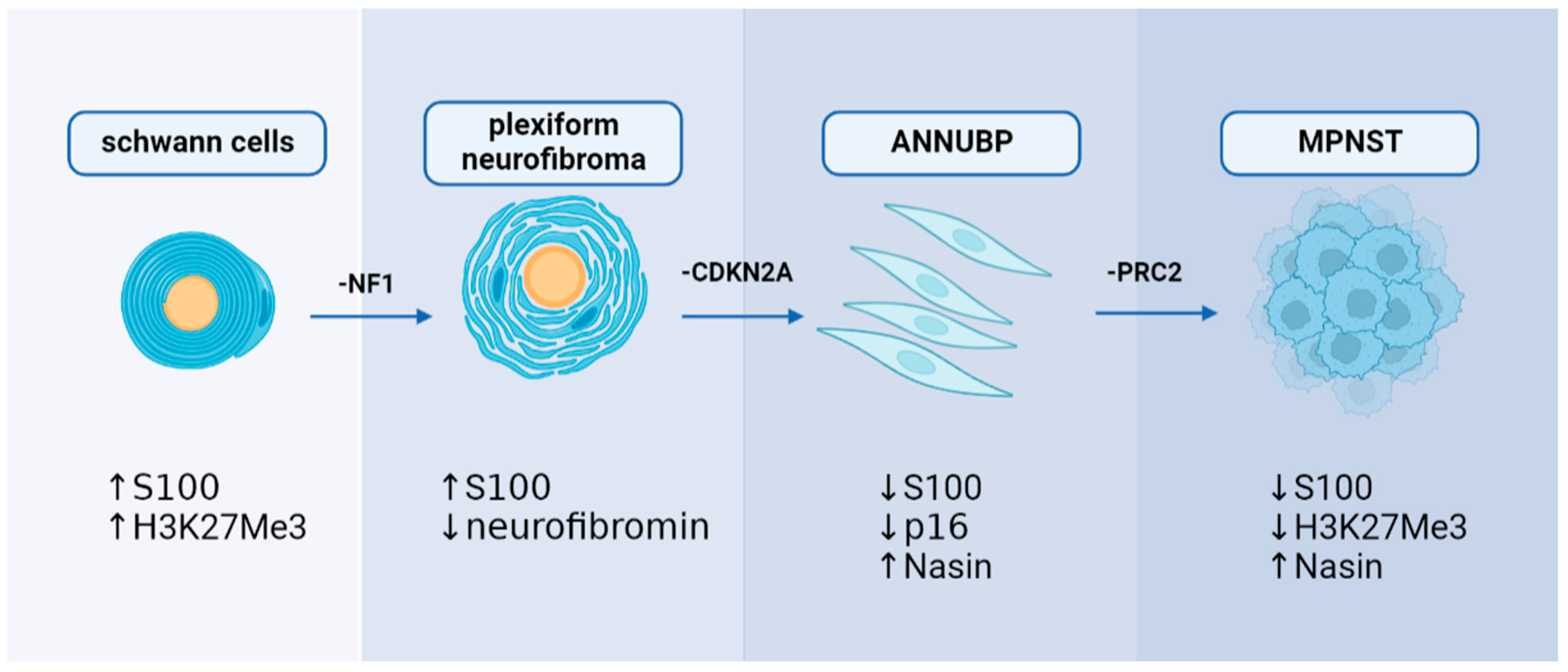
Figure 1. A hypothesis of sequential, multiple-hit genetic mutations in MPNST development: NF1, CDKN2A, and PRC2 may play a decisive role in MPNST development with alterations in immunohistochemical markers.
Several other genetic targets have been investigated concerning MPNST. Hirbe et al. illustrated that co-mutations of NF1 and TP53 can lead to MPNST in mouse models (Gfap-Cre Nf1flox/flox) without developing the PN phase [10]. This may indicate that TP53 is an independent prognostic factor for MPNST. Holtkamp et al. found increased EGFR expression, decreased ERBB2 expression, and decreased expression of the tumor suppressor gene PTEN in MPNST cells [11]. Animal experiments have also shown that only concurrent EGFR overexpression or PTEN deletion in the presence of NF1 mutations results in 100% MPNST manifestation [11].
However, only a few studies have revealed a possible pathogenesis for sporadic de novo MPNST. Some evidence suggests that sporadic MPNST has the same mutant genes, but in a different order, which may lead to atypical pathological changes and various prognoses. For example, the somatic NF1 mutation of sporadic MPNST is similar to NF1-associated MPNST [12]. CDKN2A or PRC2 mutations have also been seen in sporadic MPNST [13]. However, recent evidence indicates that some point mutations, such as BRAF V600E and NRAS Q61, are detected in sporadic MPNST. However, none of these mutations have been found in patients with NF1-associated MPNST or post-radiation MPNST [14]. Another study of 201 MPSNTs demonstrated that 11.9% of NF1-wild type MPSNTs harbored BRAF mutations compared to 2.9% of NF1-altered MPNSTs. CDKN2A is significantly altered in both NF1- and BRAF-altered MPNSTs [15]. The genetic differences between NF1-associated MPNST and sporadic MPNST may reflect different oncogenetic pathways [16].
2. Signaling Pathway and Microenvironment
The cellular signaling pathways of MPNST and the tumor microenvironment are important areas of future research. Particularly, alterations in the NF1 gene can result in abnormal activation of the RAS pathway, which can promote cell proliferation via the downstream RAF-MEK-ERK (MAPK) and PI3K-AKT-mTOR pathways. MEK is significantly activated in MPNST [17], and clinical evidence demonstrates that MEK inhibitors are significantly effective in PN [18] and MPNST [19]. The mTOR signaling pathway regulates cell growth, survival, proliferation, cytoskeletal organization, and autophagy [20]. Biplab Dasgupta et al. first reported that mTOR signaling is significantly activated in NF1-knockout cells and animals and that the rapamycin (mTOR inhibitor) can inhibit NF1-related tumor growth [21]. Johansson et al. found that everolimus inhibits the growth of 19–60% of NF1-associated or sporadic MPNST cell lines [22]; however, it is ineffective in clinical trials [23]. Wnt/β-catenin signaling is strongly associated with various human cancers [24]. A study showed that the gene expression of 20 components or regulators of the Wnt pathway are altered in MPNST compared to benign neurofibromas [25]. However, the precise role of the Wnt pathway in MPNST is still undetermined. Other signaling pathways including neuregulin-1/ErbB [26] and EGFR–STAT3 [27], which have also been reported in MPNST studies. In the MPNST microenvironment, an interesting finding is that the NF1+/− microenvironment may contribute to neurofibroma development [28]. It is possibly due to the alteration in immune cells, such as CD8+ T cells and natural killer cells, compared to wild-type counterparts [29]. NF1-heterozygous mast cells were also found to be activated by c-kit signaling and to promote tumor cell growth by releasing TGF-β [30], which leads to related clinical studies of tyrosine kinase receptors targeting KIT receptors [31]. Others, such as macrophages, are also involved in tumor formation [32]. Together, the alterations in cellular signaling pathways and the NF1+/− microenvironment promote cell survival and proliferation of MPNST (Figure 2).
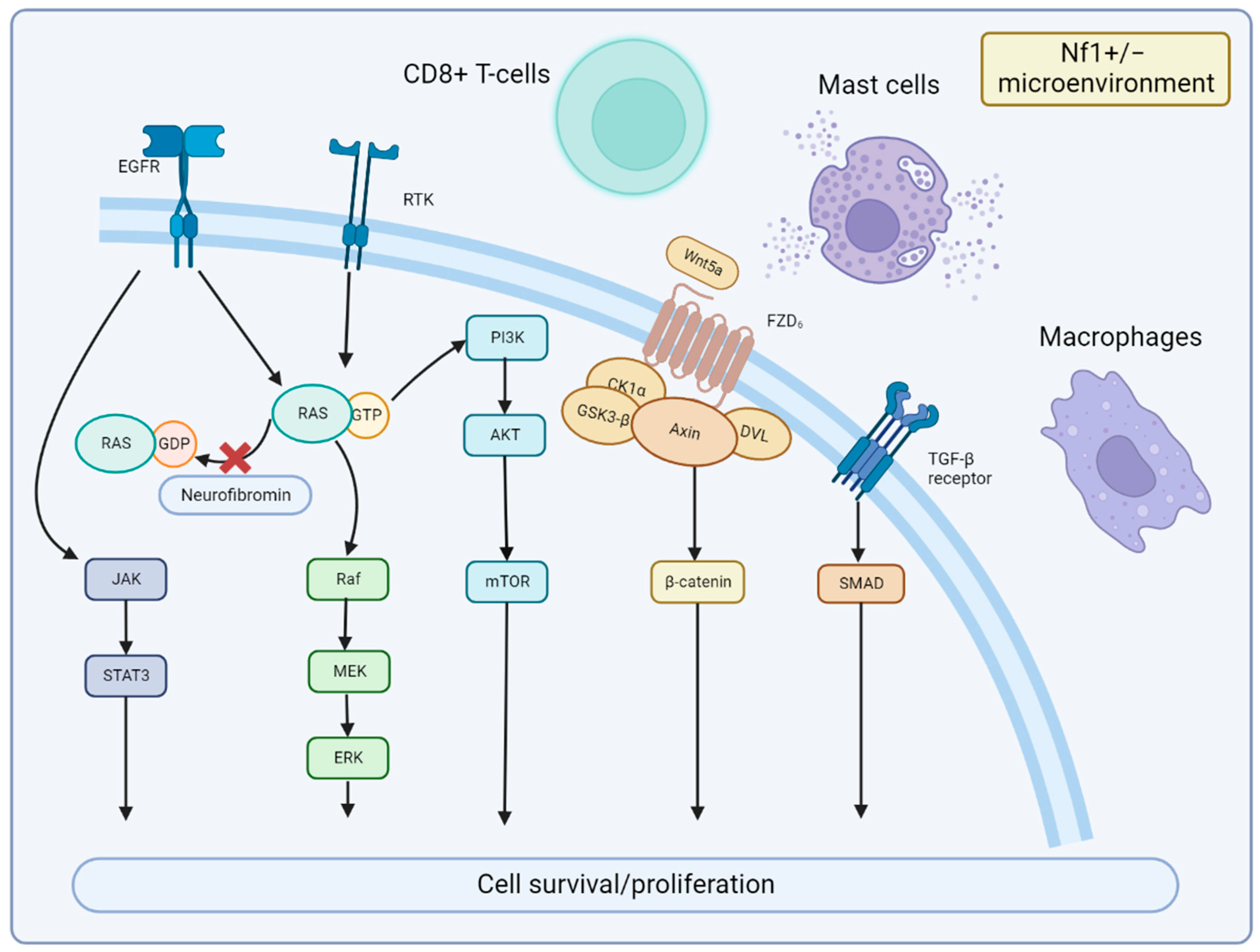
Figure 2. Signaling Pathways and Microenvironment of MPNST. Neurofibromin encoded by NF1 is lost in MPNST, which leads to the continued activation of RAS signaling and downstream MEK and mTOR effectors. JAK/STAT3 and Wnt/β-catenin signaling are also involved in MPNST development. Nf1+/− microenvironment of mast cells, CD8+ T cells, and macrophages can promote MPNST growth with cytokines or immune dysfunction.
References
- Wu, J.; Williams, J.P.; Rizvi, T.A.; Kordich, J.J.; Witte, D.; Meijer, D.; Stemmer-Rachamimov, A.O.; Cancelas, J.A.; Ratner, N. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell 2008, 13, 105–116.
- Brossier, N.M.; Carroll, S.L. Genetically engineered mouse models shed new light on the pathogenesis of neurofibromatosis type I-related neoplasms of the peripheral nervous system. Brain Res. Bull. 2012, 88, 58–71.
- Pemov, A.; Hansen, N.F.; Sindiri, S.; Patidar, R.; Higham, C.S.; Dombi, E.; Miettinen, M.M.; Fetsch, P.; Brems, H.; Chandrasekharappa, S.C.; et al. Low mutation burden and frequent loss of CDKN2A/B and SMARCA2, but not PRC2, define premalignant neurofibromatosis type 1-associated atypical neurofibromas. Neuro Oncol. 2019, 21, 981–992.
- Pemov, A.; Li, H.; Presley, W.; Wallace, M.R.; Miller, D.T. Genetics of human malignant peripheral nerve sheath tumors. Neuro-Oncol. Adv. 2020, 2, i50–i61.
- Carrió, M.; Gel, B.; Terribas, E.; Zucchiatti, A.C.; Moliné, T.; Rosas, I.; Teulé, Á.; Cajal, S.R.Y.; López-Gutiérrez, J.C.; Blanco, I.; et al. Analysis of intratumor heterogeneity in Neurofibromatosis type 1 plexiform neurofibromas and neurofibromas with atypical features: Correlating histological and genomic findings. Hum. Mutat. 2018, 39, 1112–1125.
- Rhodes, S.D.; He, Y.; Smith, A.; Jiang, L.; Lu, Q.; Mund, J.; Li, X.; Bessler, W.; Qian, S.; Dyer, W.; et al. Cdkn2a (Arf) loss drives NF1-associated atypical neurofibroma and malignant transformation. Hum. Mol. Genet. 2019, 28, 2752–2762.
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232.
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349.
- Ma, K.H.; Duong, P.; Moran, J.J.; Junaidi, N.; Svaren, J. Polycomb repression regulates Schwann cell proliferation and axon regeneration after nerve injury. Glia 2018, 66, 2487–2502.
- Hirbe, A.C.; Dahiya, S.; Friedmann-Morvinski, D.; Verma, I.M.; Clapp, D.W.; Gutmann, D.H. Spatially- and temporally-controlled postnatal p53 knockdown cooperates with embryonic Schwann cell precursor Nf1 gene loss to promote malignant peripheral nerve sheath tumor formation. Oncotarget 2016, 7, 7403–7414.
- Holtkamp, N.; Malzer, E.; Zietsch, J.; Okuducu, A.F.; Mucha, J.; Mawrin, C.; Mautner, V.F.; Schildhaus, H.U.; von Deimling, A. EGFR and erbB2 in malignant peripheral nerve sheath tumors and implications for targeted therapy. Neuro Oncol. 2008, 10, 946–957.
- Bottillo, I.; Ahiquist, T.; Brekke, H.; Danielsen, S.A.; van den Berg, E.; Mertens, F.; Lothe, R.A.; Dallapiccola, B. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J. Pathol. 2009, 217, 693–701.
- Prieto-Granada, C.N.; Wiesner, T.; Messina, J.L.; Jungbluth, A.A.; Chi, P.; Antonescu, C.R. Loss of H3K27me3 Expression Is a Highly Sensitive Marker for Sporadic and Radiation-induced MPNST. Am. J. Surg. Pathol. 2016, 40, 479–489.
- Kao, E.Y.; Wakeman, K.M.; Wu, Y.; Gross, J.M.; Chen, E.Y.; Ricciotti, R.W.; Liu, Y.J.; Mantilla, J.G. Prevalence and detection of actionable BRAF V600 and NRAS Q61 mutations in malignant peripheral nerve sheath tumor by droplet digital PCR. Hum. Pathol. 2022, 129, 90–97.
- Kaplan, H.G.; Rostad, S.; Ross, J.S.; Ali, S.M.; Millis, S.Z. Genomic Profiling in Patients With Malignant Peripheral Nerve Sheath Tumors Reveals Multiple Pathways With Targetable Mutations. J. Natl. Compr. Cancer Netw. JNCCN 2018, 16, 967–974.
- Plaat, B.E.C.; Molenaar, W.M.; Mastik, M.F.; Hoekstra, H.J.; Meerman, G.J.T.; van den Berg, E. Computer-assisted cytogenetic analysis of 51 malignant peripheral-nerve-sheath tumors: Sporadic vs. neurofibromatosis-type-1-associated malignant schwannomas. Int. J. Cancer 1999, 83, 171–178.
- Ambrosini, G.; Cheema, H.S.; Seelman, S.; Teed, A.; Sambol, E.B.; Singer, S.; Schwartz, G.K. Sorafenib inhibits growth and mitogen-activated protein kinase signaling in malignant peripheral nerve sheath cells. Mol. Cancer Ther. 2008, 7, 890–896.
- Dombi, E.; Baldwin, A.; Marcus, L.J.; Fisher, M.J.; Weiss, B.; Kim, A.; Whitcomb, P.; Martin, S.; Aschbacher-Smith, L.E.; Rizvi, T.A.; et al. Activity of selumetinib in neurofibromatosis type 1–related plexiform neurofibromas. N. Engl. J. Med. 2016, 375, 2550–2560.
- Nagabushan, S.; Lau, L.M.S.; Barahona, P.; Wong, M.; Sherstyuk, A.; Marshall, G.M.; Tyrrell, V.; Wegner, E.A.; Ekert, P.G.; Cowley, M.J.; et al. Efficacy of MEK inhibition in a recurrent malignant peripheral nerve sheath tumor. NPJ Precis. Oncol. 2021, 5, 9.
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71.
- Dasgupta, B.; Yi, Y.; Chen, D.Y.; Weber, J.D.; Gutmann, D.H. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1–associated human and mouse brain tumors. Cancer Res. 2005, 65, 2755–2760.
- Johansson, G.; Mahller, Y.Y.; Collins, M.H.; Kim, M.O.; Nobukuni, T.; Perentesis, J.; Cripe, T.P.; Lane, H.A.; Kozma, S.C.; Thomas, G.; et al. Effective in vivo targeting of the mammalian target of rapamycin pathway in malignant peripheral nerve sheath tumors. Mol. Cancer Ther. 2008, 7, 1237–1245.
- Widemann, B.C.; Meyer, C.F.; Cote, G.M.; Chugh, R.; Milhem, M.M.; Van Tine, B.A.; Kim, A.; Turpin, B.; Dombi, E.; Jayaprakash, N. SARC016: Phase II Study of Everolimus in Combination with Bevacizumab in Sporadic and Neurofibromatosis Type 1 (NF1) Related Refractory Malignant Peripheral Nerve Sheath Tumors (MPNST); American Society of Clinical Oncology: Alexandria, VA, USA, 2016.
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473.
- Luscan, A.; Shackleford, G.G.; Masliah-Planchon, J.; Laurendeau, I.; Ortonne, N.; Varin, J.; Lallemand, F.; Leroy, K.; Dumaine, V.; Hivelin, M.; et al. The Activation of the WNT Signaling Pathway Is a Hallmark in Neurofibromatosis Type 1 TumorigenesisWnt Pathway Activation in NF1 Tumorigenesis. Clin. Cancer Res. 2014, 20, 358–371.
- Stonecypher, M.S.; Byer, S.J.; Grizzle, W.E.; Carroll, S.L. Activation of the neuregulin-1/ErbB signaling pathway promotes the proliferation of neoplastic Schwann cells in human malignant peripheral nerve sheath tumors. Oncogene 2005, 24, 5589–5605.
- Wu, J.; Patmore, D.M.; Jousma, E.; Eaves, D.W.; Breving, K.; Patel, A.V.; Schwartz, E.B.; Fuchs, J.R.; Cripe, T.P.; Stemmer-O Stemmer-Rachamimov, A.; et al. EGFR–STAT3 signaling promotes formation of malignant peripheral nerve sheath tumors. Oncogene 2014, 33, 173–180.
- Brosseau, J.P.; Le, L.Q. Heterozygous Tumor Suppressor Microenvironment in Cancer Development. Trends. Cancer 2019, 5, 541–546.
- Brosseau, J.P.; Liao, C.P.; Wang, Y.; Ramani, V.; Vandergriff, T.; Lee, M.; Patel, A.; Ariizumi, K.; Le, L.Q. NF1 heterozygosity fosters de novo tumorigenesis but impairs malignant transformation. Nat. Commun. 2018, 9, 5014.
- Yang, F.-C.; Ingram, D.A.; Chen, S.; Zhu, Y.; Yuan, J.; Li, X.; Yang, X.; Knowles, S.; Horn, W.; Li, Y.; et al. Nf1-dependent tumors require a microenvironment containing Nf1+/−-and c-kit-dependent bone marrow. Cell 2008, 135, 437–448.
- Robertson, K.A.; Nalepa, G.; Yang, F.C.; Bowers, D.C.; Ho, C.Y.; Hutchins, G.D.; Croop, J.M.; Vik, T.A.; Denne, S.C.; Parada, L.F.; et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: A phase 2 trial. Lancet Oncol. 2012, 13, 1218–1224.
- Prada, C.E.; Jousma, E.; Rizvi, T.A.; Wu, J.; Dunn, R.S.; Mayes, D.A.; Cancelas, J.A.; Dombi, E.; Kim, M.O.; West, B.L.; et al. Neurofibroma-associated macrophages play roles in tumor growth and response to pharmacological inhibition. Acta Neuropathol. 2013, 125, 159–168.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
22 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No