 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Masumi Taki | -- | 4384 | 2023-02-17 04:14:51 | | | |
| 2 | Catherine Yang | + 2 word(s) | 4386 | 2023-02-17 04:31:31 | | |
Video Upload Options
Introduction of artificial structure(s) into a middle-sized therapeutic biomolecule, which would often add a superior function to the drug molecule, is easier than that into an antibody. In this direction, introducing a reactive warhead structure into such therapeutic biomolecules to create BIOmolecular Targeted Covalent Inhibitors (abbreviated as bioTCIs). It would create precise and safe covalent drugs that semi-permanently inhibit the target protein activity upon binding and the duration of the drug effect is only limited by the target protein turnover. Regardless of the modalities, all bioTCIs reduce the skepticism of small-molecule TCIs’ safety concerns because bioTCIs can stringently recognize and conjugate only to the target proteins. Among them, oligonucleotide-type bioTCIs possess unique features, such as nuclease resistance and on-demand-reversal of the drug action with the complementary-strand antidote, which circumvents another major limitation to clinical translation of the aptamer drugs.
1. History and General Principle of bioTCI
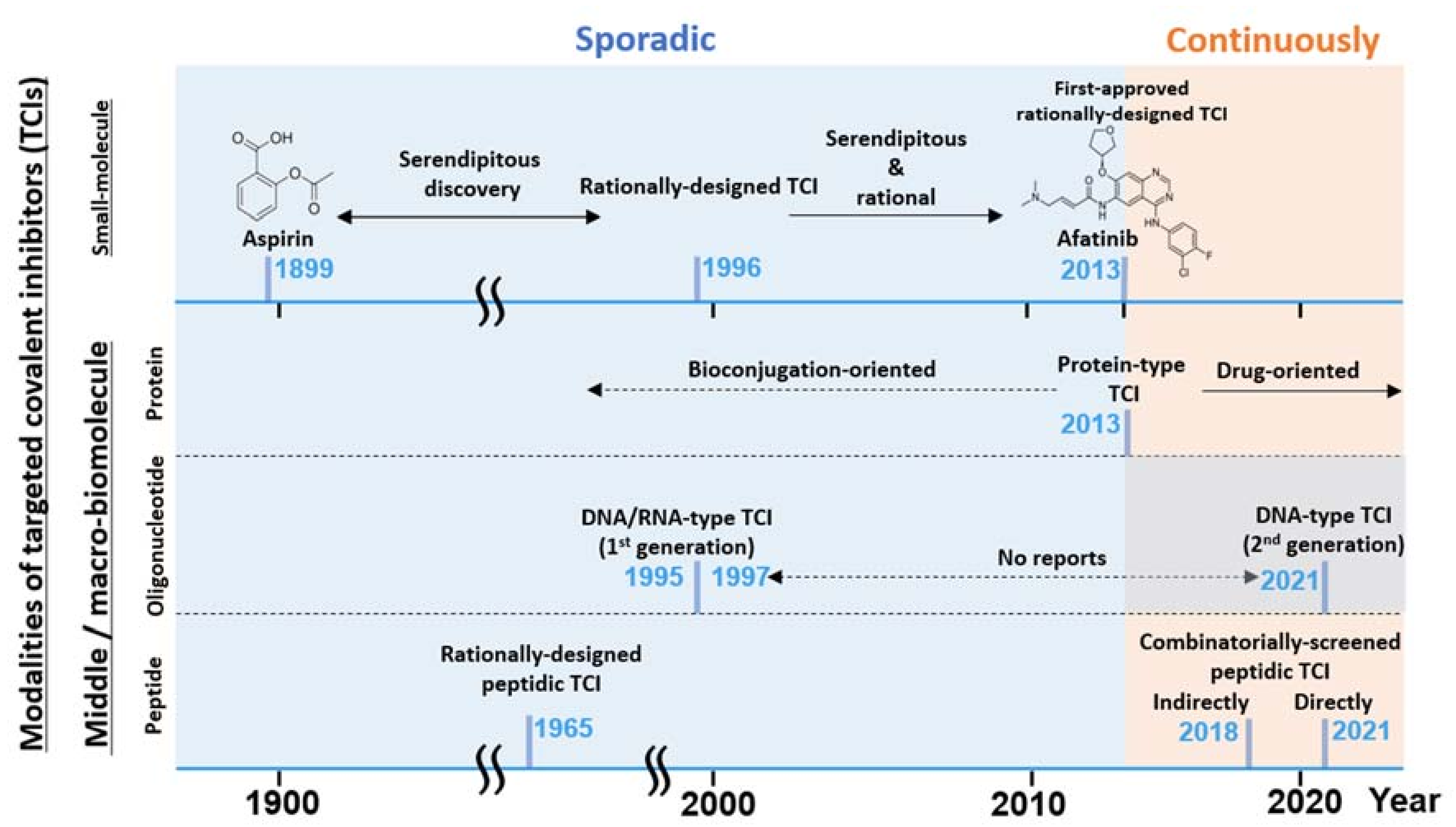
1.1. From Small Molecular TCI to bioTCI
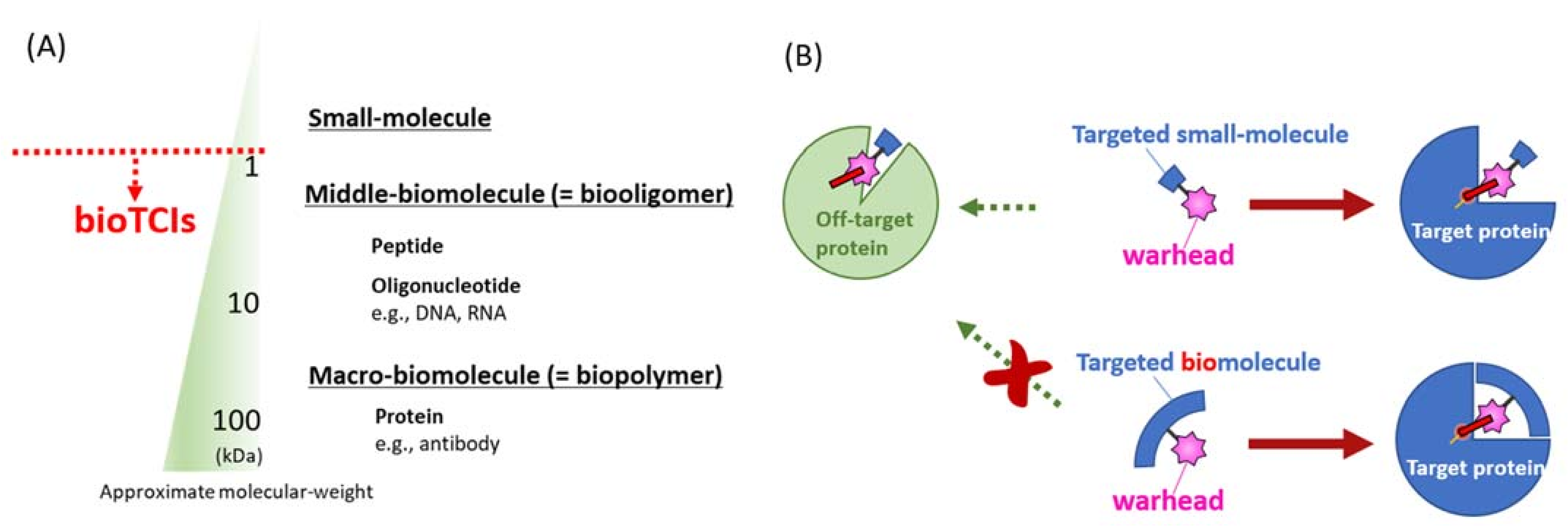
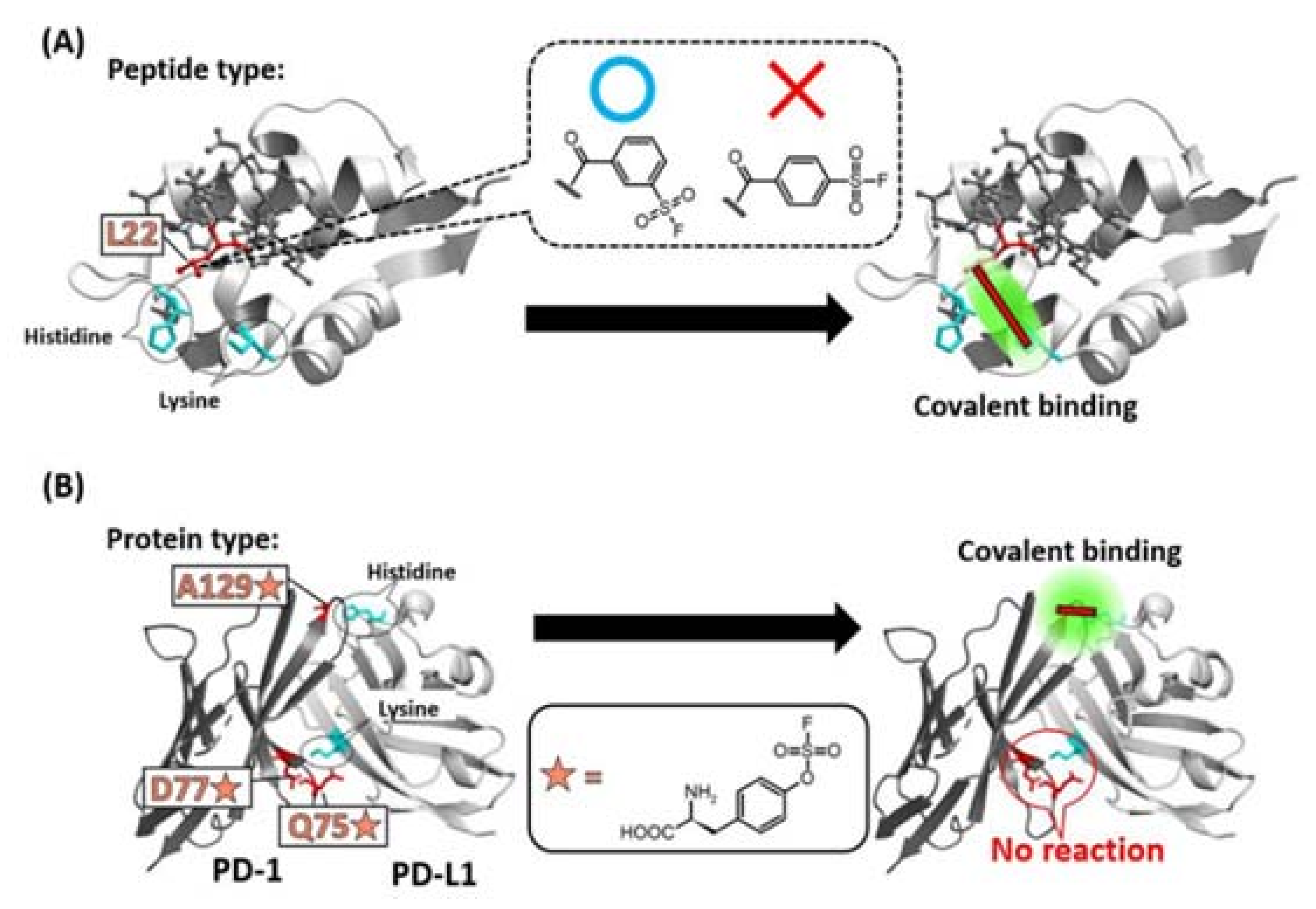
1.2. Warhead Design and Introduction into Middle/Macro-Biomolecules

1.3. Pros and Cons of bioTCI over Non-Covalent Biomolecular Targeted Inhibitors

2. Recent Hot Topics of bioTCI
2.1. Combinatorial Screening of Peptidic TCI: A Well-Developed Modality

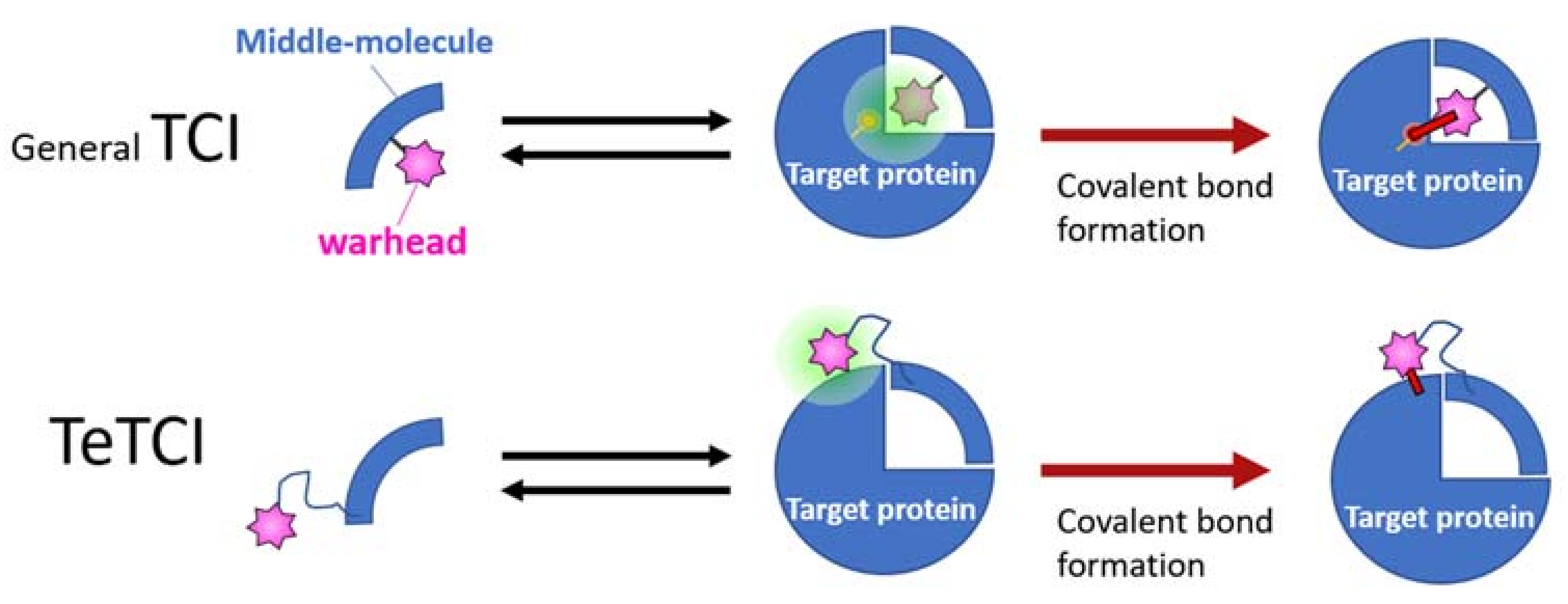
2.2. Nucleotidic TCI: A Developing Modality

References
- Potashman, M.H.; Duggan, M.E. Covalent Modifiers: An Orthogonal Approach to Drug Design. J. Med. Chem. 2009, 52, 1231–1246.
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073.
- Halford, B. Covalent drugs go from fringe field to fashionable endeavor. Chem. Eng. News 2020, 98, 28–33.
- Roth, G.J.; Majerus, P.W. The mechanism of the effect of aspirin on human platelets. I. Acetylation of a particulate fraction protein. J. Clin. Investig. 1975, 56, 624–632.
- Vane, J.R. Inhibition of Prostaglandin Synthesis as a Mechanism of Action for Aspirin-like Drugs. Nat. New Biol. 1971, 231, 232–235.
- De Vita, E. 10 years into the resurgence of covalent drugs. Futur. Med. Chem. 2021, 13, 193–210.
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317.
- Singh, J.; Dobrusin, E.M.; Fry, D.W.; Haske, T.; Whitty, A.; McNamara, D.J. Structure-Based Design of a Potent, Selective, and Irreversible Inhibitor of the Catalytic Domain of the erbB Receptor Subfamily of Protein Tyrosine Kinases. J. Med. Chem. 1997, 40, 1130–1135.
- Fry, D.W.; Bridges, A.J.; Denny, W.A.; Doherty, A.; Greis, K.D.; Hicks, J.L.; Hook, K.E.; Keller, P.R.; Leopold, W.R.; Loo, J.A.; et al. Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc. Natl. Acad. Sci. USA 1998, 95, 12022–12027.
- de Freitas, R.F.; Schapira, M. A systematic analysis of atomic protein–ligand interactions in the PDB. MedChemComm 2017, 8, 1970–1981.
- Rao, M.S.; Gupta, R.; Liguori, M.J.; Hu, M.; Huang, X.; Mantena, S.R.; Mittelstadt, S.W.; Blomme, E.A.G.; Van Vleet, T.R. Novel Computational Approach to Predict Off-Target Interactions for Small Molecules. Front. Big Data 2019, 2, 25.
- Lanning, B.R.; Whitby, L.R.; Dix, M.M.; Douhan, J.; Gilbert, A.M.; Hett, E.C.; Johnson, T.O.; Joslyn, C.M.; Kath, J.C.; Niessen, S.; et al. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat. Chem. Biol. 2014, 10, 760–767.
- Paulussen, F.M.; Grossmann, T.N. Peptide-based covalent inhibitors of protein–protein interactions. J. Pept. Sci. 2022, 29, e3457.
- Tabuchi, Y.; Yang, J.; Taki, M. Inhibition of thrombin activity by a covalent-binding aptamer and reversal by the complementary strand antidote. Chem. Commun. 2021, 57, 2483–2486.
- Berdan, V.Y.; Klauser, P.C.; Wang, L. Covalent peptides and proteins for therapeutics. Bioorganic Med. Chem. 2020, 29, 115896.
- Li, Q.; Chen, Q.; Klauser, P.C.; Li, M.; Zheng, F.; Wang, N.; Li, X.; Zhang, Q.; Fu, X.; Wang, Q.; et al. Developing Covalent Protein Drugs via Proximity-Enabled Reactive Therapeutics. Cell 2020, 182, 85–97.e16.
- Gambini, L.; Baggio, C.; Udompholkul, P.; Jossart, J.; Salem, A.F.; Perry, J.J.P.; Pellecchia, M. Covalent Inhibitors of Protein–Protein Interactions Targeting Lysine, Tyrosine, or Histidine Residues. J. Med. Chem. 2019, 62, 5616–5627.
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48.
- Fu, Z.; Xiang, J. Aptamers, the Nucleic Acid Antibodies, in Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 2793.
- Tabuchi, Y.; Katsuki, R.; Ito, Y.; Taki, M. Direct Combinatorial Screening of a Target-Specific Covalent Binding Peptide: Ac-tivation of the Warhead Reactivity in a Pseudo-Catalytic Microenvironment. Pept. Sci. 2021, 2022, 15–16.
- Tabuchi, Y.; Yang, J.; Taki, M. Relative Nuclease Resistance of a DNA Aptamer Covalently Conjugated to a Target Protein. Int. J. Mol. Sci. 2022, 23, 7778.
- Tivon, Y.; Falcone, G.; Deiters, A. Protein Labeling and Crosslinking by Covalent Aptamers. Angew. Chem. Int. Ed. 2021, 60, 15899–15904.
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2018, 62, 5673–5724.
- Tuley, A.; Fast, W. The Taxonomy of Covalent Inhibitors. Biochemistry 2018, 57, 3326–3337.
- Péczka, N.; Orgován, Z.; Ábrányi-Balogh, P.; Keserű, G.M. Electrophilic warheads in covalent drug discovery: An overview. Expert Opin. Drug Discov. 2022, 17, 413–422.
- Martin, J.S.; MacKenzie, C.J.; Fletcher, D.; Gilbert, I.H. Characterising covalent warhead reactivity. Bioorganic Med. Chem. 2019, 27, 2066–2074.
- Copeland, R.A. Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Methods Biochem. Anal. 2005, 46, 1–265.
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 345–382.
- McAulay, K.; Bilsland, A.; Bon, M. Reactivity of Covalent Fragments and Their Role in Fragment Based Drug Discovery. Pharmaceuticals 2022, 15, 1366.
- Carneiro, S.N.; Khasnavis, S.R.; Lee, J.; Butler, T.W.; Majmudar, J.D.; Ende, C.W.A.; Ball, N.D. Sulfur(vi) fluorides as tools in biomolecular and medicinal chemistry. Org. Biomol. Chem. 2023.
- Wang, D.; Zhang, J.; Huang, Z.; Yang, Y.; Fu, T.; Yang, Y.; Lyu, Y.; Jiang, J.; Qiu, L.; Cao, Z.; et al. Robust Covalent Aptamer Strategy Enables Sensitive Detection and Enhanced Inhibition of SARS-CoV-2 Proteins. ACS Central Sci. 2023, 9, 72–83.
- Baker, B.R. Tissue-Specific Irreversible Inhibitors of Dihydrofolic Reductase1. Acc. Chem. Res. 1969, 2, 129–136.
- Dong, J.; Krasnova, L.; Finn, M.G.; Sharpless, K.B. Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry. Angew. Chem. Int. Ed. 2014, 53, 9430–9448.
- Barrow, A.S.; Smedley, C.J.; Zheng, Q.; Li, S.; Dong, J.; Moses, J.E. The growing applications of SuFEx click chemistry. Chem. Soc. Rev. 2019, 48, 4731–4758.
- Zheng, Q.; Woehl, J.L.; Kitamura, S.; Santos-Martins, D.; Smedley, C.J.; Li, G.; Forli, S.; Moses, J.E.; Wolan, D.W.; Sharpless, K.B. SuFEx-enabled, agnostic discovery of covalent inhibitors of human neutrophil elastase. Proc. Natl. Acad. Sci. USA 2019, 116, 18808–18814.
- Narayanan, A.; Jones, L.H. Sulfonyl fluorides as privileged warheads in chemical biology. Chem. Sci. 2015, 6, 2650–2659.
- Mukherjee, H.; Debreczeni, J.; Breed, J.; Tentarelli, S.; Aquila, B.; Dowling, J.E.; Whitty, A.; Grimster, N.P. A study of the reactivity of S(VI)–F containing warheads with nucleophilic amino-acid side chains under physiological conditions. Org. Biomol. Chem. 2017, 15, 9685–9695.
- Hoppmann, C.; Wang, L. Proximity-enabled bioreactivity to generate covalent peptide inhibitors of p53–Mdm4. Chem. Commun. 2016, 52, 5140–5143.
- Xiang, Z.; Ren, H.; Hu, Y.S.; Coin, I.; Wei, J.; Cang, H.; Wang, L. Adding an unnatural covalent bond to proteins through proximity-enhanced bioreactivity. Nat. Methods 2013, 10, 885–888.
- Tabuchi, Y.; Watanabe, T.; Katsuki, R.; Ito, Y.; Taki, M. Direct screening of a target-specific covalent binder: Stringent regulation of warhead reactivity in a matchmaking environment. Chem. Commun. 2021, 57, 5378–5381.
- Guldalian, J., Jr.; Lawson, W.B.; Brown, R.K. Formation of a Covalent Linkage between Bromoacetylated Antigen and Its Antibody. J. Biol. Chem. 1965, 240, PC2757–PC2759.
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154.
- Hubbell, G.E.; Tepe, J.J. Natural product scaffolds as inspiration for the design and synthesis of 20S human proteasome inhibitors. RSC Chem. Biol. 2020, 1, 305–332.
- Hanada, M.; Sugawara, K.; Kaneta, K.; Toda, S.; Nishiyama, Y.; Tomita, K.; Yamamoto, H.; Konishi, M.; Oki, T. Epoxomicin, a new antitumor agent of microbial origin. J. Antibiot. 1992, 45, 1746–1752.
- Chen, Y.; McClure, R.A.; Zheng, Y.; Thomson, R.J.; Kelleher, N.L. Proteomics Guided Discovery of Flavopeptins: Anti-proliferative Aldehydes Synthesized by a Reductase Domain-Containing Non-ribosomal Peptide Synthetase. J. Am. Chem. Soc. 2013, 135, 10449–10456.
- Vagner, J.; Qu, H.; Hruby, V.J. Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 292–296.
- Lenci, E.; Trabocchi, A. Peptidomimetic toolbox for drug discovery. Chem. Soc. Rev. 2020, 49, 3262–3277.
- Boike, L.; Henning, N.J.; Nomura, D.K. Advances in covalent drug discovery. Nat. Rev. Drug Discov. 2022, 21, 881–898.
- Wang, L. Engineering the Genetic Code in Cells and Animals: Biological Considerations and Impacts. Accounts Chem. Res. 2017, 50, 2767–2775.
- Yu, B.; Li, S.; Tabata, T.; Wang, N.; Cao, L.; Kumar, G.R.; Sun, W.; Liu, J.; Ott, M.; Wang, L. Accelerating PERx reaction enables covalent nanobodies for potent neutralization of SARS-CoV-2 and variants. Chem 2022, 8, 2766–2783.
- Han, Y.; Yang, Z.; Hu, H.; Zhang, H.; Chen, L.; Li, K.; Kong, L.; Wang, Q.; Liu, B.; Wang, M.; et al. Covalently Engineered Protein Minibinders with Enhanced Neutralization Efficacy against Escaping SARS-CoV-2 Variants. J. Am. Chem. Soc. 2022, 144, 5702–5707.
- Cao, L.; Wang, L. New covalent bonding ability for proteins. Protein Sci. 2021, 31, 312–322.
- Strelow, J.M. A Perspective on the Kinetics of Covalent and Irreversible Inhibition. SLAS Discov. Adv. Sci. Drug Discov. 2017, 22, 3–20.
- Zhai, X.; Ward, R.A.; Doig, P.; Argyrou, A. Insight into the Therapeutic Selectivity of the Irreversible EGFR Tyrosine Kinase Inhibitor Osimertinib through Enzyme Kinetic Studies. Biochemistry 2020, 59, 1428–1441.
- Mons, E.; Roet, S.; Kim, R.Q.; Mulder, M.P.C. A Comprehensive Guide for Assessing Covalent Inhibition in Enzymatic Assays Illustrated with Kinetic Simulations. Curr. Protoc. 2022, 2, e419.
- Schwartz, P.A.; Kuzmic, P.; Solowiej, J.; Bergqvist, S.; Bolanos, B.; Almaden, C.; Nagata, A.; Ryan, K.; Feng, J.; Dalvie, D.; et al. Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc. Natl. Acad. Sci. USA 2013, 111, 173–178.
- Qin, Z.; Zhu, Y.; Xiang, Y. Covalent inhibition of SARS-CoV-2 RBD-ACE2 interaction by aptamers with multiple sulfur(VI) fluoride exchange modifications. ChemRxiv 2021, 1–22.
- Liu, R.; Li, X.; Lam, K.S. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126.
- Uematsu, S.; Tabuchi, Y.; Ito, Y.; Taki, M. Combinatorially Screened Peptide as Targeted Covalent Binder: Alteration of Bait-Conjugated Peptide to Reactive Modifier. Bioconjugate Chem. 2018, 29, 1866–1871.
- Chen, S.; Lovell, S.; Lee, S.; Fellner, M.; Mace, P.D.; Bogyo, M. Identification of highly selective covalent inhibitors by phage display. Nat. Biotechnol. 2020, 39, 490–498.
- Choi, E.J.; Jung, D.; Kim, J.-S.; Lee, Y.; Kim, B.M. Chemoselective Tyrosine Bioconjugation through Sulfate Click Reaction. Chem. A Eur. J. 2018, 24, 10948–10952.
- Bolding, J.E.; Martín-Gago, P.; Rajabi, N.; Gamon, L.F.; Hansen, T.N.; Bartling, C.R.O.; Strømgaard, K.; Davies, M.J.; Olsen, C.A. Aryl Fluorosulfate Based Inhibitors That Covalently Target the SIRT5 Lysine Deacylase. Angew. Chem. Int. Ed. 2022, 61, e202204565.
- van der Zouwen, A.J.; Witte, M.D. Modular Approaches to Synthesize Activity- and Affinity-Based Chemical Probes. Front. Chem. 2021, 9.
- Fukunaga, K.; Taki, M. Practical Tips for Construction of Custom Peptide Libraries and Affinity Selection by Using Commercially Available Phage Display Cloning Systems. J. Nucl. Acids 2012, 2012, 295719.
- Zheng, M.; Chen, F.-J.; Li, K.; Reja, R.M.; Haeffner, F.; Gao, J. Lysine-Targeted Reversible Covalent Ligand Discovery for Proteins via Phage Display. J. Am. Chem. Soc. 2022, 144, 15885–15893.
- Serafimova, I.M.; Pufall, M.; Krishnan, S.; Duda, K.; Cohen, M.S.; Maglathlin, R.L.; McFarland, J.M.; Miller, R.M.; Frödin, M.; Taunton, J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012, 8, 471–476.
- Bandyopadhyay, A.; Gao, J. Targeting biomolecules with reversible covalent chemistry. Curr. Opin. Chem. Biol. 2016, 34, 110–116.
- Abe, T.; Tabuchi, Y.; Taki, M. Reversal of covalent conjugation of benzoxaborole-modified targeted peptide by reduction. Pept. Sci. 2021, 2022, 17–18.
- Graham, B.J.; Windsor, I.W.; Gold, B.; Raines, R.T. Boronic acid with high oxidative stability and utility in biological contexts. Proc. Natl. Acad. Sci. USA 2021, 118.
- Byun, J. Recent Progress and Opportunities for Nucleic Acid Aptamers. Life 2021, 11, 193.
- Komarova, N.; Kuznetsov, A. Inside the Black Box: What Makes SELEX Better? Molecules 2019, 24, 3598.
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202.
- Adachi, T.; Nakamura, Y. Aptamers: A Review of Their Chemical Properties and Modifications for Therapeutic Application. Molecules 2019, 24, 4229.
- Xiao, X.; Li, H.; Zhao, L.; Zhang, Y.; Liu, Z. Oligonucleotide aptamers: Recent advances in their screening, molecular conformation and therapeutic applications. Biomed. Pharmacother. 2021, 143, 112232.
- Smith, D.; Kirschenheuter, G.P.; Charlton, J.; Guidot, D.M.; Repine, J.E. In vitro selection of RNA-based irreversible inhibitors of human neutrophil elastase. Chem. Biol. 1995, 2, 741–750.
- Charlton, J.; Kirschenheuter, G.P.; Smith, D. Highly Potent Irreversible Inhibitors of Neutrophil Elastase Generated by Selection from a Randomized DNA−Valine Phosphonate Library. Biochemistry 1997, 36, 3018–3026.
- Ueda, T.; Tamura, T.; Kawano, M.; Shiono, K.; Hobor, F.; Wilson, A.J.; Hamachi, I. Enhanced Suppression of a Protein–Protein Interaction in Cells Using Small-Molecule Covalent Inhibitors Based on an N-Acyl-N-alkyl Sulfonamide Warhead. J. Am. Chem. Soc. 2021, 143, 4766–4774.
- Corso, A.D.; Catalano, M.; Schmid, A.; Scheuermann, J.; Neri, D. Affinity Enhancement of Protein Ligands by Reversible Covalent Modification of Neighboring Lysine Residues. Angew. Chem. Int. Ed. 2018, 57, 17178–17182.
- Krauss, I.R.; Merlino, A.; Randazzo, A.; Novellino, E.; Mazzarella, L.; Sica, F. High-resolution structures of two complexes between thrombin and thrombin-binding aptamer shed light on the role of cations in the aptamer inhibitory activity. Nucleic Acids Res. 2012, 40, 8119–8128.
- Bock, L.C.; Griffin, L.C.; Latham, J.A.; Vermaas, E.H.; Toole, J.J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355, 564–566.
- Wakui, K.; Yoshitomi, T.; Yamaguchi, A.; Tsuchida, M.; Saito, S.; Shibukawa, M.; Furusho, H.; Yoshimoto, K. Rapidly Neutralizable and Highly Anticoagulant Thrombin-Binding DNA Aptamer Discovered by MACE SELEX. Mol. Ther. - Nucleic Acids 2019, 16, 348–359.
- Sun, M.; Liu, S.; Wei, X.; Wan, S.; Huang, M.; Song, T.; Lu, Y.; Weng, X.; Lin, Z.; Chen, H.; et al. Aptamer Blocking Strategy Inhibits SARS-CoV-2 Virus Infection. Angew. Chem. Int. Ed. 2021, 60, 10266–10272.
- Chrominski, M.; Ziemkiewicz, K.; Kowalska, J.; Jemielity, J. Introducing SuFNucs: Sulfamoyl-Fluoride-Functionalized Nucleosides That Undergo Sulfur Fluoride Exchange Reaction. Org. Lett. 2022, 24, 4977–4981.

