Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Renata Esposito | -- | 2201 | 2023-02-20 02:50:24 | | | |
| 2 | Sirius Huang | Meta information modification | 2201 | 2023-02-20 03:20:19 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Esposito, R.; Mirra, D.; Spaziano, G.; Panico, F.; Gallelli, L.; D'agostino, B. Matrix Metalloproteinases in Cystic Fibrosis Pathogenesis. Encyclopedia. Available online: https://encyclopedia.pub/entry/41404 (accessed on 24 June 2026).
Esposito R, Mirra D, Spaziano G, Panico F, Gallelli L, D'agostino B. Matrix Metalloproteinases in Cystic Fibrosis Pathogenesis. Encyclopedia. Available at: https://encyclopedia.pub/entry/41404. Accessed June 24, 2026.
Esposito, Renata, Davida Mirra, Giuseppe Spaziano, Francesca Panico, Luca Gallelli, Bruno D'agostino. "Matrix Metalloproteinases in Cystic Fibrosis Pathogenesis" Encyclopedia, https://encyclopedia.pub/entry/41404 (accessed June 24, 2026).
Esposito, R., Mirra, D., Spaziano, G., Panico, F., Gallelli, L., & D'agostino, B. (2023, February 20). Matrix Metalloproteinases in Cystic Fibrosis Pathogenesis. In Encyclopedia. https://encyclopedia.pub/entry/41404
Esposito, Renata, et al. "Matrix Metalloproteinases in Cystic Fibrosis Pathogenesis." Encyclopedia. Web. 20 February, 2023.
Copy Citation
Cystic fibrosis (CF) is a high-prevalence disease characterized by significant lung remodeling, responsible for high morbidity and mortality worldwide. The lung structural changes are partly due to proteolytic activity associated with inflammatory cells such as neutrophils and macrophages. Matrix metalloproteases (MMPs) are the major proteases involved in CF, and recent literature data focused on their potential role in the pathogenesis of the disease. In fact, an imbalance of proteases and antiproteases was observed in CF patients, resulting in dysfunction of protease activity and loss of lung homeostasis.
cystic fibrosis
matrix metalloproteases
lung remodeling
1. Introduction
Cystic fibrosis (CF) is an autosomal recessive disorder and the most common life-limiting fatal disease in the Caucasian population, affecting approximately 100,000 individuals around the world and over 42,000 in Europe [1]. According to the CF Foundation 2018 Registry Report, the rate of survival for CF patients is 47.4 years (people born in 2018 in the United States) [2]. The mutation characterizing the disease affects the CF gene encoding a protein called “the cystic fibrosis transmembrane conductance regulator” (CFTR) [3]. CFTR is a chloride channel included in the adenosine triphosphate (ATP)-binding cassette (ABC) transporter family, whose primary function is mediating the passage of chloride ions and other electrolytes from inside to outside the epithelial cells on which it is expressed [4]. Once the epithelial surface is reached, CFTR acts synergistically with other ion channels such as epithelial sodium channels (ENaCs). Notably, in the lungs, CFTR controls the chloride and bicarbonate ion flux across the epithelium into the airway surface liquid (ASL), playing a crucial role in regulating the ASL pH and protein composition [5].
CF patients experience a loss of bicarbonate secretion in airways and hyperabsorption of Na+ from the ASL, which induces acidification, dehydration, volume depletion, and increased viscosity of the mucus [6]. This microenvironment promotes airway inflammation with the activation of airway epithelial cells and fibroblasts, progressive thickening of the mucus layer, and impaired mucociliary clearance [7][8]. Moreover, major bacterial and viral colonization and infection happen, thus contributing to self-perpetuating and chronic inflammation and airway injury [9][10]. However, CF variants influence other organs with secretory functions such as the gastrointestinal system and pancreas; however, progressive destruction of the pulmonary tissue remains the major cause of morbidity and mortality in CF subjects [11]. Immune system imbalance was observed in CF pathogenesis, with the presence of enormous activated phagocytes without efficient antibacterial activity, as well as leukocyte and neutrophil infiltration into the lungs [12]. Thus, Haemophilus influenzae, Pseudomonas aeruginosa, and Staphylococcus aureus easily flourish in a dysfunctional airway microenvironment, leading to persistent infection, which results in toxic product releases such as proteases, reactive oxygen species, and cytokines [13]. In fact, abnormal levels of inflammatory and remodeling mediators have been found in CF patient airways [14]. Consequently, further immune cell recruitment promotes progressive lung tissue remodeling, a phenomenon in response to aberrant attempts to repair injured tissue, characterized by hyperplasia of goblet and basal cells, squamous metaplasia, and an increase in epithelial height [15]. Hence, the integrity of lung tissue is lost and replaced by damaged structural cells. These changes promote progressive impaired lung function, hypoxemia, and bronchiectasis associated with pulmonary symptom onset and ventilation/perfusion mismatch in the CF lungs. They result in so-called acute pulmonary exacerbations (APEs), which in turn contribute to CF morbidity and mortality [16]. Interestingly, lung remodeling is already observed in the early life of both small and large CF patient airways [17][18]. In fact, increased activity related to proteases, including matrix metalloproteinases (MMPs), is already seen during childhood [8]; hence, an early diagnosis of the disease is essential, as is an early start of treatment [19]. Like a systemic disease, the pharmacological treatment of CF is very complex, and several drugs have been approved, in addition to symptomatic therapy with inhaled antibiotics, airway clearance techniques, pancreatic enzyme replacement, and nutritional support [20][21]. Currently, four different groups of CFTR modulators are commercially available (potentiators, correctors, stabilizers, and amplifiers), transforming CF from a life-limiting to a lifelong chronic disease. Notably, ivafactor is the first licensed modulator by the regulatory authorities [22][23] based on real-world efficacy with a 50% reduction in pulmonary exacerbation in patients who did not show improvements in FEV1 at the beginning of therapy [24]. Furthermore, a combination of drugs with different actions was introduced to maximize efficacy. In fact, a triple combination of elexacaftor/tezacaftor/ivacaftor results in more efficacy for patients with the F508del mutation, maximizing the rescue of CFTR function [25][26][27][28]. Unfortunately, despite the steps forward in the treatment of CF, identifying new targets and drugs remains a challenge. Dysregulated MMP activity has been linked to the pathogenesis of numerous chronic lung diseases, including asthma, emphysema, and acute lung injury [29][30]. In fact, abnormal remodeling of tissue associated with the accumulation of extracellular matrix (ECM) is a hallmark of these diseases in which MMPs play a key role. However, more recently, MMPs have been shown to be linked to lung remodeling, a key driver of lung severity in CF patients, but there are not yet any specific antifibrotic treatments [31].
2. MMPs in CF Pathogenesis
One of the most important features of CF onset is progressive lung remodeling promoted by a significant increase in protease activity. In fact, CF patients have an imbalance between proteases and protease inhibitors, required physiologically for the equilibration of defense mechanisms and prevention of tissue damage [32]. The alteration of these dynamic network leads to proteolytic activity dysfunction in the lung as reported by many studies that brought to light changes in protease levels, notably in MMPs [33].
Overall, more than 20 MMPs have been described as key drivers of lung remodeling [34], and researchers report those with significant dysfunctions related to CF onset and progression, whose modulation could represent an important step forward in diagnosis, monitoring, and additional therapy for better management of the disease.
MMPs are a superfamily of metallo-endopeptidases known as metzincins, synthesized as inactive proenzymes by several structural and immune cells such as macrophages, neutrophils, epithelial cells, endothelial cells, and fibroblasts [34]. While polymorphonucleates produce MMPs in a constitutive manner, many other cells release MMPs only after an inflammatory trigger and stimulation related to tissue remodeling and wound repair by transcription factor modulation [35]. Once synthesized, they became active by specific cleavage and, in turn, can cleave targeted substrates to perform biological functions. In fact, they contribute to the destruction of connective tissue and the alveolar epithelium, the release of cytokines and growth factors, and the regulation of cell mobility and migration by extracellular matrix (ECM) remodeling [36]. Moreover, they are involved in the wound repair process thanks to their capacity to catalyze the normal turnover of the ECM. In general, elevated MMP activity results in harm to lung homeostasis with low ECM turnover associated with an impaired repair phenomenon or excessive ECM accumulation associated with tissue fibrosis [37]. Moreover, MMPs can influence CFTR and ENAC channel structures via proteolytic breakdown, contributing to CF pathogenesis and progression [38]. Regardless of the molecular mechanism via which MMPs can promote CF onset, their role in the disease is supported by huge studies. For example, MMP-1, MMP-8, and MMP-9 levels were found higher in CF patients than in healthy controls [39]. Interestingly, a major increase in MMP-1 was observed during symptom exacerbations, indicating MMPs as potential additive biomarkers of disease severity. In contrast, other studies suggested a protective role of MMP-10 against bacterial infection because of its ability to modulate macrophage inflammation. However, the changes in MMP levels are often correlated to changes in MMP inhibitors such as α2-macroglobulin and the tissue inhibitors of metalloproteinases (TIMPs) [40]. A relationship between MMP and bacterial infection in CF subjects has also been found. Lastly, neutrophil elastase (NE) activity was found to be higher in CF childhood BAL, and its levels correlated with FEV1 [41]. Overall, MMPs can exert both beneficial and deleterious effects depending on the cellular source and the disease stage, Figure 1.
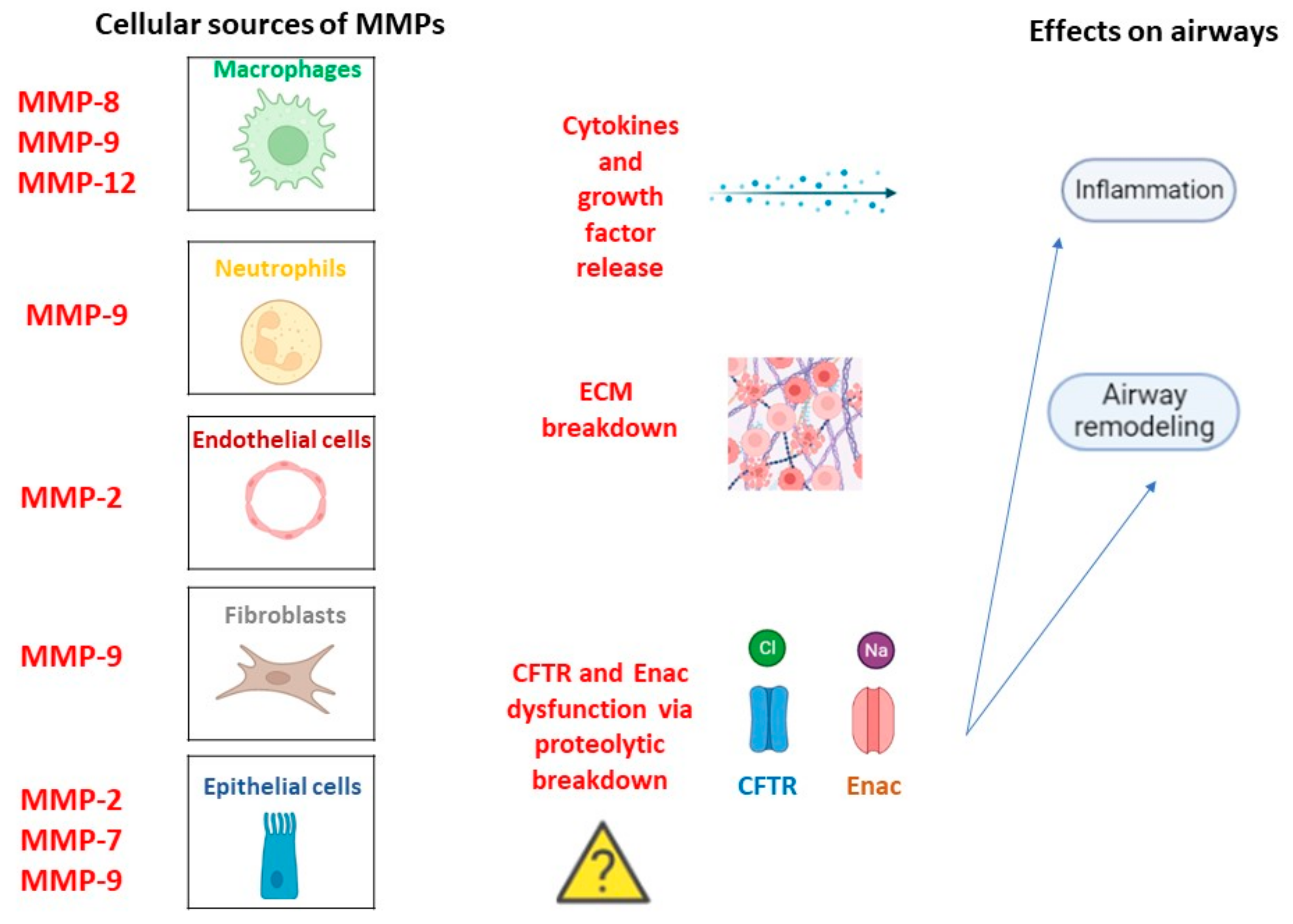
Figure 1. MMPs in CF pathogenesis. Cellular types involved in MMPs release in a constitutive manner or following inflammation trigger and related lung homeostasis dysfunctions.
MMP-9 represents the most abundant MMP in bronchopulmonary secretions derived from CF patients. MMP-9 is constitutively expressed by neutrophils, which are the main source of MMP-9 in neutrophilic inflammation characterizing CF disease. After inflammatory stimuli, other cellular types can produce MMP-9, such as macrophages and epithelial cells [42]. Once synthesized as a pro-enzyme, MMP-9 is further transformed into an active enzyme through the loss of its pro-domain. Many observational studies detected an association between BAL MMP-9 levels and CF progression, identifying elevated quantity and activity in the lower-airway secretions of CF patients [43][44][45]. Notably, BAL MMP-9 expression was related to CF severity in children with CF, pointing to its role in the earlier phase of the disease [46]. The role of NE as a key driver of MMP-9 activity is noteworthy, and researchers have referred to a causal link between the two proteases in CF as surrogated by a direct correlation between the increase in MMP-9 and NE activity [47]. Unlike NE, TIMP-1 is an endogenous inhibitor of MMP-9 activity, and a correlation between MMP-9 and TIMP-1 activity has been also detected [48]. In fact, an increase in the MMP-9/TIMP-1 ratio has been reported in the sputum and BAL of CF children and adults. The role of MMP-9 in the amplification of airway inflammation and associated lung tissue damage is supported by its ability to cleave and activate proinflammatory mediators such as IL-1-β and IL-8 release by inflammatory cells recruited into airways [49]. In turn, the proinflammatory transcription factor AP-1 is probably responsible for the increased expression of MMP-9 during inflammation. Moreover, MMP-9 levels were higher during CF progression, characterized by an acute pulmonary exacerbation associated with higher inflammation requiring systemic antibiotic treatment [50]. Overall, MMP-9 levels are positively correlated with the degradation of basement membrane collagen, decline in lung function, and onset of bronchiectasis in CF patients. Moreover, another substrate that can be activated by MMP-9 is serum surfactant (SP-D), an innate defense lectin secreted in the lungs. Previous studies identified serum SP-D as a potential biomarker of CF according to its adverse relationship with the FEV-1 parameter [51]. Therefore, the link between elevated levels of MMP-9 and SP-D is useful for supporting the role of MMP in CF pathogenesis. A recent study investigated both the levels and the activity of MMP-9, showing that blood MMP-9 activity is negatively associated with FEV-1, and that MMP-9 protein is positively associated with Staphylococcus aureus and Pseudomonas aeruginosa infections in CF. The significant correlation between FEV1 decline and MMP-9 increase could make MMP a useful additive biomarker of disease progression. However, the inhibition of MMP-9 alone did not affect goblet cell metaplasia, mucin secretion, and the emphysema onset in an animal study but reduced the bronchial obstruction by enhancing mucus clearance [52].
MMP-12 is produced by macrophages following an inflammatory trigger in CF airways. MMP-12 is known as macrophage elastase for its elastolytic capacity, and its function can be related to the migration of macrophages toward airway tissues [53]. Recently, the expression of MMP-12 in CF individuals has been demonstrated, and the association between MMP-12 increase and lung destruction has been reported [47][54]. Interestingly, the rs2276109 polymorphism, located in the MMP-12 promoter, was identified, and the decrease in MMP-12 expression linked to polymorphism was positively associated with the FEV1 percentage predicted in patients with CF [55]. To better understand the function of MMP-12 in CF pathogenesis, a βENaC-Tg mouse was used as an experimental animal model mimicking the CF disease, and the inhibition of MMP-12 significantly reduced the emphysema-like features. Taken together, these studies give interesting evidence that the proteolytic activity of MMP-12 may contribute to the pathogenesis of structural lung damage and lung function decline in patients with CF [54][56].
MMP-2 is expressed constitutively in lung cells and is involved in several biological functions such as inflammation and angiogenesis. Hence, MMP2 can boost airway injury by promoting abnormal remodeling and an impaired immune response to the infection in CF patients [57]. Interestingly, it seems able to influence CFTR activity, as reported by in vitro studies that describe a chloride flux increase after MMP-2 inhibition, supporting its involvement in ASL dehydration and increased bacterial colonization [58]. Otherwise, a recent paper found significantly low blood MMP-2 levels versus healthy subjects and even lower levels during acute CF exacerbation [59]. The authors speculated that the decreased expression of MMP-2 results from the binding site absence of proinflammatory transcription factors in the MMP-2 gene, which could hinder its activation by proinflammatory agents. Lastly, an upregulation of MMP-2 has been found during wound repair [60]. In fact, MMP-2 seems to be involved in epithelial–mesenchymal transition, in which airway epithelial cells start to produce collagen deposition [57]. However, further studies will be useful to elucidate the role of MMP-2 in CF onset and evolution.
MMP-7 is produced in constitutive manner by airway epithelial cells, and its significant increase has been observed in CF individuals [38]. Notably, MMP-7 levels were increased in migrating airway epithelial cells during human wound repair. Therefore, like other MMPs, MMP-7 can also modulate airway re-epithelialization, inflammation, host defense, and cell growth, playing a critical role in the injury lung response in CF patients [61][62].
MMP-8 is a neutrophil collagenase with several biological effects regarding the modulation of cytokines, immune activity, and repair process [63]. MMP-8 concentration has also been found augmented in the airway secretions and blood of patients with CF. Not strikingly, MMP-8 expression was significantly correlated with lung function decline (MMP-8 level vs. %FEV1, r = −0.468, p < 0.001) [59].
References
- Campagna, G.; Amato, A.; Majo, F.; Ferrari, G.; Quattrucci, S.; Padoan, R.; Floridia, G.; Salvatore, D.; Carnovale, V.; Puppo Fornaro, G.; et al. Gruppo di lavoro RIFC. Registro italiano Fibrosi Cistica (RIFC). Rapporto 2019-2020 . Epidemiol. Prev. 2022, 46 (Suppl. 2), 1–38.
- Cystic Fibrosis Foundation. 2018 Patient Registry: Annual Data Report. 2018. Available online: https://www.cff.org/sites/default/files/2021-11/Patient-Registry-Annual-Data-Report.pdf (accessed on 14 August 2020).
- O’Sullivan, B.P.; Freedman, S.D. Cystic fibrosis. Lancet 2009, 373, 1891–1904.
- Linsdell, P. Cystic fibrosis transmembrane conductance regulator (CFTR): Making an ion channel out of an active transporter structure. Channels 2018, 12, 284–290.
- Cuthbert, A.W. New horizons in the treatment of cystic fibrosis. Br. J. Pharmacol. 2011, 163, 173–183.
- Haq, I.J.; Gray, M.A.; Garnett, J.P.; Ward, C.; Brodlie, M. Airway surface liquid homeostasis in cystic fibrosis: Pathophysiology and therapeutic targets. Thorax 2016, 71, 284–287.
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in cystic fibrosis: An update. Pediatr. Pulmonol. 2018, 53 (Suppl. S3), S30–S50.
- Hilliard, T.N.; Regamey, N.; Shute, J.K.; Nicholson, A.G.; Alton, E.W.; Bush, A.; Davies, J.C. Airway remodelling in children with cystic fibrosis. Thorax 2007, 62, 1074–1080.
- De Boeck, K. Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatr. 2020, 109, 893–899.
- Li, Z.; Kosorok, M.R.; Farrell, P.M.; Laxova, A.; West, S.E.; Green, C.G.; Collins, J.; Rock, M.J.; Splaingard, M.L. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA 2005, 293, 581–588.
- McCague, A.F.; Raraigh, K.S.; Pellicore, M.J.; Davis-Marcisak, E.F.; Evans, T.A.; Han, S.T.; Lu, Z.; Joynt, A.T.; Sharma, N.; Castellani, C.; et al. Correlating Cystic Fibrosis Transmembrane Conductance Regulator Function with Clinical Features to Inform Precision Treatment of Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1116–1126.
- Hartl, D.; Gaggar, A.; Bruscia, E.; Hector, A.; Marcos, V.; Jung, A.; Greene, C.; McElvaney, G.; Mall, M.; Döring, G. Innate immunity in cystic fibrosis lung disease. J. Cyst. Fibros. 2012, 11, 363–382.
- Law, S.M.; Gray, R.D. Neutrophil extracellular traps and the dysfunctional innate immune response of cystic fibrosis lung disease: A review. J. Inflamm. 2017, 14, 29.
- Bergeron, C.; Cantin, A.M. Cystic Fibrosis: Pathophysiology of Lung Disease. Semin. Respir. Crit. Care Med. 2019, 40, 715–726.
- Montgomery, S.T.; Mall, M.A.; Kicic, A.; Stick, S.M.; Arest, C.F. Hypoxia and sterile inflammation in cystic fibrosis airways: Mechanisms and potential therapies. Eur Respir. J. 2017, 49, 1600903.
- Stenbit, A.E.; Flume, P.A. Pulmonary exacerbations in cystic fibrosis. Curr. Opin. Pulm. Med. 2011, 17, 442–447.
- Regamey, N.; Jeffery, P.K.; Alton, E.W.; Bush, A.; Davies, J.C. Airway remodelling and its relationship to inflammation in cystic fibrosis. Thorax 2011, 66, 624–629.
- Stoltz, D.A.; Meyerholz, D.K.; Welsh, M.J. Origins of cystic fibrosis lung disease. N. Engl. J. Med. 2015, 372, 1574–1575.
- Liu, G.; Philp, A.M.; Corte, T.; Travis, M.A.; Schilter, H.; Hansbro, N.G.; Burns, C.J.; Eapen, M.S.; Sohal, S.S.; Burgess, J.K.; et al. Therapeutic targets in lung tissue remodelling and fibrosis. Pharmacol. Ther. 2021, 225, 107839.
- Gentzsch, M.; Mall, M.A. Ion Channel Modulators in Cystic Fibrosis. Chest 2018, 154, 383–393.
- Proesmans, M. Best practices in the treatment of early cystic fibrosis lung disease. Ther. Adv. Respir. Dis. 2017, 11, 97–104.
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672.
- Bierlaagh, M.C.; Muilwijk, D.; Beekman, J.M.; van der Ent, C.K. A new era for people with cystic fibrosis. Eur. J. Pediatr. 2021, 180, 2731–2739.
- Heltshe, S.L.; Rowe, S.M.; Skalland, M.; Baines, A.; Jain, M. GOAL Investigators of the Cystic Fibrosis Foundation Therapeutics Development Network. Ivacaftor-treated Patients with Cystic Fibrosis Derive Long-Term Benefit Despite No Short-Term Clinical Improvement. Am. J. Respir. Crit. Care Med. 2018, 197, 1483–1486.
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819.
- Shaughnessy, C.A.; Zeitlin, P.L.; Bratcher, P.E. Elexacaftor is a CFTR potentiator and acts synergistically with ivacaftor during acute and chronic treatment. Sci. Rep. 2021, 11, 19810.
- Taylor-Cousar, J.L.; Mall, M.A.; Ramsey, B.W.; McKone, E.F.; Tullis, E.; Marigowda, G.; McKee, C.M.; Waltz, D.; Moskowitz, S.M.; Savage, J.; et al. Clinical development of triple-combination CFTR modulators for cystic fibrosis patients with one or two F508del alleles. ERJ Open Res. 2019, 5, 00082–02019.
- Bear, C.E. A Therapy for Most with Cystic Fibrosis. Cell 2020, 180, 211.
- Churg, A.; Zhou, S.; Wright, J.L. Series “matrix metalloproteinases in lung health and disease”: Matrix metalloproteinases in COPD. Eur. Respir. J. 2012, 39, 197–209.
- Chuliá-Peris, L.; Carreres-Rey, C.; Gabasa, M.; Alcaraz, J.; Carretero, J.; Pereda, J. Matrix Metalloproteinases and Their Inhibitors in Pulmonary Fibrosis: EMMPRIN/CD147 Comes into Play. Int. J. Mol. Sci. 2022, 23, 6894.
- Serra, R. Matrix Metalloproteinases in Health and Disease 2.0. Biomolecules 2022, 12, 1190.
- Oriano, M.; Amati, F.; Gramegna, A.; De Soyza, A.; Mantero, M.; Sibila, O.; Chotirmall, S.H.; Voza, A.; Marchisio, P.; Blasi, F.; et al. Protease-Antiprotease Imbalance in Bronchiectasis. Int. J. Mol. Sci. 2021, 22, 5996.
- Gaggar, A.; Hector, A.; Bratcher, P.E.; Mall, M.A.; Griese, M.; Hartl, D. The role of matrix metalloproteinases in cystic fibrosis lung disease. Eur. Respir. J. 2011, 38, 721–727.
- Greenlee, K.J.; Werb, Z.; Kheradmand, F. Matrix metalloproteinases in lung: Multiple, multifarious, and multifaceted. Physiol. Rev. 2007, 87, 69–98.
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233.
- McKelvey, M.C.; Brown, R.; Ryan, S.; Mall, M.A.; Weldon, S.; Taggart, C.C. Proteases, Mucus, and Mucosal Immunity in Chronic Lung Disease. Int. J. Mol. Sci. 2021, 22, 5018.
- Conese, M.; Di Gioia, S. Pathophysiology of Lung Disease and Wound Repair in Cystic Fibrosis. Pathophysiology 2021, 28, 155–188.
- Taggart, C.; Mall, M.A.; Lalmanach, G.; Cataldo, D.; Ludwig, A.; Janciauskiene, S.; Heath, N.; Meiners, S.; Overall, C.M.; Schultz, C.; et al. Protean proteases: At the cutting edge of lung diseases. Eur. Respir. J. 2017, 49, 1501200.
- Ratjen, F.; Hartog, C.M.; Paul, K.; Wermelt, J.; Braun, J. Matrix metalloproteases in BAL fluid of patients with cystic fibrosis and their modulation by treatment with dornase alpha. Thorax 2002, 57, 930–934.
- Yamamoto, K.; Murphy, G.; Troeberg, L. Extracellular regulation of metalloproteinases. Matrix Biol. 2015, 44–46, 255–263.
- Sly, P.D.; Gangell, C.L.; Chen, L.; Ware, R.S.; Ranganathan, S.; Mott, L.S.; Murray, C.P.; Stick, S.M.; AREST CF Investigators. Risk factors for bronchiectasis in children with cystic fibrosis. N. Engl. J. Med. 2013, 368, 1963–1970.
- Stamenkovic, I. Extracellular matrix remodelling: The role of matrix metalloproteinases. J. Pathol. 2003, 200, 448–464.
- Bergin, D.A.; Hurley, K.; Mehta, A.; Cox, S.; Ryan, D.; O’Neill, S.J.; Reeves, E.P.; McElvaney, N.G. Airway inflammatory markers in individuals with cystic fibrosis and non-cystic fibrosis bronchiectasis. J. Inflamm. Res. 2013, 6, 1–11.
- Gaggar, A.; Li, Y.; Weathington, N.; Winkler, M.; Kong, M.; Jackson, P.; Blalock, J.E.; Clancy, J.P. Matrix metalloprotease-9 dysregulation in lower airway secretions of cystic fibrosis patients. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L96–L104.
- Sagel, S.D.; Kapsner, R.K.; Osberg, I. Induced sputum matrix metalloproteinase-9 correlates with lung function and airway inflammation in children with cystic fibrosis. Pediatr. Pulmonol. 2005, 39, 224–232.
- Chakrabarti, S.; Patel, K.D. Matrix metalloproteinase-2 (MMP-2) and MMP-9 in pulmonary pathology. Exp. Lung Res. 2005, 31, 599–621.
- Garratt, L.W.; Sutanto, E.N.; Ling, K.M.; Looi, K.; Iosifidis, T.; Martinovich, K.M.; Shaw, N.C.; Kicic-Starcevich, E.; Knight, D.A.; Ranganathan, S.; et al. Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF). Matrix metalloproteinase activation by free neutrophil elastase contributes to bronchiectasis progression in early cystic fibrosis. Eur. Respir. J. 2015, 46, 384–394.
- Vandenbroucke, R.E.; Dejonckheere, E.; Libert, C. A therapeutic role for matrix metalloproteinase inhibitors in lung diseases? Eur. Respir. J. 2011, 38, 1200–1214.
- Van den Steen, P.E.; Proost, P.; Wuyts, A.; Van Damme, J.; Opdenakker, G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood 2000, 96, 2673–2681.
- Devereux, G.; Steele, S.; Jagelman, T.; Fielding, S.; Muirhead, R.; Brady, J.; Grierson, C.; Brooker, R.; Winter, J.; Fardon, T.; et al. An observational study of matrix metalloproteinase (MMP)-9 in cystic fibrosis. J. Cyst. Fibros. 2014, 13, 557–563.
- Bratcher, P.E.; Weathington, N.M.; Nick, H.J.; Jackson, P.L.; Snelgrove, R.J.; Gaggar, A. MMP-9 cleaves SP-D and abrogates its innate immune functions in vitro. PLoS ONE 2012, 7, e41881.
- Wagner, C.; Balázs, A.; Schatterny, J.; Zhou-Suckow, Z.; Duerr, J.; Schultz, C.; Mall, M.A. Genetic Deletion of Mmp9 Does Not Reduce Airway Inflammation and Structural Lung Damage in Mice with Cystic Fibrosis-like Lung Disease. Int. J. Mol. Sci. 2022, 23, 13405.
- Nighot, M.; Ganapathy, A.S.; Saha, K.; Suchanec, E.; Castillo, E.F.; Gregory, A.; Shapiro, S.; Ma, T.; Nighot, P. Matrix Metalloproteinase MMP-12 Promotes Macrophage Transmigration Across Intestinal Epithelial Tight Junctions and Increases Severity of Experimental Colitis. J. Crohns. Colitis. 2021, 15, 1751–1765.
- Wagner, C.J.; Schultz, C.; Mall, M.A. Neutrophil elastase and matrix metalloproteinase 12 in cystic fibrosis lung disease. Mol. Cell Pediatr. 2016, 3, 25.
- Dreymueller, D.; Uhlig, S.; Ludwig, A. ADAM-family metalloproteinases in lung inflammation: Potential therapeutic targets. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L325–L343.
- Trojanek, J.B.; Cobos-Correa, A.; Diemer, S.; Kormann, M.; Schubert, S.C.; Zhou-Suckow, Z.; Agrawal, R.; Duerr, J.; Wagner, C.J.; Schatterny, J.; et al. Airway mucus obstruction triggers macrophage activation and matrix metalloproteinase 12-dependent emphysema. Am. J. Respir. Cell Mol. Biol. 2014, 51, 709–720.
- Cheng, S.; Pollock, A.S.; Mahimkar, R.; Olson, J.L.; Lovett, D.H. Matrix metalloproteinase 2 and basement membrane integrity: A unifying mechanism for progressive renal injury. FASEB J. 2006, 20, 1898–1900.
- Duszyk, M.; Shu, Y.; Sawicki, G.; Radomski, A.; Man, S.F.; Radomski, M.W. Inhibition of matrix metalloproteinase MMP-2 activates chloride current in human airway epithelial cells. Can. J. Physiol. Pharmacol. 1999, 77, 529–535.
- Roderfeld, M.; Rath, T.; Schulz, R.; Seeger, W.; Tschuschner, A.; Graf, J.; Roeb, E. Serum matrix metalloproteinases in adult CF patients: Relation to pulmonary exacerbation. J. Cyst. Fibros. 2009, 8, 338–347.
- Caley, M.P.; Martins, V.L.; O’Toole, E.A. Metalloproteinases and Wound Healing. Adv. Wound Care 2015, 4, 225–234.
- Zuo, F.; Kaminski, N.; Eugui, E.; Allard, J.; Yakhini, Z.; Ben-Dor, A.; Lollini, L.; Morris, D.; Kim, Y.; DeLustro, B.; et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc. Natl. Acad. Sci. USA 2002, 99, 6292–6297.
- Rosas, I.O.; Richards, T.J.; Konishi, K.; Zhang, Y.; Gibson, K.; Lokshin, A.E.; Lindell, K.O.; Cisneros, J.; Macdonald, S.D.; Pardo, A.; et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008, 5, e93.
- Van Lint, P.; Libert, C. Matrix metalloproteinase-8: Cleavage can be decisive. Cytokine Growth Factor Rev. 2006, 17, 217–223.
More
Information
Subjects:
Respiratory System
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
892
Revisions:
2 times
(View History)
Update Date:
20 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No