Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lluís Rusiñol | -- | 4839 | 2023-02-16 18:32:20 | | | |
| 2 | Camila Xu | Meta information modification | 4839 | 2023-02-17 01:54:08 | | | | |
| 3 | Camila Xu | Meta information modification | 4839 | 2023-02-20 08:22:45 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Rusiñol, L.; Puig, L. Tyk2 Targeting in Immune-Mediated Inflammatory Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/41316 (accessed on 23 July 2026).
Rusiñol L, Puig L. Tyk2 Targeting in Immune-Mediated Inflammatory Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/41316. Accessed July 23, 2026.
Rusiñol, Lluís, Luis Puig. "Tyk2 Targeting in Immune-Mediated Inflammatory Diseases" Encyclopedia, https://encyclopedia.pub/entry/41316 (accessed July 23, 2026).
Rusiñol, L., & Puig, L. (2023, February 16). Tyk2 Targeting in Immune-Mediated Inflammatory Diseases. In Encyclopedia. https://encyclopedia.pub/entry/41316
Rusiñol, Lluís and Luis Puig. "Tyk2 Targeting in Immune-Mediated Inflammatory Diseases." Encyclopedia. Web. 16 February, 2023.
Copy Citation
Genetic linkage has related dysfunction of Tyrosine kinase 2 (Tyk2)—the first member of the Jak family that was described—to protection from psoriasis. Furthermore, Tyk2 dysfunction has been related to IMID prevention, without increasing the risk of serious infections; thus, Tyk2 inhibition has been established as a promising therapeutic target, with multiple Tyk2 inhibitors under development.
inflammatory diseases
psoriasis
treatment
1. Introduction
The intracellular Janus Kinase/signal transducer and activator of transcription (Jak-STAT) pathways play a role in intracellular signaling of cytokines in a wide variety of cellular processes and are important in both normal and pathological states such as immune-mediated inflammatory diseases (IMID), including psoriasis and psoriatic arthritis, among others) [1][2][3].
The Jak family (Jak1, Jak2, Jak3, and Tyk2) comprises receptor-associated tyrosine kinases that act within the cell as signal transducers [4][5]. Activation of the Jak pathway starts with the coupling of a circulating cytokine—e.g., interleukin (IL)-23—to its cell surface receptor, triggering a conformational change of the receptor, which leads to the activation and recruitment of two Jaks. Jak dimers are composed of two different Jak molecules, except Jak2, which can combine with itself. These Jak dimers phosphorylate the receptor, allowing the attachment, phosphorylation, and eventual dimerization of STAT proteins (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6) (Figure 1). Activated STAT proteins combine to form dimers and can translocate to the nucleus where they can act as transcription factors, upregulating the genes responsible for production of proinflammatory cytokines and growth factors, or regulate the behavior of other intracellular proteins [1][3][6][7][8].
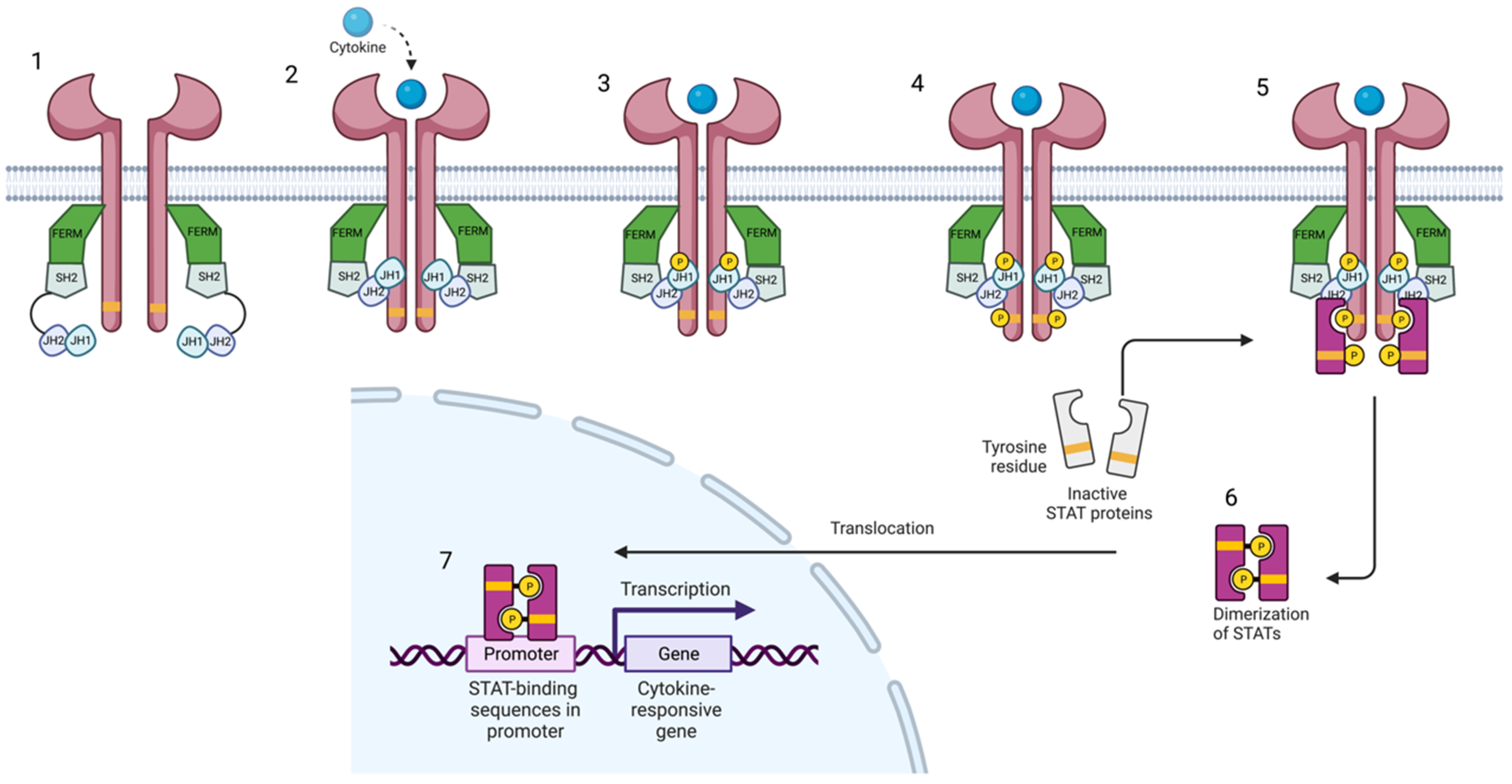
Figure 1. Janus kinase-signal transducer and activator of transcription pathway. Cytokines bind to their receptor, leading to the activation of Jak and their phosphorylation. Subsequently, STAT is phosphorylated and dimerized. Activated STAT dimers translocate to the nucleus and regulate gene transcription and expression. Created with BioRender.com (accessed on 14 January 2023). Abbreviations: FERM, 4.1 ezrin, radixin moesin domain; SH2, Src-homology 2 domain; JH1, jak homology domain 1 (kinase domain); JH2, jak homology domain 2 (pseudokinase domain).
The Jak/STAT pathway is a therapeutic target in various IMID, since the immune response is coordinated and regulated by soluble mediators corresponding to proinflammatory cytokines in most cases. Jak inhibitors are small molecules that diminish the intracellular transduction of the Jak/STAT pathway, usually by inhibiting the kinase activity of Jak. Due to their small molecular size, Jak inhibitors can be administered orally or topically [1].
Treatment with Jak inhibitors is generally associated with mild to moderate side effects, usually infections of the upper respiratory tract, urinary tract, and gastrointestinal tract, but in some patient populations the risks may overcome the benefits of treatment. Because of the relative non-selectivity of the first generation of Jak inhibitors, most adverse events are considered class specific. Inhibition of Jak1 has been associated with increases in serum levels of triglycerides, total cholesterol, low-density lipoprotein cholesterol, and high-density lipoprotein cholesterol. On the other hand, Jak2 inhibition can interfere with erythropoiesis, myelopoiesis, and platelet activation, leading to anemia, neutropenia, thrombocytopenia, and thromboembolic events. Jak3 expression is restricted to hematopoietic cells and exclusively associated with only the common γc receptor subunit of interleukins regulating lymphocyte activation, function, and proliferation. Tyk2 inhibition can increase the risk of herpesvirus, staphylococcal, and mycobacterial infections [1][9].
2. Jak-STAT Signaling and Inhibition
The Jak-STAT pathway mediates downstream signaling of receptors for type I and II cytokines, such as IL-6, IL-10, IL-12, IL-22, IL-23, and interferon (IFN)-α, IFN-β, and IFN-γ (Figure 2). As mentioned before, Jaks associate in pairs. Jak1 pairs with Jak2, Jak3, and Tyk2, leading to the downstream transduction of signals generated by receptors of cytokines such as IFN-α, IFN-γ, IL-2, IL-4, IL-6, IL-7, IL-10, and IL-15 [10][11]. Mice lacking Jak1 have highly impaired lymphopoiesis and inadequate IFN (types I and II) responses, which results in mortality. Jak3 pairs only with Jak1, transducing signals from cytokines sharing the common cytokine receptor γ chain, such as IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21; thus, Jak3 signaling is essential for lymphocyte development [10]. Jak2 forms a homodimer when pairing with itself or a heterodimer with Tyk2. Homodimers of Jak2 mediate signal transduction downstream of IL-3 and hormone-like receptors, such as erythropoietin, growth hormone, prolactin, thrombopoietin, leptin, and granulocyte-macrophage colony-stimulating factor [10], whereas heterodimers of Jak2 and Tyk2 connect with receptors for type I interferons (IFN), IL-12, and IL-23 [12][13].
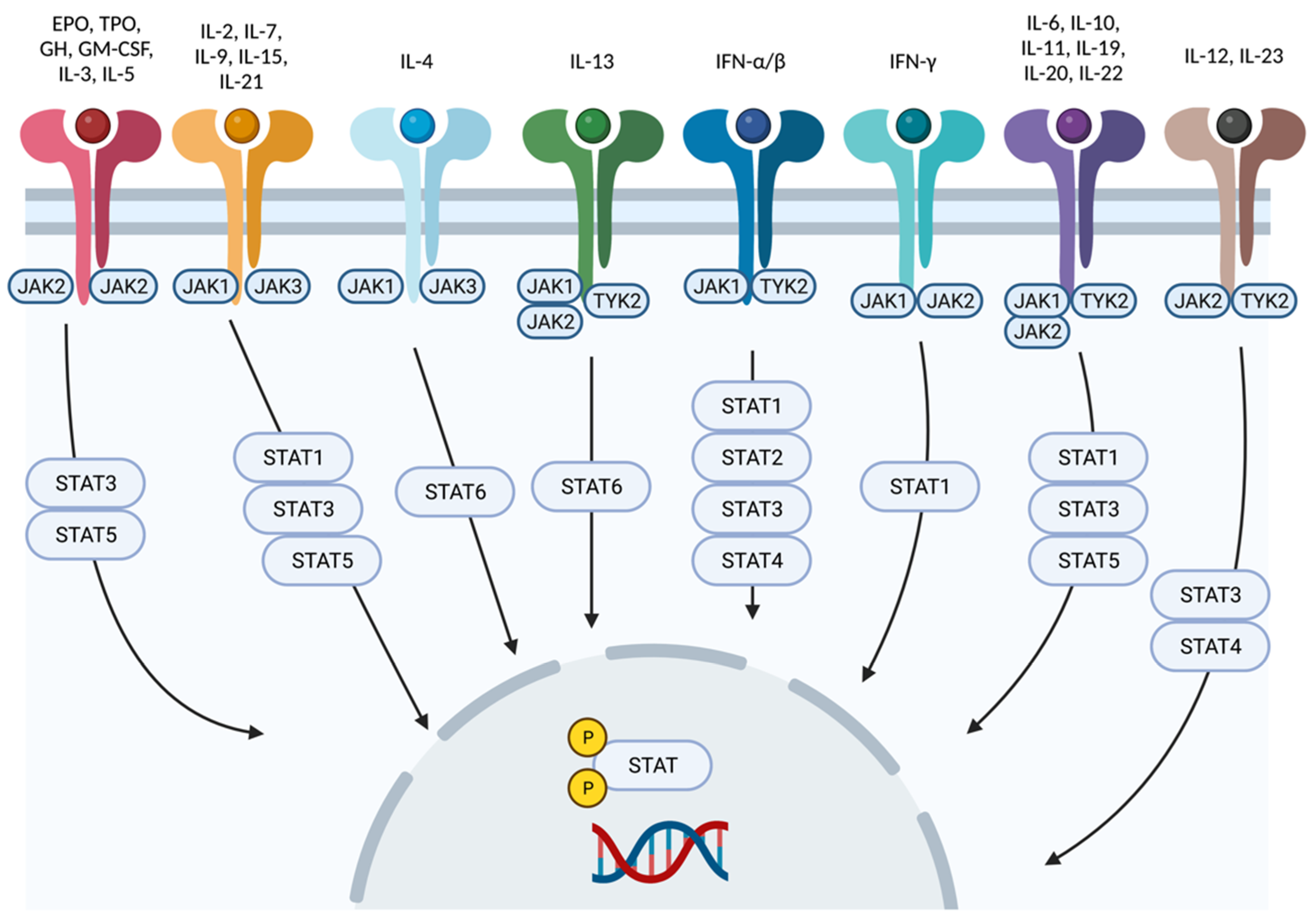
Figure 2. Cytokine signaling mediated by Jak-STAT. Each cytokine interacts with a particular combination of Jak-STAT molecules. Created with BioRender.com (accessed on 14 January 2023). Abbreviations: Jak, Janus kinase; STAT, signal transducer and activator of transcription; Tyk2, tyrosine kinase 2; IFN, interferon; IL, interleukin; EPO, erythropoietin; GH, growth hormone; TPO, thrombopoietin; GM-CSF, granulocyte macrophage colony-stimulating factor.
As shown in Figure 2, cytokine receptors interact with a particular pair of Jaks, which, in turn, interact with some of the existent STAT proteins. Hence, depending on the targeted cytokine, the choice of Jak inhibitor will be different. For example, atopic dermatitis is associated with overexpression of IL-4 and activation of its signaling pathway, which is mediated by the Jak1/Jak3 pair [14]. Conversely, signaling of IL-12 and especially IL-23, which is pathogenetically related to psoriasis, is mediated by Tyk2/Jak2; selective inhibition of Tyk2 avoids interference with multiple Jak2 mediated pathways and potential hematopoietic or thromboembolic adverse events [14][15].
Some Jak inhibitors are more selective than others in their targeting of the active regions in Jak catalytic (JH1) domains. First-generation Jak inhibitors often target two or three separate Jaks and can offer a wide range of therapeutic benefits; however, they may be more likely to cause adverse events than Jak inhibitors from later generations [13][16].
Currently approved therapeutic indications of Jak inhibitors include polycythemia vera (ruxolitinib), myelofibrosis (fedratinib, pacritinib, ruxolitinib), rheumatologic diseases such as rheumatoid arthritis (tofacitinib, baricitinib, peficitinib, filgotinib), ankylosing spondylitis (tofacitinib, upadacitinib), juvenile idiopathic arthritis (tofacitinib) and psoriatic arthritis (tofacitinib, upadacitinib), ulcerative colitis (baricitinib, upadacitinib, tofacitinib), atopic dermatitis (upadacitinib, baricitinib, abrocitinib, and topical delgocitinib in some countries), alopecia areata (baricitinib), and psoriasis (deucravacitinib) [17]. Only the indications of Jak inhibitors related to psoriatic disease will be specifically mentioned below, whereas Tyk2 inhibitors will be discussed extensively in Section 4 of this manuscript.
Tofacitinib, a pan-Jak inhibitor that predominantly targets Jak1 and Jak3, is currently approved for treatment of psoriatic arthritis at the dose of 5 mg twice daily; in phase 3 clinical trials for psoriasis, the 10 mg twice daily regimen was not inferior to etanercept [18][19], but FDA approval was declined based on long term safety issues. Filgotinib, a Jak1 inhibitor [10], and upadacitinib, a Jak1 inhibitor with partial selectivity for Jak2, have also been approved for the treatment of psoriatic arthritis [20].
3. Tyk2 Signaling and Pathogenetic Implications
Genetic linkage studies have established a connection between dysfunctional Tyk2 mutations and protection from psoriasis [3][21]. This can be explained by the implication of Tyk2 in multiple pathways related to psoriasis. The inflammatory environment of active psoriatic skin lesions is characterized by the expression of Th1- and Th17-related cytokines, and type I IFN, IL-6, IL-10, IL-12, IL-22, and IL-23 are only a few of the pathogenetically relevant cytokines with signaling pathways affected by Tyk2 loss-of-function mutations (Figure 3) [8][10][22][23]. Inhibiting Tyk2 activation might be associated with an ideal balance between efficacy and safety because individuals with deactivating genetic variants of Tyk2 are highly protected from some IMIDs but do not exhibit an increased risk of hospitalization for mycobacterial, viral, or fungal infections [23][24][25][26][27].
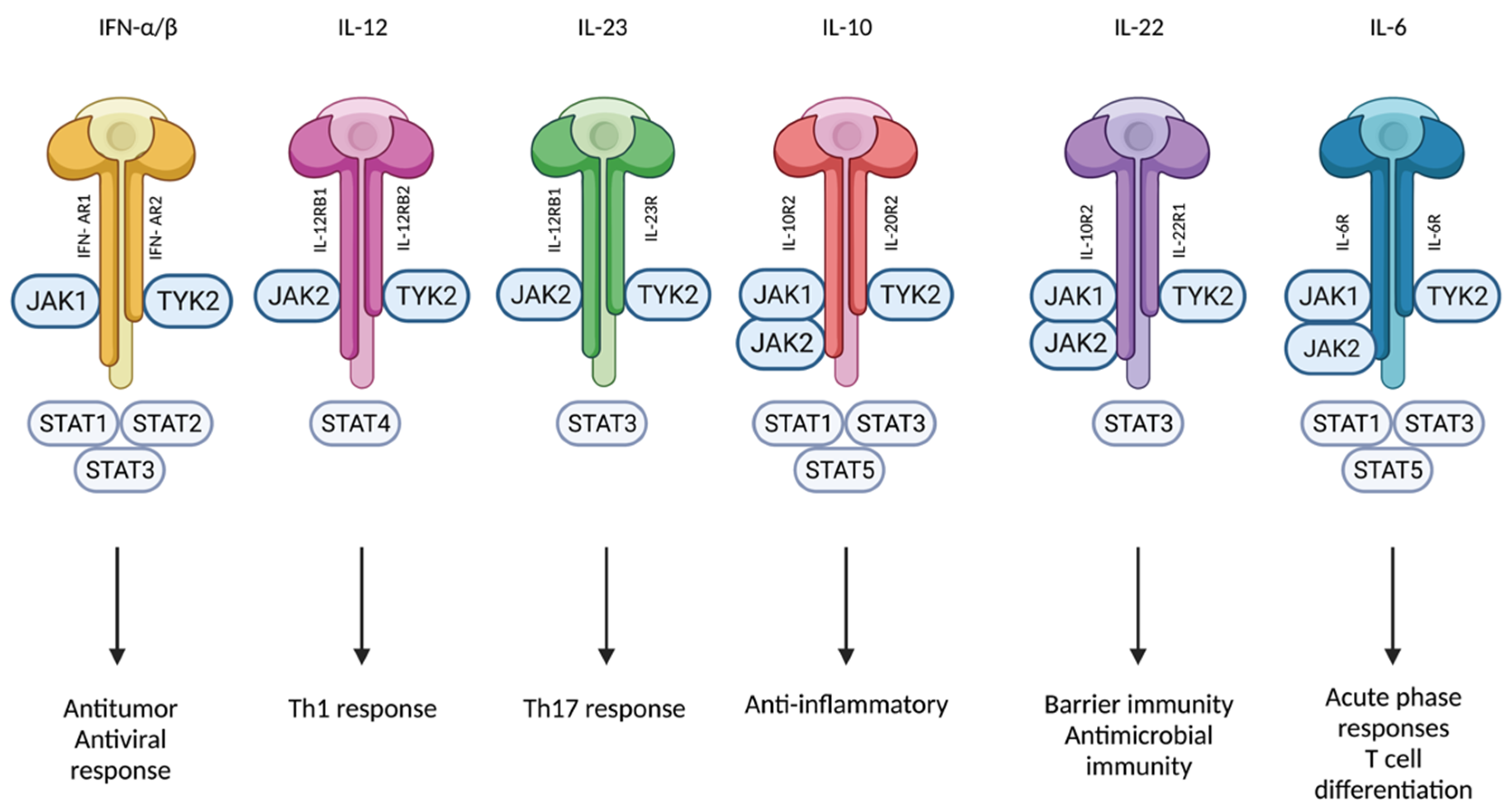
Figure 3. Tyk2 mediates signaling of IFN-α/β, IL-12, IL-23, IL-10, IL-22, and IL-6. Created with BioRender.com (accessed on 14 January 2023). Abbreviations: Tyk2, tyrosine kinase 2; IFN, interferon; IL, interleukin; Jak, Janus kinase; STAT, signal transducer and activator of transcription; Th, T-helper; R, Receptor.
In psoriasis, hidradenitis suppurativa, and other IMIDs, chronic inflammation is initiated and maintained by IL-12 and IL-23 [28][29][30]. IL-12 and IL-23 receptor signal transduction pathways are mediated by the Tyk2/Jak2 heterodimer. IL-12 is necessary for the growth of Th1 cells, which generate pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α and IFN-γ. Th17 cell growth and survival are regulated by IL-23 and keratinocyte growth and activation are enhanced by a combination of cytokines produced by Th1 and Th17 cells [28][31][32].
Tyk2 can also pair with Jak1 to participate in downstream pathway signal transduction of type I IFN receptors, which induce potent antiviral mechanisms [10]. IFN-α and IFN-β are produced rapidly and in great quantities by a variety of cell types, particularly plasmacytoid dendritic cells, during proinflammatory conditions such viral infections [33]. They are also implicated in proinflammatory processes relevant in psoriasis, such as dendritic cell maturation and activation, polarization of Th1 and Th17 cells, impairment of the T-cell regulatory function, and enhancement of the activation of B cells and their antibody production [10][33].
Redundancy in Jak/STAT pathways is common to all Jak combinations except homodimeric Jak2, but the activation of Tyk2 due to IL-12, IL-23, and type I IFN results in STAT-dependent transcription and inflammatory responses different from those occurring subsequent to activation of Jak1, Jak2, and Jak3 [33][34][35][36].
Multiple studies have established that Tyk2 is mandatory for IL-12 and IL-23 signaling. Tyk2-knockout mice do not show epidermal hyperplasia after IL-23 activation, as opposed to Tyk2-positive wild mice; moreover, it has been noted that IL-23 in Tyk2-positive lymphocytes induces dose-dependent secretion of IL-17 and IL-22, which has not been observed in Tyk2-knockout lymphocytes [37].
The need for Tyk2 for type I IFN signaling is less clear, with some contradictory data [38][39]. Majoros et al. reported that Tyk2 was essential for IFN-α signaling, since Tyk2-deficient human cells did not respond to IFN-α [40]. However, in Tyk2-knockout mice, type I IFN signaling was reduced but not suppressed [39]. Therefore, Tyk2 seems to be necessary for type I IFN signaling in human cells, but not in mice cells.
Additionally, Tyk2 signaling has been linked to the immune system response to the IL-10 family of cytokines. Numerous immune cell types release IL-10, which upon binding to its receptor activates Jak1 and Tyk2, leading to a wide range of immunosuppressive and immunostimulatory effects [38][41][42]. Some of the immunosuppressive effects include inhibition of multiple processes, such as nuclear translocation of the nuclear factor kappa light chain enhancer of activated B cells (NF-kB), IFN-α- and IFN-γ-induced gene transcription, expression of major histocompatibility complex class II molecules by activated dendritic cells and macrophages, and T-cell activation and proliferation [38][41][42][43]. Therefore, clinical studies were performed trying to assess the efficacy of recombinant IL-10 to treat autoimmune diseases. Unfortunately, results were less than encouraging [41].
On the other hand, according to several studies, IL-10 promotes humoral immune responses by enabling B cells to differentiate, proliferate, and survive, as well as by stimulating them to produce antibodies [41][42][44]. In systemic lupus erythematosus, high levels of IL-10 expression are considered pathogenic, and its inhibition would be beneficial [41]. Furthermore, IL-10 has been linked to contradictory effects in some cell types, such as natural killer cells, depending on the cellular context [41]. IL-22, an IL-10 family member generated in skin and gastrointestinal tract epithelium, activates Jak1 and Tyk2 and contributes to the epithelial integrity, primarily by enhancing barrier function and triggering the synthesis of antimicrobial peptides. On the other hand, IL-22 also promotes the synthesis by epithelial cells of chemokines that may contribute to tissue damage and gastrointestinal inflammation. This, together with the pathogenetic involvement of IL-12 and IL-13, provides the mechanistic bases for clinical research of Tyk2 inhibitors in the treatment of bowel inflammatory diseases [38][43][44].
Type I IFNs can cause monocytes to differentiate into antigen-presenting dendritic cells, the purported key mechanism by which these cells control the activity of autoreactive B and T cells in autoimmune disorders such as lupus and dermatomyositis [34][44]. Additionally, blocking type I IFN receptor (IFNAR) activation with an anti-IFNAR antibody (anifrolumab) lowers disease activity in systemic lupus erythematosus (SLE) patients [34][45]. Whole blood from 31 SLE patients was treated for 5 h with either deucravacitinib or an anti-IFNAR antibody, and the effect on type I IFN-regulated genes was assessed by quantitative PCR. Deucravacitinib decreased the expression of type I IFN-regulated genes that are part of the IFN profile enhanced in SLE patients in this ex vivo test [45]. Deucravacitinib was as effective at the blocking the anti-IFNAR antibody and the response was dose-dependent, with 12 mg twice daily providing almost complete inhibition of CXCL10, ISG20, and IFI27 [45]. These types of I IFN-regulated genes are overexpressed in diseases such as SLE, Sjögren syndrome, and systemic sclerosis [34][44][46].
4. Tyk2 Pharmacologic Inhibition
As previously mentioned, Tyk2 is an important mediator in pro-inflammatory signaling, and its inhibition does not seem to carry an increased risk of serious infections [3][33]. Hence, multiple Tyk2 inhibitors are being evaluated for the treatment of inflammatory diseases such as psoriasis, psoriatic arthritis, hidradenitis suppurativa, inflammatory bowel disease, dermatomyositis, and SLE [10][29][30]. Tyk2 inhibitors currently include 21 different molecules, but only 3 of them are selective or predominantly Tyk2 inhibitors; the first includes deucravacitinib (Bristol Myers Squibb, New york, New York, US), ropsacitinib (Priovant Therapeutics, New york, New York, US), BMS-986202 (Bristol Myers Squibb, New york, New York, US), whereas brepocitinib (Priovant Therapeutics, New york, New York, US) and SAR-20347 (ChemScene, Monmouth Junction, NJ, US), albeit potent, are not considered to be selective Tyk2 inhibitors [47] (Table 1). Ghoreschi and colleagues published, in 2021, a Tyk2 revision, focalizing on deucravacitinib [15]. Hereby, a wider revision of Tyk2 inhibitors and their latest results are provided.
Table 1. Summary of the main characteristics of the Tyk2 inhibitors discussed here.
| Treatment | Mechanism of Action | Oral and/or Topical | Approved Indications | Tested Indications | Most Frequent Adverse Events | Current Clinical Trials |
|---|---|---|---|---|---|---|
| Brepocitinib | Tyk2/Jak1 inhibitor (binds to the catalytic domain, JH1) | Oral and topical | Not yet |
|
Nasopharyngitis, upper respiratory tract infection, and headache |
|
| Ropsacitinib | Tyk2/Jak2 inhibitor (binds to the catalytic domain, JH1) | Oral | Not yet |
|
Increased serum creatinine levels, elevated alanine aminotransferase levels, and headache |
|
| Deucravacitinib | Tyk2 inhibitor (binds to the regulatory domain, JH2) | Oral |
|
|
Nasopharyngitis, and upper respiratory tract infection * No dyslipidemia |
|
| BMS-986202 | Tyk2 inhibitor (binds to the regulatory domain, JH2) | Oral | Not yet | No | Not available |
|
| SAR-20347 | Tyk2/Jak1 inhibitor (binds to the catalytic domain, JH1) | Oral | Not Yet | No | Not available |
|
* No dyslipidemia: Deucravacitinib demonstrated no dyslipidemia, conversely to the remaining Tyk2 inhibitors tested.
4.1. Brepocitinib (PF-06700841)
Brepocitinib is a dual Tyk2/Jak1 inhibitor that binds to the active sites in the catalytic domains of Tyk2 and Jak1, with less affinity for Jak2, and even minor selectivity for Jak3 [48][49]. Therefore, adverse events related to Jak2 and Jak3 inhibition are less likely to occur [48]. In a phase 1 trial, brepocitinib was well tolerated by both healthy volunteers and patients with psoriasis, but decreases in platelet and reticulocyte counts occasionally occurred and have been accounted for by some degree of Jak2 inhibition [10][48].
4.1.1. Psoriasis
Following a phase 1 clinical trial [50], the efficacy and safety of brepocitinib in psoriasis were evaluated in a phase 2a trial with a four-week induction period of 30 mg once daily, 60 mg once daily, or placebo, followed by an eight-week maintenance phase with brepocitinib 10 mg once day, 30 mg once daily, 100 mg once weekly, or placebo (ClinicalTrials.gov identifier: NCT02969018) [51]. Decreases in Psoriasis Area and Severity Index (PASI) at week 12 (the primary endpoint) were significantly greater in patients who received brepocitinib than in those who received placebo; the greatest change from baseline was observed in the 30 mg once daily continuous treatment group. Treatment was well tolerated and no herpes zoster infections were reported.
Brepocitinib treatment appeared to lower the expression of several inflammatory genes and cellular pathways associated with the pathogenesis of psoriasis, according to biomarker analyses. In psoriasis skin biopsy specimens, brepocitinib inhibited IL-17A/F and IL-12B expression more rapidly than tofacitinib (2 weeks vs. 4 weeks) [44]. The clinical and molecular improvement with brepocitinib was faster and more complete, with significant reduction in markers of keratinocyte activation, epidermal thickness, KRT16, and Ki-67 expression; furthermore, clinical improvement of psoriasis was associated with a decrease in the immune cell infiltrates CD3+/CD8+ T cells and CD11c dendritic cells [44][52].
4.1.2. Alopecia Areata
Brepocitinib has also been compared to ritlecitinib—an inhibitor of Jak3 and members of the Tec tyrosine kinase family—and placebo for the treatment of alopecia areata. Severity of Alopecia Tool (SALT)30 at 24-weeks of treatment was achieved by 64% of the patients under brepocitinib treatment vs. 50% of patients under ritlecitinib and only 2% of patients on placebo [53]. The improvement in SALT scores was positively associated with expression of Th1 markers and negatively associated with expression of hair keratins [54][55].
4.1.3. Atopic Dermatitis
Finally, topical brepocitinib has been tested for the treatment of atopic dermatitis. In 2019, a phase 2b clinical trial evaluated the efficacy and safety of topical brepocitinib in mild to moderate atopic dermatitis [48][56]. A total of 292 individuals were enrolled and randomized. Brepocitinib 1% once daily and brepocitinib 1% twice daily achieved significant reductions in Eczema and Area Severity Index (EASI) score, compared to the respective vehicle, with no serious adverse events [56].
4.1.4. New Potential Therapeutic Indications
Development of oral brepocitinib has been discontinued for most of its potential indications, including psoriasis, psoriatic arthritis, vitiligo, ulcerative colitis, hidradenitis suppurativa, and Crohn’s disease. In June 2022, Pfizer licensed oral and topical global development rights and US and Japan commercial rights of brepocitinib to Priovant; a phase 2b trial of oral brepocitinib in SLE [57] (trialsearch.who.int/EUCTR2018-004175-12-PL), a phase 2 trial of oral brepocitinib in adults with active non-infectious non-anterior uveitis (NCT05523765), a phase 3 trial of oral brepocitinib in dermatomyositis (NCT05437263), and an investigator-initiated trial of oral brepocitinib in cicatricial alopecia (NCT05076006) are currently ongoing. Phase 2 clinical trials of topical brepocitinib have also been stopped [10][52][57].
4.2. Ropsacitinib (PF-06826647)
The oral Tyk2/Jak2 inhibitor ropsacitinib binds to the active site in the catalytic domain (JH1) of each kinase. Despite inhibiting Jak2, ropsacitinib possesses more selectivity for Tyk2 than brepocitinib.
4.2.1. Phase 1 Clinical Trials
Ropsacitinib had an acceptable safety profile in two phase 1 randomized, double-blind, placebo-controlled trials in healthy volunteers and patients with moderate to severe plaque psoriasis [58][59][60]. Ropsacitinib significantly decreased disease activity, as determined by the PASI 75 (≥75% reduction from baseline in Psoriasis Area and Severity Index) response, body surface area, and target plaque severity score after 4 weeks of treatment in the previously mentioned phase 1 study. In addition, this study detected a significant reduction in the IL-17A and IL-17F expression at week 4 of treatment [58]. Increased serum creatinine levels, elevated alanine aminotransferase levels, and headache were the most frequent adverse events in patients with plaque psoriasis. No significant adverse events, fatalities, dose reductions, or temporary discontinuations occurred in individuals receiving ropsacitinib treatment; all side effects were minor [59][60].
4.2.2. Phase 2 Clinical Trials
Ropsacitinib phase 2 clinical trials on psoriasis and hidradenitis suppurativa have been completed, but results of the latter (NCT04092452) have not yet been published. The phase 2b clinical trial evaluating ropsacitinib efficacy and safety compared to placebo enrolled and treated up to 178 participants. They were randomized to receive once-daily oral placebo, or ropsacitinib at 50 mg, 100 mg, 200 mg, and 400 mg [61]. The 200 mg and 400 mg groups showed a better proportion of PASI90 (≥90% reduction from baseline in Psoriasis Area and Severity Index) response compared to placebo at week 16 [61]. Ropsacitinib was well tolerated at week 40 of follow-up and only 18 patients discontinued the treatment due to treatment-related adverse events [61].
The phase 2b clinical trial of ropsacitinib in patients with ulcerative colitis was withdrawn (NCT04209556) [57]. Currently, there is no ongoing clinical trial of ropsacitinib, which has also been licensed by Pfizer to Priovant [62].
4.3. Deucravacitinib
Deucravacitinib is an oral Tyk2 inhibitor that binds to the regulatory (JH2 pseudokinase) domain rather than the active (ATP-binding) site in the catalytic (JH1) domain of Tyk2, where other Tyk2/Jak1-3 inhibitors attach (Figure 4) [3][34][63][64]. The allosteric binding of deucravacitinib locks the regulatory (JH2) domain into an inhibitory contact with the catalytic domain. This leads to the inactivation of Tyk2, inhibiting receptor-mediated activation and subsequent signal transduction [34].
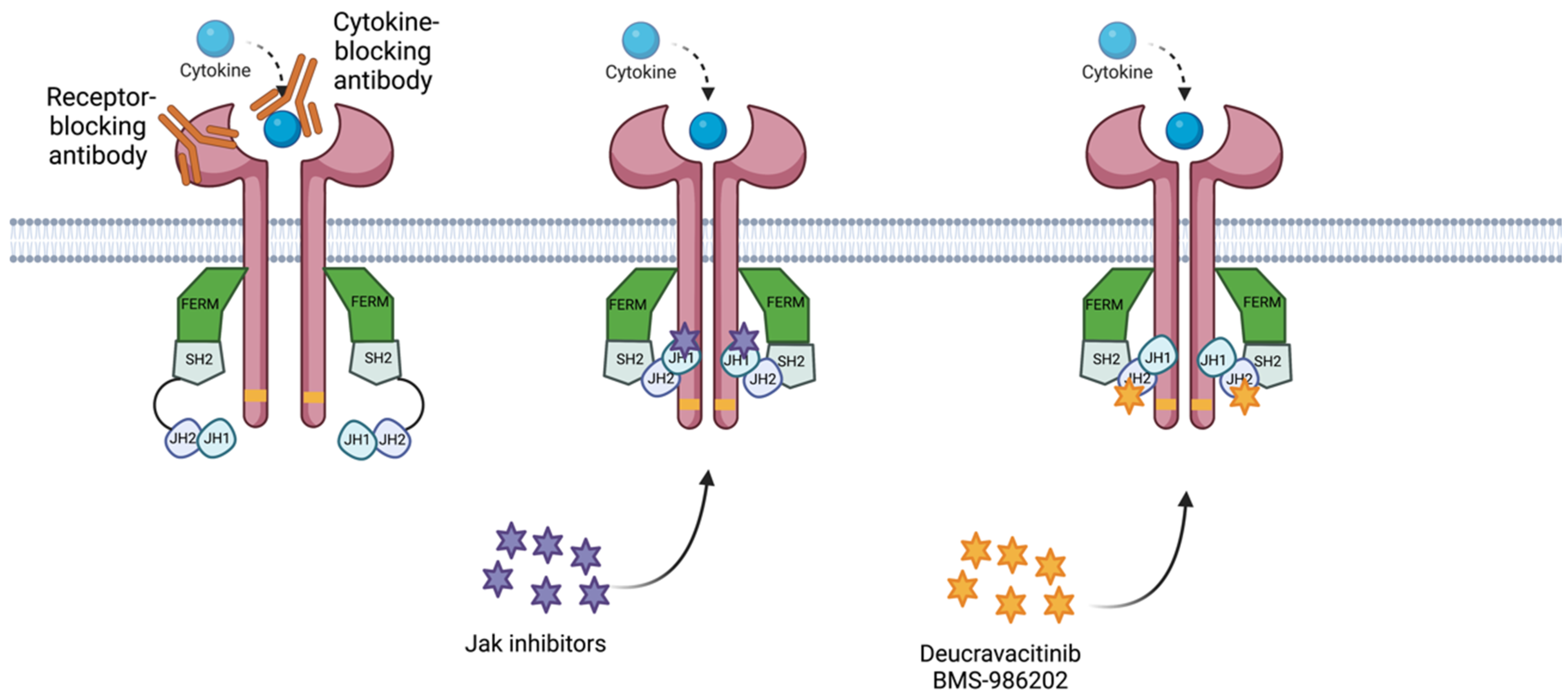
Figure 4. Diagram of mechanism of action for the treatments for IMIDs. The receptor-blocking antibody binds to the receptor, preventing its capacity to bind to the cytokine. The. cytokine-blocking antibody binds to the cytokine, blocking its capacity to bind to the receptor. Jak inhibitors bind to the JH1 domain—kinase domain—impeding adenosine triphosphate (ATP) binding to the JH1 catalytic domain. Deucravacitinib and BMS-986202 bind to the JH2 domain, locking the regulatory (JH2) domain into an inhibitory contact with the catalytic domain (JH1). Created with BioRender.com (accessed on 14 January 2023). Abbreviations: FERM, 4.1 ezrin, radixin moesin domain; SH2, Src-homology 2 domain; JH1, jak homology domain 1 (kinase domain); JH2, jak homology domain 2 (pseudokinase domain).
Deucravacitinib is highly selective for Tyk2, with little to no activity against Jak1–3. In cell-based assays, deucravacitinib has more than 100-fold greater selectivity for Tyk2 over Jak1/3 and more than 2000-fold greater selectivity for Tyk2 over Jak2 (half maximal inhibitory concentration [IC50] data) [34][64][65].
4.3.1. Psoriasis
Deucravacitinib was evaluated in a randomized, placebo-controlled, dose-ranging 12 week phase 2 trial including 267 adult patients (NCT02931838); the PASI75 response rates at week 12 (the primary endpoint) for deucravacitinib were 68.9% (3 mg twice daily), 66.7% (6 mg twice daily), and 75% (12 mg once daily) vs. placebo (6.7%) [66].
Skin biopsy specimens taken after deucravacitinib treatment in this phase 2 trial demonstrated normalization of inflammatory gene expression and inhibition of biologic markers of disease activity, such as the expression of the IL-23/Th17 and type I IFN signaling pathways, which are involved in keratinocyte activation and inflammation in psoriasis [67].
The most common adverse events were mild and included nasopharyngitis, headaches, diarrhea, nausea, and upper respiratory tract infections. No herpes zoster infections or cardiovascular events, which are adverse events of special interest for Jak inhibitors, were reported. One patient was diagnosed of melanoma during treatment [66]. Patients receiving deucravacitinib had no significant alterations of hematologic parameters (lymphocyte, natural killer cell, neutrophil, and platelet counts and hemoglobin levels), serum lipids (high-density lipoprotein and low-density lipoprotein cholesterol), creatinine, immunoglobulins, or liver enzymes [10][66][68]. Regarding dyslipidemia, its development as an adverse event is partially related to the impact on IL-6 signaling, which is regulated by Jak; since deucravacitinib does not interact with Jak1 in human cells, this adverse event should not be expected and the same applies to hematologic alterations occurring when Jak2 is inhibited. Therefore, deucravacitinib reduces the potential risk of abnormalities in laboratory parameters [12][16][34][66].
A post-hoc analysis of the same phase 2 clinical trial assessed additional clinical and quality of life (QoL) outcomes of the three most efficacious regimens (3 mg twice daily, 6 mg twice daily and 12 mg once daily) and placebo [69]. Early improvement was noted at week 4 in patients under deucravacitinib, and clinical responses, including PASI and Body surface Area (BSA), showed similar trends to QoL measured by Dermatology Life Quality Index (DLQI) [69]. In patients combined from the three deucravacitinib regimens, DLQI 0/1 response was achieved by 17.9% at week 4, 41.8% at week 8, and 55.2% at week 12. DLQI 0/1 response rates on the placebo group were 6.7%, 4.4%, and 4.4%, respectively [69].
These results indicated significant commercial potential for this new oral treatment for psoriasis, where apremilast has a leading market position in the USA and other countries; apparently apremilast had to be sold to expedite the Federal Trade Commission approval of the merger between Bristol Myers Squibb and Celgene [70].
Results from two phase 3 clinical trials comparing deucravacitinib to placebo and apremilast have been published. POETyk PSO-1 was a 52 week phase 3 trial in which participants were randomized 2:1:1 to receive deucravacitinib 6 mg every day (n = 332), placebo (n = 166), or apremilast 30 mg twice a day (n = 168) (NCT03624127) [71]. At week 16, response rates were significantly higher with deucravacitinib versus placebo or apremilast for PASI75 194 [58.4%] vs. 21 [12.7%] vs. 59 [35.1%] and static Physician’s Global Assessment score of 0 or 1 (sPGA 0/1): 178 [53.6%] vs. 12 [7.2%] vs. 54 [32.1%]. Efficacy improved beyond week 16 and was maintained through week 52. The most common adverse events related to deucravacitinib were nasopharyngitis and upper respiratory tract infection [71].
In POETyk PSO-2 (NCT03611751), a total of 1020 patients were randomized: 511 received deucravacitinib 6 mg once daily, 255 placebo, and 254 patients were treated with apremilast 30 mg twice daily [72]. Patients on deucravacitinib treatment showed better results than placebo and apremilast. At week 16, PASI75 was achieved by 53% of the patients treated with deucravacitinib, compared to 9.4% and 39.8% of the patients treated with placebo and apremilast, respectively. At week 52, efficacy with continuous deucravacitinib was maintained. The most frequent adverse event was nasopharyngitis, no meaningful laboratory changes were detected, and discontinuations due to adverse events were infrequent [72].
The efficacy and safety of deucravacitinib in treating moderate to severe psoriasis, scalp psoriasis, nail psoriasis, and in pediatric patients with moderate to severe psoriasis are being examined in phase 3 and 4 clinical trials (NCT04036435, NCT05478499, NCT05124080, and NCT04772079, respectively). Moreover, a phase 4 observational post-marketing surveillance of adverse events in patients with psoriasis in Japan is expected to start recruiting patients in the near future (NCT05633264). Finally, an adherence clinical trial in patients with psoriasis is expected to start recruiting soon (NCT05570955).
Deucravacitinib was first approved in the USA on 9 September 2022 for the treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy. Subsequently, on 26 September 2022, it was approved in Japan for the treatment of plaque psoriasis, generalized pustular psoriasis, and erythrodermic psoriasis. Currently, deucravacitinib is under consideration by the European Medicines Agency [73].
4.3.2. Psoriatic Arthritis
A phase 2 clinical trial (NCT03881059) evaluated the efficacy and safety of deucravacitinib compared to placebo in patients with active psoriatic arthritis [74]. A total of 203 participants were randomized in three groups: placebo, deucravacitinib 6 mg once daily, and deucravacitinib 12 mg once a day. The primary endpoint was American College of Rheumatology-20 (ACR-20) response at week 16. The ACR-20 response was significantly higher in the 6 mg (52.9%) and 12 mg (62.7%) groups compared to placebo. No serious adverse events, herpes zoster, or cardiovascular events were reported. Laboratory measures were not different to placebo group. The most common adverse events reported in deucravacitinib-treated patients were nasopharyngitis, upper respiratory tract infection, sinusitis, bronchitis, rash, headache, and diarrhea [74]. Currently, two phase 3 clinical trials for active PsA are recruiting patients (NCT04908202 and NCT04908189).
4.3.3. Other Therapeutic Indications
Deucravacitinib is being evaluated in a number of phase 2 and 3 trials for alopecia areata (NCT05556265), moderate to severe ulcerative colitis (NCT03934216), Crohn’s disease (NCT04877990), active SLE (NCT05617677), active discoid, and subacute cutaneous lupus erythematosus (NCT04857034). Results of these clinical trials are not available yet, except for those of the phase 2 clinical trial in ulcerative colitis, where deucravacitinib failed to meet its primary or secondary efficacy endpoints at week 12 [75].
4.4. BMS-986202
Structural and molecular variations applied on deucravacitinib led to multiple possible new drugs, and pharmacokinetic studies eventually led to further development of BMS-986202 as a potential new oral Tyk2 inhibitor binding to the regulatory JH2 pseudokinase domain [76]. To evaluate the pharmacodynamic responses of BMS-986202, its ability to inhibit IFN-γ production induced by the stimulation with IL-12/IL-18 was tested in mice. A dose-dependent reduction in IFN-γ production was detected, with an up to 80% reduction of IFN-γ levels with the highest dose [76].
Subsequently, the potential efficacy of BMS-986202 was evaluated on psoriasis, ulcerative colitis, and SLE. A well-established psoriasis-like model in mice is the induction of skin acanthosis with IL-23 injections. In this model, BMS-986202 inhibited acanthosis in a dose–response manner and the 30 mg/kg once-daily dose showed at least equivalent efficacy to ustekinumab, which was used as a positive control in the study [76]. The murine model of ulcerative colitis consists of a single injection of an anti-CD40 antibody in severe combined immune-deficient (SCID) mice, resulting in induced colitis. BMS-986202 was orally dosed in these mice for 6 days, and histopathologic studies showed a dose-dependent inhibition of colitis, with 25 and 60 mg/kg doses yielding equivalent efficacy to the anti-p40 antibody that was used as a positive control [76]. In a spontaneous lupus model (NZB/W mice), BMS-986202 inhibited development of anti-dsDNA titers and severe proteinuria in a dose-dependent manner [76].
As regards human studies, a phase 1 clinical trial evaluating BMS-986202 safety, tolerability, pharmacokinetics, and pharmacodynamics has been completed, but results have not been posted yet (NCT02763969).
4.5. SAR-20347
SAR-20347 is an oral inhibitor of Tyk2 and Jak1 with selectivity over Jak2 and Jak3. Preclinical assays demonstrated that SAR-20347 inhibited IL-12, IL-23 and IFN-α signaling [22]. Both Tyk2 mutant mice and mice treated with SAR-20347 showed significant reduction of IL-6 and IL-17 in imiquimod-induced skin lesions, but only SAR-20347-treated mice presented reduced levels of IL-23, decreased keratinocyte proliferation and improved clinical score [22]. In addition, SAR-20347-treated mice manifested lower IL-17 gene expression compared to Tyk2 mutant mice [22]. In this model, Works et al. demonstrated that SAR-20347 treated mice showed an almost complete loss of IL-22 gene expression in skin lesions, and they postulate that SAR-20347 would impair the ability of Th17 and γδ cells to induce IL-22 [22]. IL-22 is required for development of autoreactive Th17 cells [77][78]. However, not only IL-22 production was impaired, since IL-22 signaling was also affected in vitro in a human colonic cell line, and STAT3 phosphorylation dependent of IL-22 was completely blocked [22].
Blocking both Tyk2 and Jak1 in this research was more effective than inhibition of Tyk2 alone at reducing psoriasis-like disease severity, keratinocyte proliferation, as well as IL-23, IL-17, IL-6, IL-22, and antimicrobial peptide gene expression, and the authors postulate that targeting a combination of Jak1 and Tyk2 using an orally available inhibitor may be a viable approach for treating psoriasis, but currently there are no ongoing or completed clinical trials for SAR-20347 [22].
References
- Garcia-Melendo, C.; Cubiró, X.; Puig, L. Janus Kinase Inhibitors in Dermatology: Part 1—General Considerations and Applications in Vitiligo and Alopecia Areata. Actas Dermo-Sifiliográficas Engl. Ed. 2021, 112, 503–515.
- Garcia-Melendo, C.; Cubiró, X.; Puig, L. Janus Kinase Inhibitors in Dermatology: Part 2: Applications in Psoriasis, Atopic Dermatitis, and Other Dermatoses. Actas Dermo-Sifiliográficas Engl. Ed. 2021, 112, 586–600.
- Nogueira, M.; Puig, L.; Torres, T. JAK Inhibitors for Treatment of Psoriasis: Focus on Selective TYK2 Inhibitors. Drugs 2020, 80, 341–352.
- Harrison, D.A. The JAK/STAT Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011205.
- Choy, E.H. Clinical Significance of Janus Kinase Inhibitor Selectivity. Rheumatology 2019, 58, 953–962.
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT Pathway: Impact on Human Disease and Therapeutic Intervention. Annu. Rev. Med. 2015, 66, 311–328.
- Villarino, A.V.; Kanno, Y.; Ferdinand, J.R.; O’Shea, J.J. Mechanisms of Jak/STAT Signaling in Immunity and Disease. J. Immunol. 2015, 194, 21–27.
- Shang, L.; Cao, J.; Zhao, S.; Zhang, J.; He, Y. TYK2 in Immune Responses and Treatment of Psoriasis. J. Inflamm. Res. 2022, 15, 5373–5385.
- Kreins, A.Y.; Ciancanelli, M.J.; Okada, S.; Kong, X.-F.; Ramírez-Alejo, N.; Kilic, S.S.; El Baghdadi, J.; Nonoyama, S.; Mahdaviani, S.A.; Ailal, F.; et al. Human TYK2 Deficiency: Mycobacterial and Viral Infections without Hyper-IgE Syndrome. J. Exp. Med. 2015, 212, 1641–1662.
- Krueger, J.G.; McInnes, I.B.; Blauvelt, A. Tyrosine Kinase 2 and Janus Kinase—signal Transducer and Activator of Transcription Signaling and Inhibition in Plaque Psoriasis. J. Am. Acad. Dermatol. 2022, 86, 148–157.
- Velazquez, L.; Fellous, M.; Stark, G.R.; Pellegrini, S. A protein tyrosine kinase in the interferon αβ signaling pathway. Cell 1992, 70, 313–322.
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862.
- Kvist-Hansen, A.; Hansen, P.R.; Skov, L. Systemic Treatment of Psoriasis with JAK Inhibitors: A Review. Dermatol. Ther. 2019, 10, 29–42.
- Nogueira, M.; Torres, T. Janus Kinase Inhibitors for the Treatment of Atopic Dermatitis: Focus on Abrocitinib, Baricitinib, and Upadacitinib. Dermatol. Pract. Concept. 2021, 11, e2021145.
- Ghoreschi, K.; Augustin, M.; Baraliakos, X.; Krönke, G.; Schneider, M.; Schreiber, S.; Schulze-Koops, H.; Zeißig, S.; Thaçi, D. TYK2 inhibition and its potential in the treatment of chronic inflammatory immune diseases. JDDG J. Dtsch. Dermatol. Ges. 2021, 19, 1409–1420.
- Winthrop, K.L. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat. Rev. Rheumatol. 2017, 13, 234–243.
- Shawky, A.M.; Almalki, F.A.; Abdalla, A.N.; Abdelazeem, A.H.; Gouda, A.M. A Comprehensive Overview of Globally Approved JAK Inhibitors. Pharmaceutics 2022, 14, 1001.
- Bachelez, H.; van de Kerkhof, P.C.M.; Strohal, R.; Kubanov, A.; Valenzuela, F.; Lee, J.-H.; Yakusevich, V.; Chimenti, S.; Papacharalambous, J.; Proulx, J.; et al. Tofacitinib versus Etanercept or Placebo in Moderate-to-Severe Chronic Plaque Psoriasis: A Phase 3 Randomised Non-Inferiority Trial. Lancet 2015, 386, 552–561.
- Krueger, J.; Clark, J.D.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Cueto, I.; Wang, C.Q.; Tan, H.; Wolk, R.; Rottinghaus, S.T.; Whitley, M.Z.; et al. Tofacitinib Attenuates Pathologic Immune Pathways in Patients with Psoriasis: A Randomized Phase 2 Study. J. Allergy Clin. Immunol. 2016, 137, 1079–1090.
- McInnes, I.B.; Kato, K.; Magrey, M.; Merola, J.F.; Kishimoto, M.; Haaland, D.; Chen, L.; Duan, Y.; Liu, J.; Lippe, R.; et al. Efficacy and Safety of Upadacitinib in Patients with Psoriatic Arthritis: 2-Year Results from the Phase 3 SELECT-PsA 1 Study. Rheumatol. Ther. 2022.
- Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2 A Genome-Wide Association Study Identifies New Psoriasis Susceptibility Loci and an Interaction between HLA-C and ERAP1. Nat. Genet. 2010, 42, 985–990.
- Works, M.G.; Yin, F.; Yin, C.C.; Yiu, Y.; Shew, K.; Tran, T.-T.; Dunlap, N.; Lam, J.; Mitchell, T.; Reader, J.; et al. Inhibition of TYK2 and JAK1 Ameliorates Imiquimod-Induced Psoriasis-like Dermatitis by Inhibiting IL-22 and the IL-23/IL-17 Axis. J. Immunol. 2014, 193, 3278–3287.
- Strobl, B. Tyrosine Kinase 2 (TYK2) in Cytokine Signalling and Host Immunity. Front Biosci 2011, 16, 3224.
- Minegishi, Y.; Saito, M.; Morio, T.; Watanabe, K.; Agematsu, K.; Tsuchiya, S.; Takada, H.; Hara, T.; Kawamura, N.; Ariga, T.; et al. Human Tyrosine Kinase 2 Deficiency Reveals Its Requisite Roles in Multiple Cytokine Signals Involved in Innate and Acquired Immunity. Immunity 2006, 25, 745–755.
- Dendrou, C.A.; Cortes, A.; Shipman, L.; Evans, H.G.; Attfield, K.E.; Jostins, L.; Barber, T.; Kaur, G.; Kuttikkatte, S.B.; Leach, O.A.; et al. Resolving TYK2 locus genotype-to-phenotype differences in autoimmunity. Sci. Transl. Med. 2016, 8, 363ra149.
- Seto, Y.; Nakajima, H.; Suto, A.; Shimoda, K.; Saito, Y.; Nakayama, K.I.; Iwamoto, I. Enhanced Th2 Cell-Mediated Allergic Inflammation in Tyk2-Deficient Mice. J. Immunol. 2003, 170, 1077–1083.
- Spach, K.M.; Noubade, R.; McElvany, B.; Hickey, W.F.; Blankenhorn, E.P.; Teuscher, C. A Single Nucleotide Polymorphism in Tyk2 Controls Susceptibility to Experimental Allergic Encephalomyelitis. J. Immunol. 2009, 182, 7776–7783.
- Xie, J.; LeBaron, M.J.; Nevalainen, M.T.; Rui, H. Role of Tyrosine Kinase Jak2 in Prolactin-Induced Differentiation and Growth of Mammary Epithelial Cells. J. Biol. Chem. 2002, 277, 14020–14030.
- Amat-Samaranch, V.; Agut-Busquet, E.; Vilarrasa, E.; Puig, L. New Perspectives on the Treatment of Hidradenitis Suppurativa. Ther. Adv. Chronic Dis. 2021, 12, 204062232110559.
- Markota Čagalj, A.; Marinović, B.; Bukvić Mokos, Z. New and Emerging Targeted Therapies for Hidradenitis Suppurativa. Int. J. Mol. Sci. 2022, 23, 3753.
- Ghoreschi, K.; Laurence, A.; O’Shea, J.J. Janus Kinases in Immune Cell Signaling. Immunol. Rev. 2009, 228, 273–287.
- Tokarski, J.S.; Zupa-Fernandez, A.; Tredup, J.A.; Pike, K.; Chang, C.; Xie, D.; Cheng, L.; Pedicord, D.; Muckelbauer, J.; Johnson, S.R.; et al. Tyrosine Kinase 2-Mediated Signal Transduction in T Lymphocytes Is Blocked by Pharmacological Stabilization of Its Pseudokinase Domain. J. Biol. Chem. 2015, 290, 11061–11074.
- Baker, K.F.; Isaacs, J.D. Novel therapies for immune-mediated inflammatory diseases: What can we learn from their use in rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease and ulcerative colitis? Ann. Rheum. Dis. 2017, 77, 175–187.
- Burke, J.R.; Cheng, L.; Gillooly, K.M.; Strnad, J.; Zupa-Fernandez, A.; Catlett, I.M.; Zhang, Y.; Heimrich, E.M.; McIntyre, K.W.; Cunningham, M.D.; et al. Autoimmune Pathways in Mice and Humans Are Blocked by Pharmacological Stabilization of the TYK2 Pseudokinase Domain. Sci. Transl. Med. 2019, 11, eaaw1736.
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT Signaling: From Interferons to Cytokines. J. Biol. Chem. 2007, 282, 20059–20063.
- Jiang, L.; Li, Z.; Rui, L. Leptin Stimulates Both JAK2-Dependent and JAK2-Independent Signaling Pathways. J. Biol. Chem. 2008, 283, 28066–28073.
- Ishizaki, M.; Muromoto, R.; Akimoto, T.; Sekine, Y.; Kon, S.; Diwan, M.; Maeda, H.; Togi, S.; Shimoda, K.; Oritani, K.; et al. Tyk2 Is a Therapeutic Target for Psoriasis-like Skin Inflammation. Int. Immunol. 2014, 26, 257–267.
- Sohn, S.J.; Barrett, K.; Van Abbema, A.; Chang, C.; Kohli, P.B.; Kanda, H.; Smith, J.; Lai, Y.; Zhou, A.; Zhang, B.; et al. A Restricted Role for TYK2 Catalytic Activity in Human Cytokine Responses Revealed by Novel TYK2-Selective Inhibitors. J. Immunol. 2013, 191, 2205–2216.
- Karaghiosoff, M.; Neubauer, H.; Lassnig, C.; Kovarik, P.; Schindler, H.; Pircher, H.; McCoy, B.; Bogdan, C.; Decker, T.; Brem, G.; et al. Partial Impairment of Cytokine Responses in Tyk2-Deficient Mice. Immunity 2000, 13, 549–560.
- Majoros, A.; Platanitis, E.; Kernbauer-Hölzl, E.; Rosebrock, F.; Müller, M.; Decker, T. Canonical and Non-Canonical Aspects of JAK–STAT Signaling: Lessons from Interferons for Cytokine Responses. Front. Immunol. 2017, 8, 29.
- Saxena, A.; Khosraviani, S.; Noel, S.; Mohan, D.; Donner, T.; Hamad, A.R.A. Interleukin-10 Paradox: A Potent Immunoregulatory Cytokine That Has Been Difficult to Harness for Immunotherapy. Cytokine 2015, 74, 27–34.
- Ouyang, W.; O’Garra, A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 2019, 50, 871–891.
- Pérez-Jeldres, T.; Tyler, C.J.; Boyer, J.D.; Karuppuchamy, T.; Yarur, A.; Giles, D.A.; Yeasmin, S.; Lundborg, L.; Sandborn, W.J.; Patel, D.R.; et al. Targeting Cytokine Signaling and Lymphocyte Traffic via Small Molecules in Inflammatory Bowel Disease: JAK Inhibitors and S1PR Agonists. Front. Pharmacol. 2019, 10, 212.
- Danese, S.; Peyrin-Biroulet, L. Selective Tyrosine Kinase 2 Inhibition for Treatment of Inflammatory Bowel Disease: New Hope on the Rise. Inflamm. Bowel Dis. 2021, 27, 2023–2030.
- Furie, R.; Khamashta, M.; Merrill, J.T.; Werth, V.P.; Kalunian, K.; Brohawn, P.; Illei, G.G.; Drappa, J.; Wang, L.; Yoo, S.; et al. Anifrolumab, an Anti-Interferon-α Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus: Anifrolumab in moderate-to-severe SLE. Arthritis Rheumatol. 2017, 69, 376–386.
- Yao, Y.; Higgs, B.W.; Morehouse, C.; de los Reyes, M.; Trigona, W.; Brohawn, P.; White, W.; Zhang, J.; White, B.; Coyle, A.J.; et al. Development of Potential Pharmacodynamic and Diagnostic Markers for Anti-IFN-α Monoclonal Antibody Trials in Systemic Lupus Erythematosus. Hum. Genom. Proteom. 2009, 17, 374312.
- Tyk2 Selective Inhibitors. Dec 2022. Available online: https://www.Selleckchem.Com/Subunits/Tyk2_JAK_selpan.Html (accessed on 4 January 2023).
- Jo, C.E.; Gooderham, M.; Beecker, J. TYK 2 Inhibitors for the Treatment of Dermatologic Conditions: The Evolution of JAK Inhibitors. Int J Dermatol. 2022, 61, 139–147.
- Fensome, A.; Ambler, C.M.; Arnold, E.; Banker, M.E.; Brown, M.F.; Chrencik, J.; Clark, J.D.; Dowty, M.E.; Efremov, I.V.; Flick, A.; et al. Dual Inhibition of TYK2 and JAK1 for the Treatment of Autoimmune Diseases: Discovery of ((S)-2,2-Difluorocyclopropyl)((1R,5S)-3-(2-((1-methyl-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)-3,8-diazabicyclooctan-8-yl)methanone (PF-06700841). J. Med. Chem. 2018, 61, 8597–8612.
- Banfield, C.; Scaramozza, M.; Zhang, W.; Kieras, E.; Page, K.M.; Fensome, A.; Vincent, M.; Dowty, M.E.; Goteti, K.; Winkle, P.J.; et al. The Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of a TYK2/JAK1 Inhibitor (PF-06700841) in Healthy Subjects and Patients with Plaque Psoriasis. J. Clin. Pharmacol. 2018, 58, 434–447.
- Forman, S.B.; Pariser, D.M.; Poulin, Y.; Vincent, M.S.; Gilbert, S.A.; Kieras, E.M.; Qiu, R.; Yu, D.; Papacharalambous, J.; Tehlirian, C.; et al. TYK2/JAK1 Inhibitor PF-06700841 in Patients with Plaque Psoriasis: Phase IIa, Randomized, Double-Blind, Placebo-Controlled Trial. J. Investig. Dermatol. 2020, 140, 2359–2370.e5.
- Page, K.M.; Suarez-Farinas, M.; Suprun, M.; Zhang, W.; Garcet, S.; Fuentes-Duculan, J.; Li, X.; Scaramozza, M.; Kieras, E.; Banfield, C.; et al. Molecular and Cellular Responses to the TYK2/JAK1 Inhibitor PF-06700841 Reveal Reduction of Skin Inflammation in Plaque Psoriasis. J. Investig. Dermatol. 2020, 140, 1546–1555.e4.
- King, B.; Guttman-Yassky, E.; Peeva, E.; Banerjee, A.; Sinclair, R.; Pavel, A.B.; Zhu, L.; Cox, L.A.; Craiglow, B.; Chen, L.; et al. A Phase 2a Randomized, Placebo-Controlled Study to Evaluate the Efficacy and Safety of the Oral Janus Kinase Inhibitors Ritlecitinib and Brepocitinib in Alopecia Areata: 24-Week Results. J. Am. Acad. Dermatol. 2021, 85, 379–387.
- Guttman-Yassky, E.; Pavel, A.B.; Diaz, A.; Zhang, N.; Del Duca, E.; Estrada, Y.; King, B.; Banerjee, A.; Banfield, C.; Cox, L.A.; et al. Ritlecitinib and Brepocitinib Demonstrate Significant Improvement in Scalp Alopecia Areata Biomarkers. J. Allergy Clin. Immunol. 2022, 149, 1318–1328.
- Peeva, E.; Guttman-Yassky, E.; Banerjee, A.; Sinclair, R.; Cox, L.A.; Zhu, L.; Zhu, H.; Vincent, M.; King, B. Maintenance, Withdrawal, and Re-Treatment with Ritlecitinib and Brepocitinib in Patients with Alopecia Areata in a Single-Blind Extension of a Phase 2a Randomized Clinical Trial. J. Am. Acad. Dermatol. 2022, 87, 390–393.
- Landis, M.N.; Arya, M.; Smith, S.; Draelos, Z.; Usdan, L.; Tarabar, S.; Pradhan, V.; Aggarwal, S.; Banfield, C.; Peeva, E.; et al. Efficacy and Safety of Topical Brepocitinib for the Treatment of Mild-to-moderate Atopic Dermatitis: A Phase IIb, Randomized, Double-blind, Vehicle-controlled, Dose-ranging and Parallel-group Study*. Br. J. Derm. 2022, 187, 878–887.
- Pfizer Bows out of the Tyk2 Race. Available online: http://www.evaluate.com/vantage/articles/news/snippets/pfizer-bows-out-tyk2-race (accessed on 8 January 2023).
- Tehlirian, C.; Peeva, E.; Kieras, E.; Scaramozza, M.; Roberts, E.S.; Singh, R.S.P.; Pradhan, V.; Banerjee, A.; Garcet, S.; Xi, L.; et al. Safety, Tolerability, Efficacy, Pharmacokinetics, and Pharmacodynamics of the Oral TYK2 Inhibitor PF-06826647 in Participants with Plaque Psoriasis: A Phase 1, Randomised, Double-Blind, Placebo-Controlled, Parallel-Group Study. Lancet Rheumatol. 2021, 3, e204–e213.
- Gerstenberger, B.S.; Ambler, C.; Arnold, E.P.; Banker, M.-E.; Brown, M.F.; Clark, J.D.; Dermenci, A.; Dowty, M.E.; Fensome, A.; Fish, S.; et al. Discovery of Tyrosine Kinase 2 (TYK2) Inhibitor (PF-06826647) for the Treatment of Autoimmune Diseases. J. Med. Chem. 2020, 63, 13561–13577.
- Singh, R.S.P.; Pradhan, V.; Roberts, E.S.; Scaramozza, M.; Kieras, E.; Gale, J.D.; Peeva, E.; Vincent, M.S.; Banerjee, A.; Fensome, A.; et al. Safety and Pharmacokinetics of the Oral TYK2 Inhibitor PF-06826647: A Phase I, Randomized, Double-Blind, Placebo-Controlled, Dose-Escalation Study. Clin Transl Sci 2021, 14, 671–682.
- Tehlirian, C.; Singh, R.S.P.; Pradhan, V.; Roberts, E.S.; Tarabar, S.; Peeva, E.; Vincent, M.S.; Gale, J.D. Oral Tyrosine Kinase 2 Inhibitor PF-06826647 Demonstrates Efficacy and an Acceptable Safety Profile in Participants with Moderate-to-Severe Plaque Psoriasis in a Phase 2b, Randomized, Double-Blind, Placebo-Controlled Study. J. Am. Acad. Dermatol. 2022, 87, 333–342.
- Taylor, N.P. Pfizer Sells Midphase Inflammatory Drugs to Mystery Startup, Exiting Race against Bristol Myers. Available online: https://www.fiercebiotech.com/biotech/pfizer-sells-midphase-inflammatory-drugs-to-mystery-startup-exiting-race-against-bristol (accessed on 8 January 2023).
- Moslin, R.; Zhang, Y.; Wrobleski, S.T.; Lin, S.; Mertzman, M.; Spergel, S.; Tokarski, J.S.; Strnad, J.; Gillooly, K.; McIntyre, K.W.; et al. Identification of N -Methyl Nicotinamide and N -Methyl Pyridazine-3-Carboxamide Pseudokinase Domain Ligands as Highly Selective Allosteric Inhibitors of Tyrosine Kinase 2 (TYK2). J. Med. Chem. 2019, 62, 8953–8972.
- Wrobleski, S.T.; Moslin, R.; Lin, S.; Zhang, Y.; Spergel, S.; Kempson, J.; Tokarski, J.S.; Strnad, J.; Zupa-Fernandez, A.; Cheng, L.; et al. Highly Selective Inhibition of Tyrosine Kinase 2 (TYK2) for the Treatment of Autoimmune Diseases: Discovery of the Allosteric Inhibitor BMS-986165. J. Med. Chem. 2019, 62, 8973–8995.
- Chimalakonda, A.; Burke, J.; Cheng, L.; Catlett, I.; Tagen, M.; Zhao, Q.; Patel, A.; Shen, J.; Girgis, I.G.; Banerjee, S.; et al. Selectivity Profile of the Tyrosine Kinase 2 Inhibitor Deucravacitinib Compared with Janus Kinase 1/2/3 Inhibitors. Derm. (Heidelb) 2021, 11, 1763–1776.
- Papp, K.; Gordon, K.; Thaçi, D.; Morita, A.; Gooderham, M.; Foley, P.; Girgis, I.G.; Kundu, S.; Banerjee, S. Phase 2 Trial of Selective Tyrosine Kinase 2 Inhibition in Psoriasis. N. Engl. J. Med. 2018, 379, 1313–1321.
- Catlett, I.M.; Hu, Y.; Gao, L.; Banerjee, S.; Gordon, K.; Krueger, J.G. Molecular and Clinical Effects of Selective Tyrosine Kinase 2 Inhibition with Deucravacitinib in Psoriasis. J. Allergy Clin. Immunol. 2022, 149, 2010–2020.e8.
- Gordon, K.; Papp, K.; Gooderham, M.; Morita, A.; Foley, P.; Thaçi, D.; Kundu, S.; Kisa, R.; Wei, L.; Banergee, S. BMS-986165, an Oral, Selective Tyrosine Kinase 2 (TYK2) Inhibitor: Evaluation of Changes in Laboratory Parameters in Response to Treatment in a Phase 2 Trial in Psoriasis Patients. Ski. J. Cutan. Med. 2020, 4, s28.
- Thaçi, D.; Strober, B.; Gordon, K.B.; Foley, P.; Gooderham, M.; Morita, A.; Papp, K.A.; Puig, L.; Menter, M.A.; Colombo, M.J.; et al. Deucravacitinib in Moderate to Severe Psoriasis: Clinical and Quality-of-Life Outcomes in a Phase 2 Trial. Dermatol. Ther. 2022, 12, 495–510.
- Blankenship, K. Celgene, Say Goodbye to Otezla: BMS Agrees to Sell Psoriasis Drug to Clear $74B Merger. Available online: https://www.fiercepharma.com/pharma/say-goodbye-to-otezla-bms-agress-to-sell-psoriasis-med-as-part-celgene-merger-deal (accessed on 8 January 2023).
- Armstrong, A.W.; Gooderham, M.; Warren, R.B.; Papp, K.A.; Strober, B.; Thaçi, D.; Morita, A.; Szepietowski, J.C.; Imafuku, S.; Colston, E.; et al. Deucravacitinib versus Placebo and Apremilast in Moderate to Severe Plaque Psoriasis: Efficacy and Safety Results from the 52-Week, Randomized, Double-Blinded, Placebo-Controlled Phase 3 POETYK PSO-1 Trial. J. Am. Acad. Dermatol. 2023, 88, 29–39.
- Strober, B.; Thaçi, D.; Sofen, H.; Kircik, L.; Gordon, K.B.; Foley, P.; Rich, P.; Paul, C.; Bagel, J.; Colston, E.; et al. Deucravacitinib versus Placebo and Apremilast in Moderate to Severe Plaque Psoriasis: Efficacy and Safety Results from the 52-Week, Randomized, Double-Blinded, Phase 3 Program fOr Evaluation of TYK2 Inhibitor Psoriasis Second Trial. J. Am. Acad. Dermatol. 2023, 88, 40–51.
- Hoy, S.M. Deucravacitinib: First Approval. Drugs 2022, 82, 1671–1679.
- Mease, P.J.; Deodhar, A.A.; van der Heijde, D.; Behrens, F.; Kivitz, A.J.; Neal, J.; Kim, J.; Singhal, S.; Nowak, M.; Banerjee, S. Efficacy and Safety of Selective TYK2 Inhibitor, Deucravacitinib, in a Phase II Trial in Psoriatic Arthritis. Ann. Rheum. Dis. 2022, 81, 815–822.
- Danese, S.; Panaccione, R.; D’Haens, G.; Peyrin-Biroulet, L.; Schreiber, S.; Kobayashi, T.; Gawdis-Wojnarska, B.; Korga, P.; Aguilar, H.; Sharkey, B.; et al. DOP42 Efficacy and Safety of Deucravacitinib, an Oral, Selective Tyrosine Kinase 2 Inhibitor, in Patients with Moderately-to-Severely Active Ulcerative Colitis: 12-Week Results from the Phase 2 LATTICE-UC Study. J. Crohn’s Colitis 2022, 16, i091–i092.
- Liu, C.; Lin, J.; Langevine, C.; Smith, D.; Li, J.; Tokarski, J.S.; Khan, J.; Ruzanov, M.; Strnad, J.; Zupa-Fernandez, A.; et al. Discovery of BMS-986202: A Clinical Tyk2 Inhibitor That Binds to Tyk2 JH2. J. Med. Chem. 2021, 64, 677–694.
- Ma, H.-L.; Liang, S.; Li, J.; Napierata, L.; Brown, T.; Benoit, S.; Senices, M.; Gill, D.; Dunussi-Joannopoulos, K.; Collins, M.; et al. IL-22 is required for Th17 cell–mediated pathology in a mouse model of psoriasis-like skin inflammation. J. Clin. Investig. 2008, 118, 597–607.
- Van Belle, A.B.; de Heusch, M.; Lemaire, M.M.; Hendrickx, E.; Warnier, G.; Dunussi-Joannopoulos, K.; Fouser, L.A.; Renauld, J.-C.; Dumoutier, L. IL-22 Is Required for Imiquimod-Induced Psoriasiform Skin Inflammation in Mice. J. Immunol. 2012, 188, 462–469.
More
Information
Subjects:
Dermatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
3.7K
Revisions:
3 times
(View History)
Update Date:
20 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No