 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shintaro Komatsu | -- | 2029 | 2023-02-17 10:03:24 | | | |
| 2 | Lindsay Dong | Meta information modification | 2029 | 2023-02-20 02:02:57 | | | | |
| 3 | Lindsay Dong | -1 word(s) | 2028 | 2023-02-20 06:34:02 | | |
Video Upload Options
MicroRNAs (miRNAs) are versatile, post-transcriptional regulators of gene expression. Canonical miRNAs are generated through the two-step DROSHA- and DICER-mediated processing of primary miRNA (pri-miRNA) transcripts with optimal or suboptimal features for DROSHA and DICER cleavage and loading into Argonaute (AGO) proteins, whereas multiple hairpin-structured RNAs are encoded in the genome and could be a source of non-canonical miRNAs. Advances in miRNA biogenesis research have revealed details of the structural basis of miRNA processing and cluster assistance mechanisms that facilitate the processing of suboptimal hairpins encoded together with optimal hairpins in polycistronic pri-miRNAs. In addition, a deeper investigation of miRNA–target interaction has provided insights into the complexity of target recognition with distinct outcomes, including target-mediated miRNA degradation (TDMD) and cooperation in target regulation by multiple miRNAs.
1. Introduction
2. Biogenesis of Canonical miRNAs
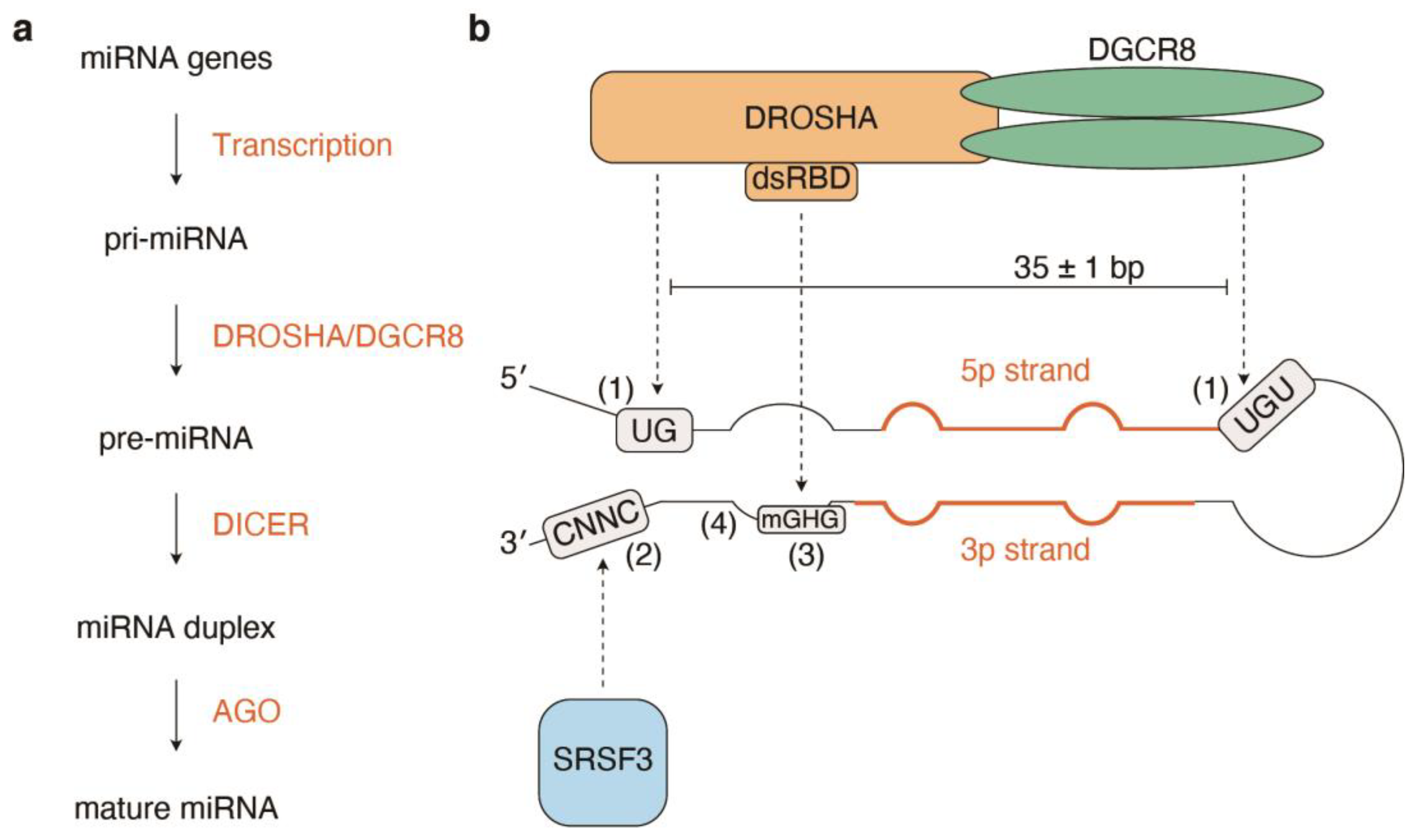
3. Structural Basis of Pri-miRNA Processing
4. Cluster Assistance in Pri-miRNA Processing
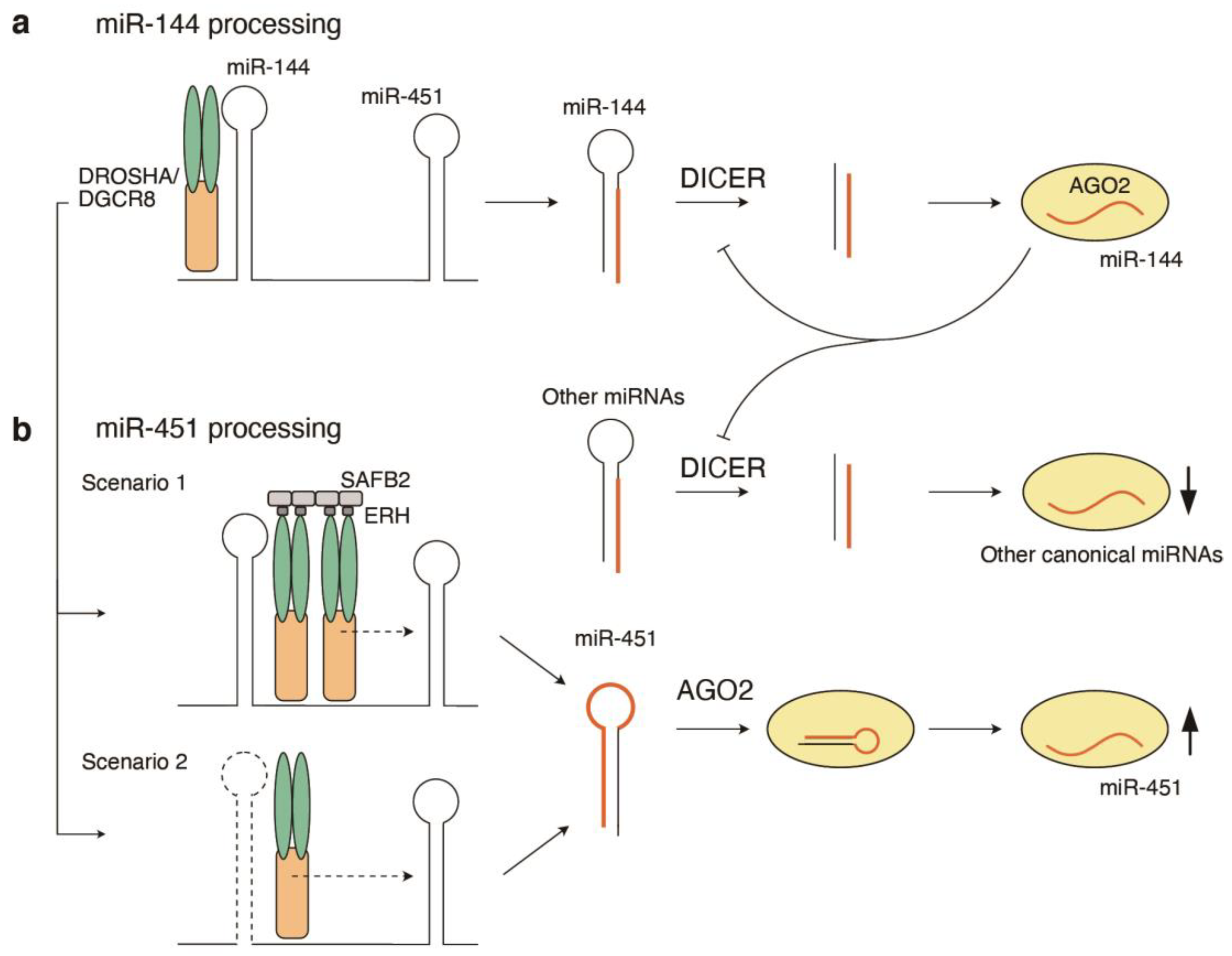
5. Structural Basis of Pre-miRNA Processing
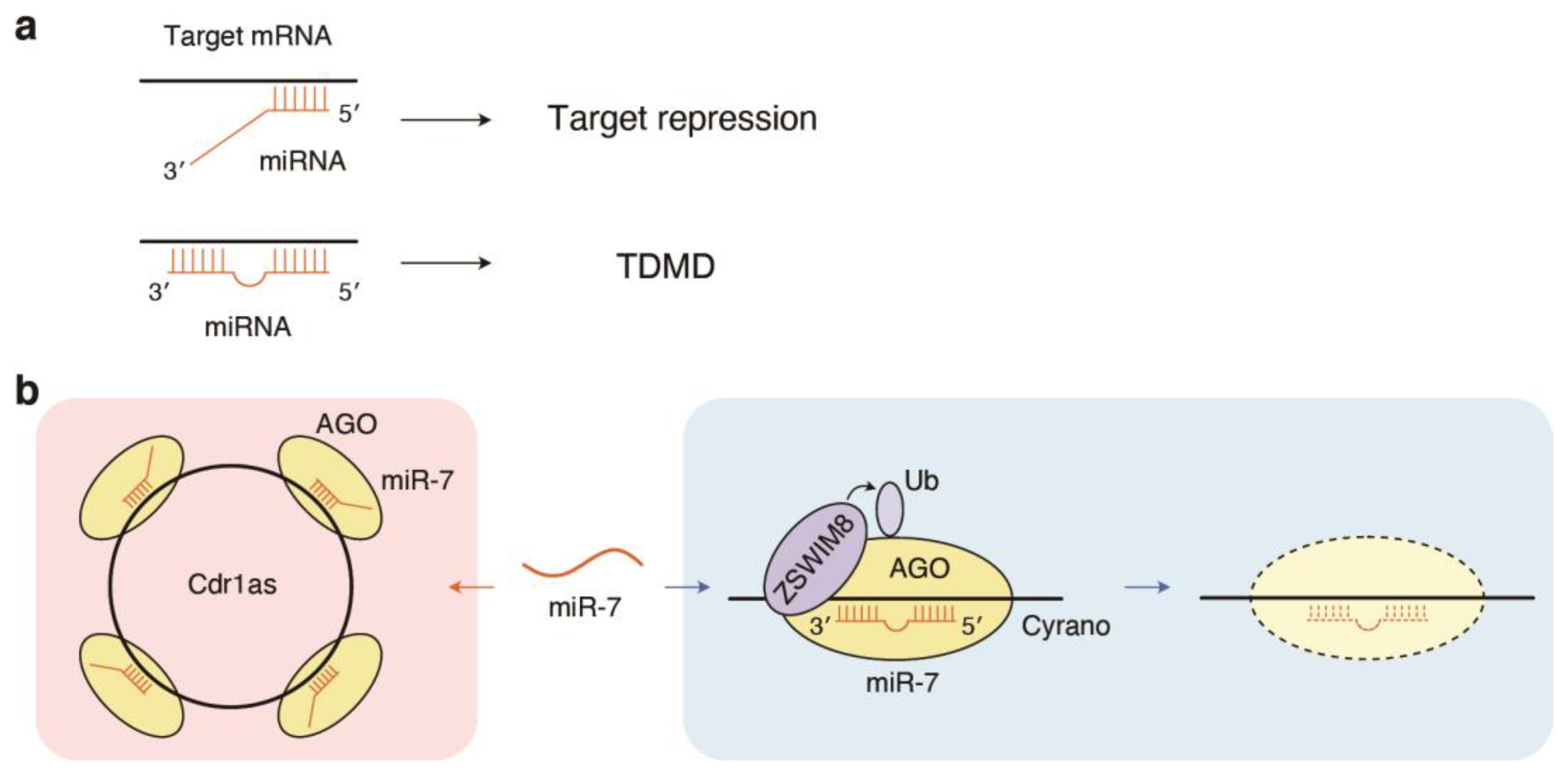
6. Inverse Regulation of miRNAs by Target RNAs: Target-Directed miRNA Degradation (TDMD)
7. miRNA Dosage Control by Fine-Tuning of miRNA Biogenesis Pathways
8. Impacts of Target Site Properties on Target Regulation
References
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51.
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37.
- Matsuyama, H.; Suzuki, H.I. Systems and Synthetic microRNA Biology: From Biogenesis to Disease Pathogenesis. Int. J. Mol. Sci. 2019, 21, 132.
- Wang, Y.; Luo, J.; Zhang, H.; Lu, J. microRNAs in the Same Clusters Evolve to Coordinately Regulate Functionally Related Genes. Mol. Biol. Evol. 2016, 33, 2232–2247.
- Suzuki, H.I.; Young, R.A.; Sharp, P.A. Super-Enhancer-Mediated RNA Processing Revealed by Integrative MicroRNA Network Analysis. Cell 2017, 168, 1000–1014.e15.
- Kim, Y.K.; Kim, B.; Kim, V.N. Re-evaluation of the roles of DROSHA, Export in 5, and DICER in microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E1881–E1889.
- Liu, Z.; Wang, J.; Cheng, H.; Ke, X.; Sun, L.; Zhang, Q.C.; Wang, H.W. Cryo-EM Structure of Human Dicer and Its Complexes with a Pre-miRNA Substrate. Cell 2018, 173, 1191–1203.e12.
- Suzuki, H.I.; Katsura, A.; Yasuda, T.; Ueno, T.; Mano, H.; Sugimoto, K.; Miyazono, K. Small-RNA asymmetry is directly driven by mammalian Argonautes. Nat. Struct. Mol. Biol. 2015, 22, 512–521.
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003, 115, 209–216.
- Schwarz, D.S.; Hutvagner, G.; Du, T.; Xu, Z.; Aronin, N.; Zamore, P.D. Asymmetry in the assembly of the RNAi enzyme complex. Cell 2003, 115, 199–208.
- Frank, F.; Sonenberg, N.; Nagar, B. Structural basis for 5′-nucleotide base-specific recognition of guide RNA by human AGO2. Nature 2010, 465, 818–822.
- Schirle, N.T.; Sheu-Gruttadauria, J.; MacRae, I.J. Structural basis for microRNA targeting. Science 2014, 346, 608–613.
- Baronti, L.; Guzzetti, I.; Ebrahimi, P.; Friebe Sandoz, S.; Steiner, E.; Schlagnitweit, J.; Fromm, B.; Silva, L.; Fontana, C.; Chen, A.A.; et al. Base-pair conformational switch modulates miR-34a targeting of Sirt1 mRNA. Nature 2020, 583, 139–144.
- Auyeung, V.C.; Ulitsky, I.; McGeary, S.E.; Bartel, D.P. Beyond secondary structure: Primary-sequence determinants license pri-miRNA hairpins for processing. Cell 2013, 152, 844–858.
- Fang, W.; Bartel, D.P. The Menu of Features that Define Primary MicroRNAs and Enable De Novo Design of MicroRNA Genes. Mol. Cell 2015, 60, 131–145.
- Kim, K.; Baek, S.C.; Lee, Y.Y.; Bastiaanssen, C.; Kim, J.; Kim, H.; Kim, V.N. A quantitative map of human primary microRNA processing sites. Mol. Cell 2021, 81, 3422–3439.e11.
- Kang, W.; Fromm, B.; Houben, A.J.; Hoye, E.; Bezdan, D.; Arnan, C.; Thrane, K.; Asp, M.; Johnson, R.; Biryukova, I.; et al. MapToCleave: High-throughput profiling of microRNA biogenesis in living cells. Cell Rep. 2021, 37, 110015.
- Nguyen, T.A.; Jo, M.H.; Choi, Y.G.; Park, J.; Kwon, S.C.; Hohng, S.; Kim, V.N.; Woo, J.S. Functional Anatomy of the Human Microprocessor. Cell 2015, 161, 1374–1387.
- Herbert, K.M.; Sarkar, S.K.; Mills, M.; Delgado De la Herran, H.C.; Neuman, K.C.; Steitz, J.A. A heterotrimer model of the complete Microprocessor complex revealed by single-molecule subunit counting. RNA 2016, 22, 175–183.
- Jin, W.; Wang, J.; Liu, C.P.; Wang, H.W.; Xu, R.M. Structural Basis for pri-miRNA Recognition by Drosha. Mol. Cell 2020, 78, 423–433.e5.
- Partin, A.C.; Zhang, K.; Jeong, B.C.; Herrell, E.; Li, S.; Chiu, W.; Nam, Y. Cryo-EM Structures of Human Drosha and DGCR8 in Complex with Primary MicroRNA. Mol. Cell 2020, 78, 411–422.e4.
- Kwon, S.C.; Baek, S.C.; Choi, Y.G.; Yang, J.; Lee, Y.S.; Woo, J.S.; Kim, V.N. Molecular Basis for the Single-Nucleotide Precision of Primary microRNA Processing. Mol. Cell 2019, 73, 505–518.e5.
- Kretov, D.A.; Walawalkar, I.A.; Mora-Martin, A.; Shafik, A.M.; Moxon, S.; Cifuentes, D. Ago2-Dependent Processing Allows miR-451 to Evade the Global MicroRNA Turnover Elicited during Erythropoiesis. Mol. Cell 2020, 78, 317–328.e6.
- Shang, R.; Baek, S.C.; Kim, K.; Kim, B.; Kim, V.N.; Lai, E.C. Genomic Clustering Facilitates Nuclear Processing of Suboptimal Pri-miRNA Loci. Mol. Cell 2020, 78, 303–316.e4.
- Fang, W.; Bartel, D.P. MicroRNA Clustering Assists Processing of Suboptimal MicroRNA Hairpins through the Action of the ERH Protein. Mol. Cell 2020, 78, 289–302.e6.
- Kwon, S.C.; Jang, H.; Shen, S.; Baek, S.C.; Kim, K.; Yang, J.; Kim, J.; Kim, J.S.; Wang, S.; Shi, Y.; et al. ERH facilitates microRNA maturation through the interaction with the N-terminus of DGCR8. Nucleic Acids Res. 2020, 48, 11097–11112.
- Hutter, K.; Lohmuller, M.; Jukic, A.; Eichin, F.; Avci, S.; Labi, V.; Szabo, T.G.; Hoser, S.M.; Huttenhofer, A.; Villunger, A.; et al. SAFB2 Enables the Processing of Suboptimal Stem-Loop Structures in Clustered Primary miRNA Transcripts. Mol. Cell 2020, 78, 876–889.e6.
- Vilimova, M.; Contrant, M.; Randrianjafy, R.; Dumas, P.; Elbasani, E.; Ojala, P.M.; Pfeffer, S.; Fender, A. Cis regulation within a cluster of viral microRNAs. Nucleic Acids Res. 2021, 49, 10018–10033.
- Zapletal, D.; Taborska, E.; Pasulka, J.; Malik, R.; Kubicek, K.; Zanova, M.; Much, C.; Sebesta, M.; Buccheri, V.; Horvat, F.; et al. Structural and functional basis of mammalian microRNA biogenesis by Dicer. Mol. Cell 2022, 82, 4064–4079.e13.
- Jouravleva, K.; Golovenko, D.; Demo, G.; Dutcher, R.C.; Hall, T.M.T.; Zamore, P.D.; Korostelev, A.A. Structural basis of microRNA biogenesis by Dicer-1 and its partner protein Loqs-PB. Mol. Cell 2022, 82, 4049–4063.e6.
- Su, S.; Wang, J.; Deng, T.; Yuan, X.; He, J.; Liu, N.; Li, X.; Huang, Y.; Wang, H.W.; Ma, J. Structural insights into dsRNA processing by Drosophila Dicer-2-Loqs-PD. Nature 2022, 607, 399–406.
- Yamaguchi, S.; Naganuma, M.; Nishizawa, T.; Kusakizako, T.; Tomari, Y.; Nishimasu, H.; Nureki, O. Structure of the Dicer-2-R2D2 heterodimer bound to a small RNA duplex. Nature 2022, 607, 393–398.
- Park, J.E.; Heo, I.; Tian, Y.; Simanshu, D.K.; Chang, H.; Jee, D.; Patel, D.J.; Kim, V.N. Dicer recognizes the 5′ end of RNA for efficient and accurate processing. Nature 2011, 475, 201–205.
- Heo, I.; Ha, M.; Lim, J.; Yoon, M.J.; Park, J.E.; Kwon, S.C.; Chang, H.; Kim, V.N. Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell 2012, 151, 521–532.
- Gu, S.; Jin, L.; Zhang, Y.; Huang, Y.; Zhang, F.; Valdmanis, P.N.; Kay, M.A. The loop position of shRNAs and pre-miRNAs is critical for the accuracy of dicer processing in vivo. Cell 2012, 151, 900–911.
- Tsutsumi, A.; Kawamata, T.; Izumi, N.; Seitz, H.; Tomari, Y. Recognition of the pre-miRNA structure by Drosophila Dicer-1. Nat. Struct. Mol. Biol. 2011, 18, 1153–1158.
- Liu, Z.; Wang, J.; Li, G.; Wang, H.W. Structure of precursor microRNA’s terminal loop regulates human Dicer’s dicing activity by switching DExH/D domain. Protein Cell 2015, 6, 185–193.
- Zhang, X.; Zeng, Y. The terminal loop region controls microRNA processing by Drosha and Dicer. Nucleic Acids Res. 2010, 38, 7689–7697.
- Nguyen, T.D.; Trinh, T.A.; Bao, S.; Nguyen, T.A. Secondary structure RNA elements control the cleavage activity of DICER. Nat. Commun. 2022, 13, 2138.
- Reichholf, B.; Herzog, V.A.; Fasching, N.; Manzenreither, R.A.; Sowemimo, I.; Ameres, S.L. Time-Resolved Small RNA Sequencing Unravels the Molecular Principles of MicroRNA Homeostasis. Mol. Cell 2019, 75, 756–768.e7.
- Kingston, E.R.; Bartel, D.P. Global analyses of the dynamics of mammalian microRNA metabolism. Genome Res. 2019, 29, 1777–1790.
- De, N.; Young, L.; Lau, P.W.; Meisner, N.C.; Morrissey, D.V.; MacRae, I.J. Highly complementary target RNAs promote release of guide RNAs from human Argonaute2. Mol. Cell 2013, 50, 344–355.
- Jo, M.H.; Shin, S.; Jung, S.R.; Kim, E.; Song, J.J.; Hohng, S. Human Argonaute 2 Has Diverse Reaction Pathways on Target RNAs. Mol. Cell 2015, 59, 117–124.
- Park, J.H.; Shin, S.Y.; Shin, C. Non-canonical targets destabilize microRNAs in human Argonautes. Nucleic Acids Res. 2017, 45, 1569–1583.
- Ameres, S.L.; Horwich, M.D.; Hung, J.H.; Xu, J.; Ghildiyal, M.; Weng, Z.; Zamore, P.D. Target RNA-directed trimming and tailing of small silencing RNAs. Science 2010, 328, 1534–1539.
- Cazalla, D.; Yario, T.; Steitz, J.A. Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science 2010, 328, 1563–1566.
- Piwecka, M.; Glazar, P.; Hernandez-Miranda, L.R.; Memczak, S.; Wolf, S.A.; Rybak-Wolf, A.; Filipchyk, A.; Klironomos, F.; Cerda Jara, C.A.; Fenske, P.; et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 2017, 357, eaam8526.
- Kleaveland, B.; Shi, C.Y.; Stefano, J.; Bartel, D.P. A Network of Noncoding Regulatory RNAs Acts in the Mammalian Brain. Cell 2018, 174, 350–362.e17.
- Bitetti, A.; Mallory, A.C.; Golini, E.; Carrieri, C.; Carreno Gutierrez, H.; Perlas, E.; Perez-Rico, Y.A.; Tocchini-Valentini, G.P.; Enright, A.J.; Norton, W.H.J.; et al. MicroRNA degradation by a conserved target RNA regulates animal behavior. Nat. Struct. Mol. Biol. 2018, 25, 244–251.
- Han, J.; Mendell, J.T. MicroRNA turnover: A tale of tailing, trimming, and targets. Trends Biochem. Sci. 2022, 48, 26–39.
- Martello, G.; Rosato, A.; Ferrari, F.; Manfrin, A.; Cordenonsi, M.; Dupont, S.; Enzo, E.; Guzzardo, V.; Rondina, M.; Spruce, T.; et al. A MicroRNA targeting dicer for metastasis control. Cell 2010, 141, 1195–1207.
- Han, J.; Pedersen, J.S.; Kwon, S.C.; Belair, C.D.; Kim, Y.K.; Yeom, K.H.; Yang, W.Y.; Haussler, D.; Blelloch, R.; Kim, V.N. Posttranscriptional crossregulation between Drosha and DGCR8. Cell 2009, 136, 75–84.
- Cui, Y.; Lyu, X.; Ding, L.; Ke, L.; Yang, D.; Pirouz, M.; Qi, Y.; Ong, J.; Gao, G.; Du, P.; et al. Global miRNA dosage control of embryonic germ layer specification. Nature 2021, 593, 602–606.
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005.
- Zhang, K.; Zhang, X.; Cai, Z.; Zhou, J.; Cao, R.; Zhao, Y.; Chen, Z.; Wang, D.; Ruan, W.; Zhao, Q.; et al. A novel class of microRNA-recognition elements that function only within open reading frames. Nat. Struct. Mol. Biol. 2018, 25, 1019–1027.
- Yang, A.; Bofill-De Ros, X.; Shao, T.J.; Jiang, M.; Li, K.; Villanueva, P.; Dai, L.; Gu, S. 3′ Uridylation Confers miRNAs with Non-canonical Target Repertoires. Mol. Cell 2019, 75, 511–522.e4.


