Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Viana Jacquline Copeland | -- | 2916 | 2023-02-17 17:19:28 | | | |
| 2 | Rita Xu | Meta information modification | 2916 | 2023-02-20 06:06:40 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Copeland, V.J.; Fardman, A.; Furer, A. Right Ventricle-Predominant Cardiogenic Shock. Encyclopedia. Available online: https://encyclopedia.pub/entry/41370 (accessed on 24 July 2026).
Copeland VJ, Fardman A, Furer A. Right Ventricle-Predominant Cardiogenic Shock. Encyclopedia. Available at: https://encyclopedia.pub/entry/41370. Accessed July 24, 2026.
Copeland, Viana Jacquline, Alexander Fardman, Ariel Furer. "Right Ventricle-Predominant Cardiogenic Shock" Encyclopedia, https://encyclopedia.pub/entry/41370 (accessed July 24, 2026).
Copeland, V.J., Fardman, A., & Furer, A. (2023, February 17). Right Ventricle-Predominant Cardiogenic Shock. In Encyclopedia. https://encyclopedia.pub/entry/41370
Copeland, Viana Jacquline, et al. "Right Ventricle-Predominant Cardiogenic Shock." Encyclopedia. Web. 17 February, 2023.
Copy Citation
Cardiogenic shock (CS) remains a highly lethal condition despite many efforts and new interventions. Patients presenting with a rapid onset of hemodynamic instability and subsequent collapse require prompt and appropriate multimodality treatment. Multiple etiologies can lead to heart failure and subsequent shock. As the case prevalence of heart failure increases worldwide, it is of great importance to explore all manners and protocols of presentation and treatment present.
cardiogenic shock
right ventricle
right heart failure
1. Introduction
Cardiogenic shock (CS) describes a life-threatening circulatory failure resulting in high rates of mortality, despite a profound advancement in treatments that has been seen in recent years. In fact, with in-hospital mortality reaching up to 50% in some studies [1] and the incidence of CS constantly on the rise, this remains an important clinical condition to understand and study [2].
A wide array of etiologies could lead to the manifestation of CS, including both mechanical and structural abnormalities starting with acute myocardial infarction (AMI), as well as other causes leading to acute and chronic heart failure (HF). While there is an abundance of scientific literature assessing the association between AMI and CS relating to its pathogenesis, management and prognosis, there is relatively scarce knowledge when it concerns other etiologies. In particular, causes manifesting as right heart failure (RHF) resulting in CS have gained less focus, with fewer in-depth assessments conducted.
HF is described as a state of either inadequate cardiac contraction or the impairment of ventricular filling that leads to elevated intracardiac pressure and/or failure of the heart to adequately perfuse organ systems. Some relate to HF and CS as a continuum of the same disease spectrum, even at times approaching CS as an exacerbation of HF. HF could be dissected into subcategories by acuity and by the predominant chamber of the heart that is involved, with each subtype characterized by different symptomatology and prognostic qualities. In fact, this condition is now considered one of the leading causes of morbidity and mortality in the world, with an estimated prevalence of up to 2% in developed countries; this is expected to be on the rise with an aging population globally [3].
Though CS associated with RHF represents only 5% of CS patients, based on recent data, RHF rates are rising. This is thought to be due to the increased life expectancy, the improvement in diagnostic modalities and the increased awareness. Additionally, a substantial quantity of RHF cases are a result of left ventricular (LV) dysfunction, thus further increasing the prevalence [4][5]. It is difficult to estimate the exact prevalence of RHF, as RV-predominant HF remains the final common result in many disease states, and the true prevalence of right ventricular dysfunction (RVD) is therefore most likely underreported [6]. It is thought that, in some HF subtypes, RVD is present in up to 50% of patients, while assessment with cardiac magnetic resonance (CMR) imaging yielded a figure reaching around 19% [7][8]. Though the LV dysfunction associated with CS is more common, representing the majority of cases, right ventricle (RV) damage and subsequent CS are characterized by a more rapid deterioration, reaching hemodynamic collapse earlier than that of LV dysfunction and a higher short-term mortality [9]. However, the RV has better short-term recovery potential, making it critical that patients receive prompt and appropriate treatment, ensuring the opportunity for recovery [8].
2. The Right Ventricle—Hemodynamics and Anatomical Characteristics
The high sensitivity of the RV to changes in pressure originates from the nature of the RV, with its high-compliance, low-resistance pulmonary circulation, which makes it suited to adapt to changes in volume rather than pressure [10]. In fact, while RV output approximates that of the left, it is actually attained with a myocardial energy demand of approximately one-fifth that of the left [11]. Pulmonary vascular resistance is estimated to be less than a tenth of the systemic vascular resistance, thus explaining the RV’s milder work force and also its fragility when abrupt changes in pressure occur. It is also thought that the hydrodynamic cycle of the RV differs from the left, lacking an isovolumic period. A phase of isovolumic contraction, where ejection from the RV begins during the pressure upstroke, is hard to define. The RV’s pressure–volume loop lacks the isovolumic phases of contraction and relaxation during systole and diastole [10][12][13][14]. There is a substantial difference between ventricles in the pressure–volume (PV) relationship, as can be seen by a schematic curve of the normal RV. A phenomenon, known as the ‘hangout period’, is unique to the right chamber and is defined by the time lagging from the point in the cycle where the pressure in the pulmonary artery rises above that in the RV and up to the point where the pulmonic valve actually closes. This does not happen with the aortic valve, which closes earlier subsequently to the much higher systemic resistance in comparison with the pulmonary vascular resistance. The meaning of this in terms of the pressure–volume relationship is the practical absence of a clearly defined isovolumic relaxation time. The RV PV curve has a more triangular shape as compared to the square shape of the LV curve [11][13]. Nonetheless, the relationship of instantaneous pressure to volume is as linear as that found in the LV in varying physiological ranges [13] (Figure 1).
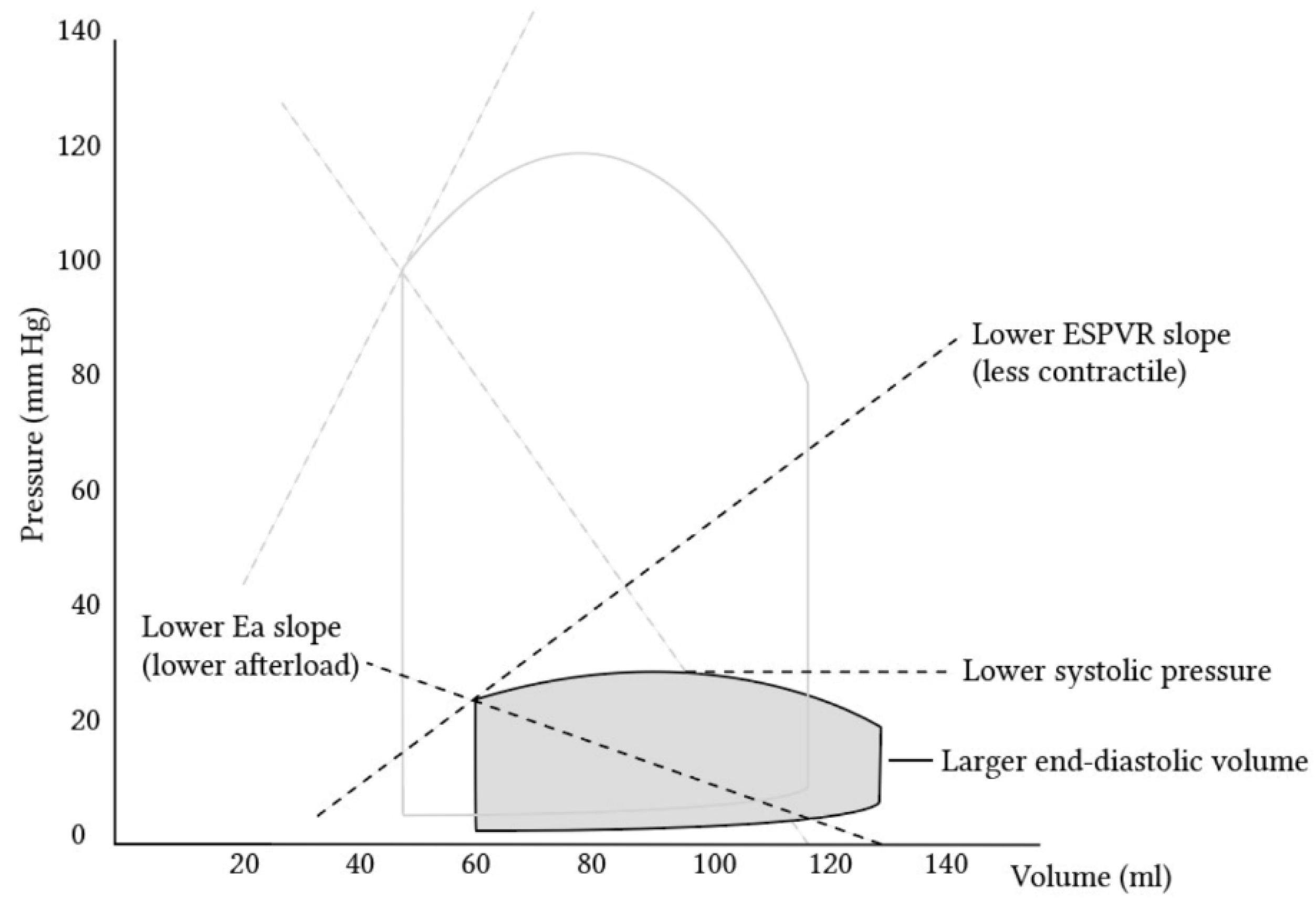
Figure 1. Pressure–volume loops comparing the left ventricle (transparent) and right ventricle (gray). It is visible that the right ventricle creates substantially lower pressures, among other differences. (Adopted with permission from https://derangedphysiology.com by Alex Yartsev [15]).
Additionally, the ventricular shapes of the two ventricles differ—the LV has an ellipsoidal shape, while the RV has a more triangular shape when assessed from the anterior frontal view and a crescent shape when viewed in a cross-sectional cut. Other factors influence the reduced strength generated by the RV, such as the different consistency of the ventricle compared to that of the left. The RV consists of circumferential and longitudinal orientations, while the LV obliquely arranges myofibers superficially and longitudinal myofibers in the sub-endocardium, with predominantly circular myofibers in between. This arrangement makes the LV more durable, with reduced points of frailty due to the interchanging fiber arrangement [14]. Another aspect would be the thickness of the RV, which is known to be substantially thinner compared to the LV. While the LV wall thickness is between 6 and 10 mm, the RV is 3–5 mm thick (without trabeculations) [16], making it substantially more difficult to generate a counterforce to the pressure and thus weaker. Wall tension is another factor of force that differs among the ventricles, with the thickness influencing the tension capacity. The RV, being thinner, has a milder surface tension, not being able to inhibit backflow during the systole. This would explain the RV’s inability to withstand a high afterload facing a high pressure, but it is very adapted to withstand a high preload and its volume, particularly as it pumps blood to a low-pressure system [14][17]. This would also explain the difficulty in accurately assessing the RV volume, particularly among different stages of the cardiac cycle, yielding overestimating values many times. Common methodologies for calculating the volume include Simpson’s rule and gated blood pool radionuclide angiography [12][18].
It Is important to note that, with the two ventricles sharing a common septum, this union affects the function of each ventricle, particularly that of the right. With the contraction of the oblique muscle of the LV, with a circular rotation and an LV contractile twist, a longitudinal movement of the heart occurs, with an anchoring source of the septum. This sends force to the RV, meaning that the LV is responsible for a portion of the contractile force of the RV.
3. Etiology and Pathophysiology of CS
As mentioned above, hypoperfusion is the landmark of cardiogenic shock, and once it is sensed by carotid baroreceptors and juxtaglomerular cells in the kidney, a reflexive sympathetic and neurohormonal response occurs. An increase in catecholamines leads to vascular endothelial constriction. With low perfusion, the renin–angiotensin–aldosterone axis is activated, consequently promoting salt and water retention. These steps result in an increased myocardial afterload and circulating plasma volume. If treatment is not promptly applied, a cycle of decreasing cardiac output (CO) and progressive volume overload ensues, leading to a reduction in coronary artery perfusion pressure, myocardial ischemia, worsening cardiac function and circulatory collapse [14].
When examining an RV insult and its progression to shock, acute damage and subsequent failure lead to a reduction in the LV preload, reduced CO and shock [10]. This is despite normal or only mildly reduced LV contractility. A reduction in RV contractility can also cause reduced right heart output. This is often a result of myocardial fibrosis due to either an infarction or myocarditis. With decreased RV SV, RV dilation occurs, which increases counter-pressure on the tricuspid valve, resulting in tricuspid regurgitation (TR), exacerbates RV dilation and drives a ventricular-interdependent effect on LV filling. This interdependence is described as forces directly transmitted from one ventricle to the other through the myocardium and pericardium [10]. RV dilation and the shift of the interventricular septum toward the LV can damage LV function and exacerbate CS. This is also known as the ‘paradoxical’ movement of the interventricular septum and is accompanied by increased intrapericardial pressure, all of which lead to increased LV end-diastolic pressure and reduced LV transmural filling pressure, depriving LV diastolic filling, which further reduces CO. Subsequently, this state is greatly influenced by the loading of both ventricles (circulating volume), the interventricular septal function and the contribution of the right atrium to RV filling. The latter is very sensitive to disturbances in the normal cardiac rhythm, which can be altered by arrhythmias such as atrial fibrillation. Overall, all of this results in decreased coronary perfusion. Consequently, this state of reduced CO results in tissue hypoxemia and end organ damage [10][18][19][20].
Volume overload can also contribute to RV failure and subsequent CS in special circumstances involving the use of mechanical support devices. A common instance would be a congested state after LVAD implantation. LVAD unloads the LV and derives an increased venous return to the right side of the heart, which can exacerbate pre-existing RV failure [21].
With the volume expansion and the subsequent rise in pressure, the pressure–volume loop of the right ventricle shifts, causing suboptimal ejection and cardiac function (Figure 2).
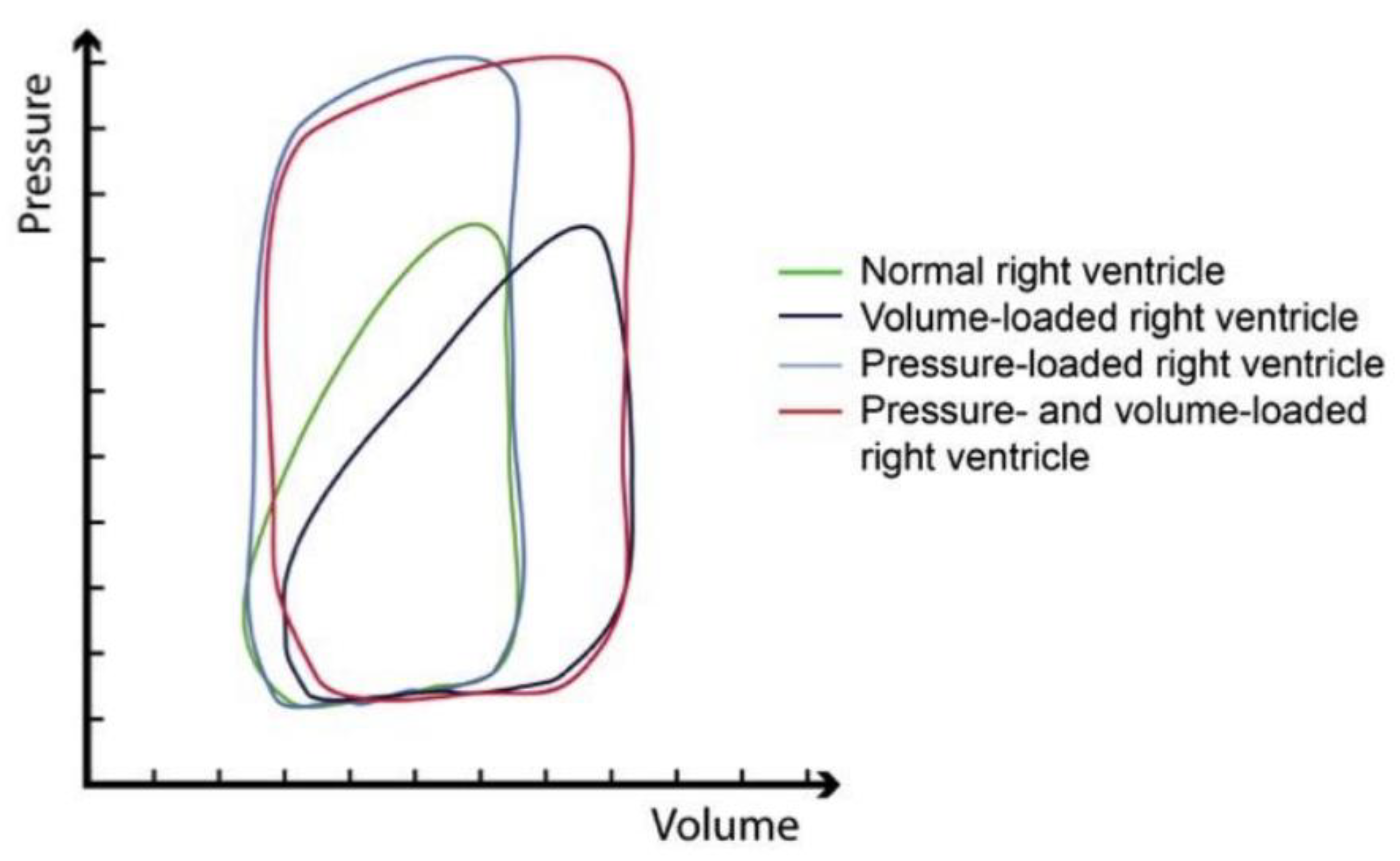
Figure 2. Pressure–volume loops of the right ventricle in a normal state (green); conditions of volume overload (dark blue); condition of pressure overload (light blue); mixed pressure and volume overload (red). Notably, while the shape of the curve changes in a very minor manner with higher volumes, the curve changes substantially and resembles that of a left ventricle when the right ventricle is required to cope with higher pressure. (Adopted with permission from De Meester et al. [22]).
Right heart involvement in CS could also be approached by dividing the pathophysiology according to the higher afterload the ventricle needs to work against, the declined contractility or the reduced preload of the RV. While, physiologically, the RV has an advantage of pumping blood into a low-compliance pulmonary vasculature, in diseased states such as heart failure, pulmonary hypertension or acute pulmonary embolism, a rise in the RV afterload increases RV pressures and volumes and consequently reduces the RV stroke volume [9]. It is noteworthy to remember that RV afterload is related not just to pulmonary vascular compliance but also to pulmonary parenchymal compliance and intrathoracic pressures [6]. Thus, once congestion occurs, the right overwhelming pressure leads to a reduction in myocardial contractility, insufficient blood oxygenation and output and subsequent congestion [6][23]. It is important to note that LV failure is also a cause of RV failure, mostly affecting it by increasing the RV afterload caused due to the higher LV filling pressures, the lower PA compliance and, therefore, the higher PA resistance and impedance [9]. Notably, the difference between RVD and RVF is that RVD is the anatomical and physiological malfunction of the RV, either due to over-relaxation or hyper-constriction, while RVF is the clinical manifestation of the RVD (Figure 3).
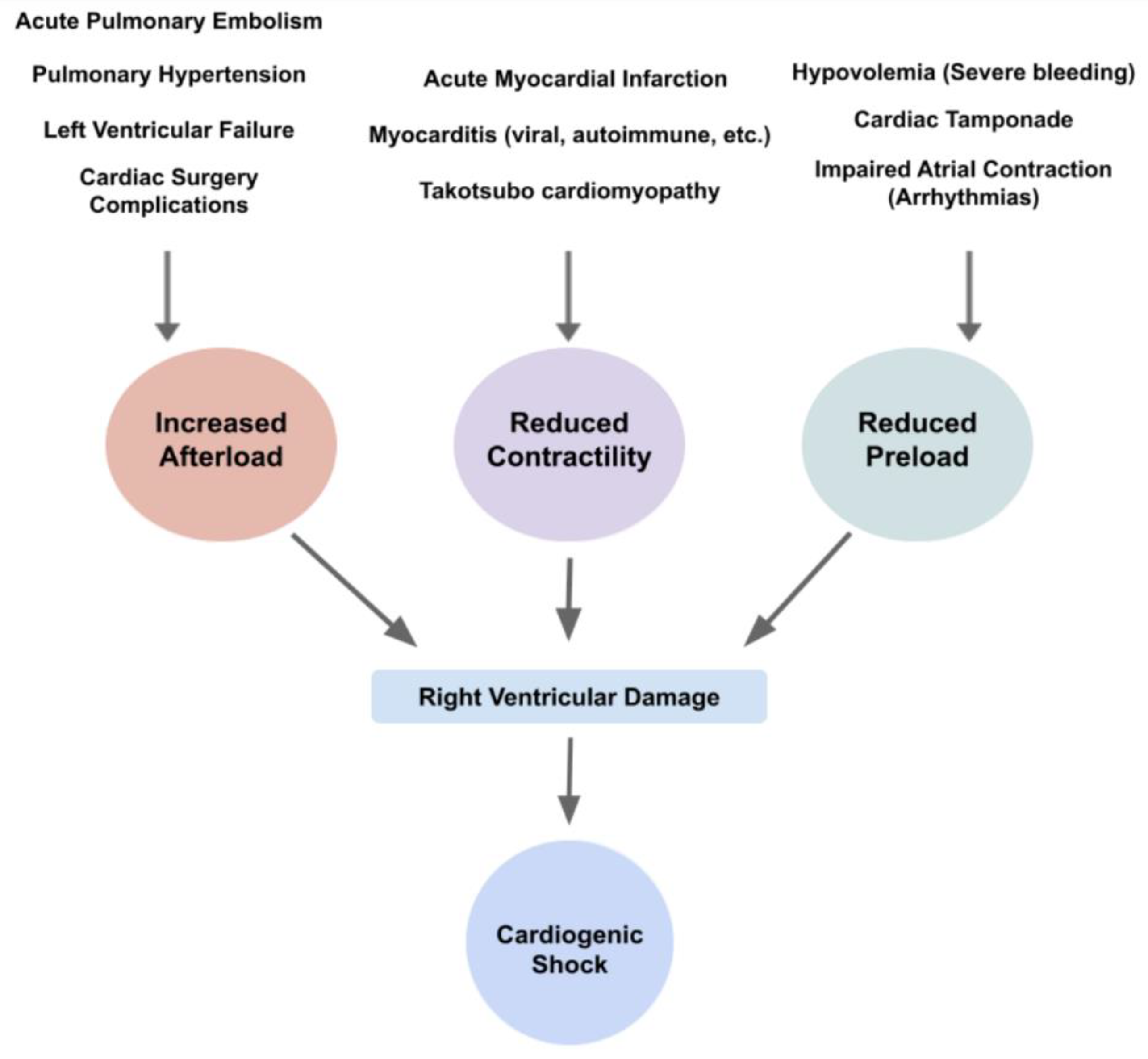
Figure 3. Diagram of etiologies and their physiological changes, leading to right ventricular damage and subsequent cardiogenic shock.
Additionally, it is essential to note that acute insults to an already ailing heart, such as chronic heart failure, can lead to CS. With the gradual increase in pressure and the more indolent decline in stability, the RV can become engorged, particularly in cases where the pericardium is intact. This can subsequently lead to the impairment of the LV chamber, impeding LV filling and equalizing biventricular diastolic pressures. RV systolic and biventricular diastolic dysfunction reduces CO, reducing coronary blood flow and worsening peripheral and abdominal volume accumulation. This could potentially lead to biventricular heart failure. This state is a deadly cycle, as, by disturbing the LV, reduced left-sided heart (LH) filling is more likely to cause RV dilation and ventricular interdependence and further hamper the RV output [10].
3.1. Acute RHF
RV–AMI, which occurs acutely, has strong hemodynamic implications, with approximately 25% to 50% of RV infractions being hemodynamically significant due to reduced contractility [24]. The occlusion of supplying coronary arteries causes oxygendeprived cardiomyocytes, leading to tissue damage and necrosis with subsequent alterations in tissue elasticity and contractility [2].
Hemodynamic instability depends upon the extent of the ischemia and infarcted myocardium. Furthermore, elevated filling pressures of the right side of the heart also cause coronary sinus congestion, which deprives the myocardium from coronary blood flow and can provoke RV ischemia and worsen RV function [10]. Subsequent CS development is dependent upon the extent of the damage, with isolated right ventricular infarction representing 5.5% of total CS cases, though some research shows up to 7% [14][25]. This is supported by recent data [26] indicating that isolated RVF is rarer, involving 7% of AMI patients, of which 88% had global RVD on echocardiography and 50% initially had a CVP > 15 mmHg. Additionally, it is thought that a substantial number of total AMI patients have some form of RV impairment, regardless of left-sided integrity. Furthermore, ischemic RVD is observed in up to 50% of patients with inferior AMI. Consequently, acute hemodynamic compromise is evident in less than half of these patients [6]. In the case of acute pulmonary embolism (PE), acute RV involvement is evident in 25–60% of patients [6]. PE occurs as a result of an obstruction in the pulmonary arteries. Obstruction is most often a result of a thrombus originating in the distal venous system, which dislodges and embolizes the pulmonary arterial system. With thrombus sizes differing, obstructive shock can occur due to large occlusions. Some retrospective assessments show that CS prevalence due to acute PE can reach as high as 18.4%. Among patients with acute PE, the total mortality is as high as 15% in the subgroup of patients who present with severe hypotension or cardiogenic shock [27]. Around 8% of acute PEs cause sudden cardiac arrest [28].
Additionally, cardiac tamponade, a pericardial emergency with fluid accumulation, is a worrisome form of CS which can impair RV function [8]. The rapid accumulation of either pus, blood, clots or gas within the pericardial space that compresses the heart leads to a reduction in diastolic filling, preload and contraction [8]. Some causes of cardiac tamponade include transmural myocardial infarction, malignancy, idiopathic, bacterial or tuberculous pericarditis and myocarditis with pericardial involvement [29].
Inflammation of the myocardium, also known as myocarditis, can potentially damage the integrity of the muscle, leading to a decline in RV contractility. There are several causes that lead to myocarditis, most prominently viral, microbial and autoimmune. Viruses represent a major pathogenic factor, which include enteroviruses such as Coxsackie B virus, erythroviruses (most prominently, Parvo B19), adenoviruses and herpes viruses [30]. In the last few years, severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) has also caused a prominent disease burden rise due to the COVID-19 pandemic and has been a concerning sequela and complication of the infection. It is thought that approximately 20% of hospitalized patients with SARS-CoV-2 have evidence of cardiac injury, as indicated by elevated levels of high-sensitivity troponin [31]. Generally, it is thought that RV involvement may reflect a greater burden of inflammation compared to LV, though it could also be due to pre-existing vulnerability to an acute process or an increased afterload caused by LHF [10]. Furthermore, a subsequent exposure to the myocardial antigen can progress to autoimmune myocarditis, either post-infectious or idiopathic [30]. In cases of autoimmune myocarditis, some studies have shown that RVD was present in 39% of patients with anti-heart autoantibodies; this is compared to 17% of those without anti-heart autoantibodies [10].
Other infectious causes of acute functional impairment include systemic infection and sepsis, which have been shown to increase pulmonary vascular pressure and propose both to HF and CS [10]. This is thought to be due to cytokine-mediated myocardial depression [32].
Acute LV failure can cause congestion and, in severe cases, lead to acute RV decompensation and shock [8]. Similarly to the causes of acute RHF, the most prominent cause of acute LV dysfunction is a consequence of AMI or the exacerbation of chronic LV conditions [33].
Additional causes, though less prevalent, include various cardiac surgeries that can lead to CS. RV failure in these cases is caused by a volume overload through complications of myocardial ischemia and arrhythmias due to surgery [8]. Furthermore, thyrotoxicosis has also been documented as a cause of acute RHF [34]. Lastly, it has been documented that a rare form of Takotsubo cardiomyopathy solely affecting the RV can cause CS. This condition is thought to be due to emotional strain and can cause cardiac insults [35].
3.2. Acute on Chronic RHF
For patients suffering from chronic right HF from various causes (valvular, pulmonary HTN, etc.), even a slight insult can lead to CS, especially among chronic RVD with an elevated pulmonary afterload. A second acute hit due to either an ischemic event, volume overload or intra/post-operative complications could definitely lead to RV-predominant CS [10].
References
- Chioncel, O.; Mebazaa, A.; Harjola, V.-P.; Coats, A.J.; Piepoli, M.F.; Crespo-Leiro, M.G.; Laroche, C.; Seferovic, P.M.; Anker, S.D.; Ferrari, R.; et al. Clinical phenotypes and outcome of patients hospitalized for acute heart failure: The ESC Heart Failure Long-Term Registry. Eur. J. Heart Fail. 2017, 19, 1242–1254.
- Ordonez, C.P.; Garan, A.R. The landscape of cardiogenic shock: Epidemiology and current definitions. Curr. Opin. Cardiol. 2022, 37, 236–240.
- Roger, V.L. Epidemiology of Heart Failure. Circ. Res. 2013, 113, 646–659.
- Mandras, S.A.; Desai, S. Right Heart Failure. . In StatPearls ; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459381/ (accessed on 18 December 2022).
- Albakri, A. Right heart failure: A review of clinical status and meta-analysis of diagnosis and clinical management methods. Intern. Med. Care 2018, 2, 1–12.
- Kanwar, M.K.; Everett, K.D.; Gulati, G.; Brener, M.I.; Kapur, N.K. Epidemiology and management of right ventricular-predominant heart failure and shock in the cardiac intensive care unit. Eur. Heart J. Acute Cardiovasc. Care 2022, 11, 584–594.
- Gorter, T.M.; Hoendermis, E.S.; Van Veldhuisen, D.J.; Voors, A.A.; Lam, C.S.P.; Geelhoed, B.; Willems, T.P.; Van Melle, J.P. Right ventricular dysfunction in heart failure with preserved ejection fraction: A systematic review and meta-analysis. Eur. J. Heart Fail. 2016, 18, 1472–1487.
- Harjola, V.P.V.; Mebazaa, A.; Čelutkiene, J.J.; Bettex, D.D.; Bueno, H.; Chioncel, O.O.; Crespo-Leiro, M.G.; Falk, V.; Filippatos, G.; Gibbs, S.S.; et al. Contemporary management of acute right ventricular failure: A statement from the Heart Failure Association and the Working Group on Pulmonary Circulation and Right Ventricular Function of the European Society of Cardiology. Eur. J. Heart Fail. 2016, 18, 226–241.
- Kapur, N.K.; Esposito, M.L.; Bader, Y.; Morine, K.J.; Kiernan, M.S.; Pham, D.T.; Burkhoff, D. Mechanical Circulatory Support Devices for Acute Right Ventricular Failure. Circulation 2017, 136, 314–326.
- Konstam, M.A.; Kiernan, M.S.; Bernstein, D.; Bozkurt, B.; Jacob, M.; Kapur, N.K.; Kociol, R.D.; Lewis, E.F.; Mehra, M.R.; Pagani, F.D.; et al. Evaluation and Management of Right-Sided Heart Failure: A Scientific Statement from the American Heart Association. Circulation 2018, 137, e578–e622.
- Sheehan, F.; Redington, A. The right ventricle: Anatomy, physiology and clinical imaging. Heart 2008, 94, 1510–1515.
- Redington, A.N.; Gray, H.H.; Hodson, M.E.; Rigby, M.L.; Oldershaw, P.J. Characterisation of the normal right ventricular pres-sure-volume relation by biplane angiography and simultaneous micromanometer pressure measurements. Br. Heart J. 1988, 59, 23–30.
- Maughan, W.L.; Shoukas, A.A.; Sagawa, K.; Weisfeldt, M.L. Instantaneous pressure-volume relationship of the canine right ventricle. Circ. Res. 1979, 44, 309–315.
- Ho, S.Y.; Nihoyannopoulos, P. Anatomy, echocardiography, and normal right ventricular dimensions. Heart 2006, 92, i2–i13.
- A Website Edited by Alex Yartsev. Available online: https://derangedphysiology.com (accessed on 14 June 2022).
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for Cardiac Chamber Quantification by Echocardiography in Adults: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2015, 28, 1–39.e14.
- Whiteman, S.; Alimi, Y.; Carrasco, M.; Gielecki, J.; Zurada, A.; Loukas, M. Anatomy of the cardiac chambers: A review of the left ventricle. Transl. Res. Anat. 2021, 23, 100095.
- Samsky, M.D.; Morrow, D.A.; Proudfoot, A.G.; Hochman, J.S.; Thiele, H.; Rao, S.V. Cardiogenic Shock After Acute Myocardial Infarction: A Review. JAMA 2021, 326, 1840–1850.
- Pfisterer, M. Right ventricular involvement in myocardial infarction and cardiogenic shock. Lancet 2003, 362, 392–394.
- Topalian, S.; Ginsberg, F.; Parrillo, J.E. Cardiogenic shock. Crit. Care Med. 2008, 36, S66–S74.
- DeFilippis, E.M.; Topkara, V.K.; Kirtane, A.J.; Takeda, K.; Naka, Y.; Garan, A.R. Mechanical Circulatory Support for Right Ventricular Failure. Card. Fail. Rev. 2022, 8, e14.
- De Meester, P. Tricuspid Regurgitation in Different Loading Conditions. Epidemiology, Determinants and Management. Ph.D. Thesis, University of Leuven, Leuven, Belgium, December 2014.
- van Diepen, S.; Katz, J.N.; Albert, N.M.; Henry, T.D.; Jacobs, A.K.; Kapur, N.K.; Kilic, A.; Menon, V.; Ohman, E.M.; Sweitzer, N.K.; et al. Contemporary Management of Cardiogenic Shock: A Scientific Statement from the American Heart Association. Circulation 2017, 136, e232–e268.
- Ondrus, T.; Kanovsky, J.; Novotny, T.; Andrsova, I.; Spinar, J.; Kala, P. Right ventricular myocardial infarction: From pathophysi-ology to prognosis. Exp. Clin. Cardiol. 2013, 18, 27–30.
- Jain, P.; Thayer, K.L.; Abraham, J.; Everett, K.D.; Pahuja, M.; Whitehead, E.H.; Schwartz, B.P.; Lala, A.; Sinha, S.S.; Kanwar, M.K.; et al. Right Ventricular Dysfunction Is Common and Identifies Patients at Risk of Dying in Cardiogenic Shock. J. Card. Fail. 2021, 27, 1061–1072.
- Josiassen, J.; Helgestad, O.K.L.; Møller, J.E.; Schmidt, H.; Jensen, L.O.; Holmvang, L.; Ravn, H.B.; Hassager, C. Cardiogenic shock due to predominantly right ventricular failure complicating acute myocardial infarction. Eur. Heart J. Acute Cardiovasc. Care 2020, 10, 33–39.
- Kukla, P.; McIntyre, W.F.; Fijorek, K.; Mirek-Bryniarska, E.; Bryniarski, L.; Krupa, E.; Jastrzębski, M.; Bryniarski, K.L.; Zhong-Qun, Z.; Baranchuk, A. Electrocardiographic abnormalities in patients with acute pulmonary embolism complicated by cardiogenic shock. Am. J. Emerg. Med. 2014, 32, 507–510.
- Vyas, V.; Goyal, A. Acute Pulmonary Embolism. . In StatPearls ; StatPearls Publishing: Treasure Island, FL, USA, 2022.
- Appleton, C.; Gillam, L.; Koulogiannis, K. Cardiac Tamponade. Cardiol. Clin. 2017, 35, 525–537.
- Sagar, S.; Liu, P.P.; Cooper, L.T., Jr. Myocarditis. Lancet 2012, 379, 738–747.
- Kawakami, R.; Sakamoto, A.; Kawai, K.; Gianatti, A.; Pellegrini, D.; Nasr, A.; Kutys, B.; Guo, L.; Cornelissen, A.; Mori, M.; et al. Pathological Evidence for SARS-CoV-2 as a Cause of Myocarditis. J. Am. Coll. Cardiol. 2021, 77, 314–325.
- King, C.; May, C.W.; Williams, J.; Shlobin, O.A. Management of Right Heart Failure in the Critically Ill. Crit. Care Clin. 2014, 30, 475–498.
- Arrigo, M.; Jessup, M.; Mullens, W.; Reza, N.; Shah, A.M.; Sliwa, K.; Mebazaa, A. Acute heart failure. Nat. Rev. Dis. Prim. 2020, 6, 1–15.
- McDonough, R.J.; Moul, M.S.; Beckman, D.; Slim, A.M. Isolated Right Ventricular Failure in Hyperthyroidism: A Clinical Dilemma. Heart Int. 2011, 6, e11.
- Brailovsky, Y.; Sayer, G. The “Right” Side of Cardiogenic Shock. JACC Case Rep. 2020, 2, 370–371.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
956
Revisions:
2 times
(View History)
Update Date:
20 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No