 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Eliška Čermáková | -- | 6230 | 2023-02-16 10:27:27 | | | |
| 2 | Lindsay Dong | Meta information modification | 6230 | 2023-02-17 03:12:20 | | | | |
| 3 | Lindsay Dong | -1 word(s) | 6229 | 2023-02-20 08:43:06 | | |
Video Upload Options
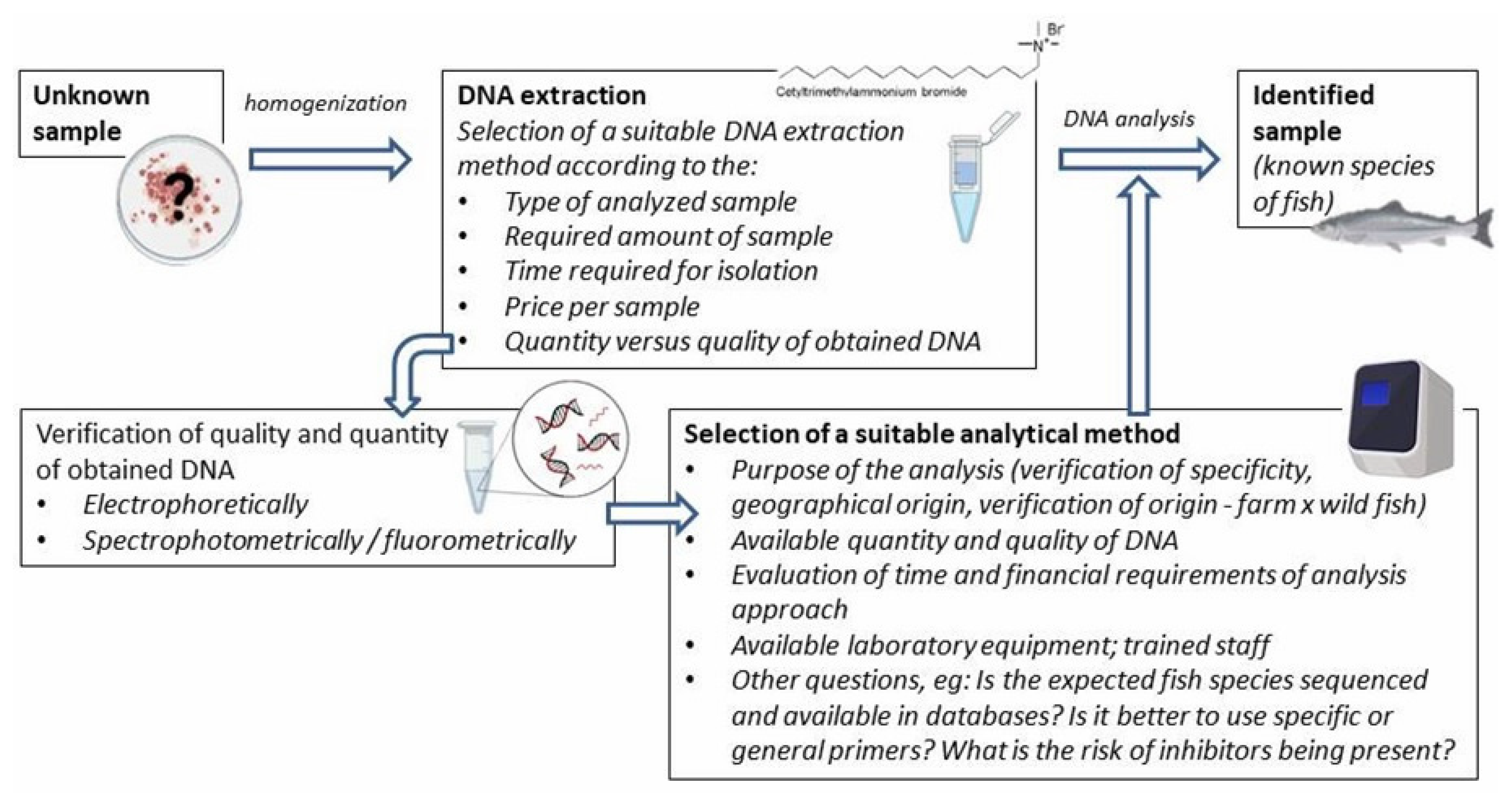
Various methods, differing in their principle and detected target molecules, can be used for fish species identification and adulteration detection. Parvalbumin detection has been preferred, but this is quite demanding compared to other food allergens because of its high biochemical and immunological variability among fish species and differing thermostability. This in itself sidelines the use of some types of methods, e.g., proteomic ones, and favors DNA-based methods, whose target, DNA, is not destroyed by the processing. Using DNA analysis, for example, a common form of adulteration—replacing the species (a more expensive one with a cheaper one)—can be detected. Additionally, DNA-based methods have the potential to detect the consumer being misled about the fish’s origin, another common type of adulteration. This is even more relevant, since genetically modified (GM) fish can be bought on the world market. Even though GM organisms do not pose a threat to public health and their quality is no different, consumers should be informed about the content of GM products, and because the public can be skeptical about GM products, sellers may want to hide this information and deliberately deceive the consumer.
1. Introduction
2. Identification of Fish Species
3. DNA-Based Methods
3.1. Nucleic Acid Databases
3.2. Mitochondrial and Nuclear Identification Markers
3.3. DNA-Based Methods
3.3.1. Polymerase Chain Reaction (PCR)
3.3.2. Sequencing Methods
3.3.3. DNA Hybridization
3.3.4. Methods Using Restriction Enzyme Cleavage
Restriction Fragment Length Polymorphism (RFLP)
Amplified Fragment Length Polymorphism (AFLP)
AFLP is a highly sensitive PCR-based method suitable for population studies and for finding genetic variation between closely related species. It is based on four steps: (i) restriction (specific cleavage of total DNA by two restriction endonucleases, most often MseI and EcoRI); (ii) ligation (adapters are attached to all fragments by T4 ligase); (iii) amplification (pre-selective and selective amplification to reduce the number of fragments); and (iv) fragment visualization. AFLP requires initial screening to find the optimal primer combination; specific primers are complementary to the adapters and have an overhang of one to three bases within the studied fragment. Obtained fragments can be separated by electrophoresis on a polyacrylamide gel and visualized autoradiographically. The final visualization can also be performed in an automatic sequencer if fluorescently labeled EcoRI primers are used.
3.3.5. Polymerase Chain Reaction–Single-Strand Conformation Polymorphism (PCR-SSCP)
3.3.6. Random Amplification of Polymorphic DNA (RAPD)
3.3.7. Loop-Mediated Isothermal Amplification (LAMP)
3.3.8. Multi-Analyte Profiling (xMAP)
3.4. Data Analysis
3.4.1. DNA Barcoding
3.4.2. Forensically Informative Nucleotide Sequencing (FINS)
3.5. Advantages and Limitations
The methods using restriction cleavage (RFLP, AFLP) benefit from their simplicity, reliability and robustness. However, the high risk of incorrect identification needs to be taken into account due to the insufficient study of related species, which may form the same restriction profile or polymorphism in the analyzed DNA, leading to a change in the restriction site and thus incorrect evaluation of the analysis. As a result, some authors tend to recommend the use of sequencing and phylogenetic analysis techniques [60][119], such as DNA barcoding or FINS. These methods provide a high power of diagnosis, which minimizes the risk of misidentification of the species. When analyzing a species that has not been studied yet, it will be included in the appropriate node in the phylogenetic tree based on its nucleotide sequence. The phylogenetic tree also contains reference sequences of several species; the assignment to a node corresponding to a certain species thus allows the identification of blank samples [117]. However, sequencing-based methods run into limitations in the analysis of processed and/or pooled samples. In this case, species-specific PCR or LAMP seems appropriate. The advantages of the LAMP method compared to PCR are the lower demand for lab equipment and the possibility of shortening the amplification time by up to a third [120], but PCR is a more robust method. PCR and FINS also have high potential for building nucleotide databases. However, the above-mentioned methods (PCR, LAMP, FINS, RFLP) require prior knowledge of the DNA sequence of the analyzed species for the analysis itself, unlike RAPD and AFLP, for example. Next, RAPD and AFLP can be used for the analysis of multiple loci at once. AFLP markers also exhibit much higher variability than, for example, isozymes. However, neither method is very robust against DNA degradation, and they both have low potential for interlaboratory reproducibility. Compared to AFLP and RFLP, RAPD is considered the most reliable method for species identification when the genome sequence is unknown [63][121]. However, for both RAPD and RFLP, intraspecific variation can be problematic, in contrast to the SSCP method, for example. On the other hand, SSCP is more demanding and always requires the presence of a reference sample on the gel together with the analyzed samples. Despite the high sensitivity of SSCP or PCR-SSCP and its capability of intraspecific differentiation, the amount of information obtained from these methods is much lower than with sequencing [30][122][123][124].
3.6. Evidence of Adulteration in Fish by DNA Analysis
3.7. Detection of Genetic Modifications in Fish
4. Conclusions
References
- Goyal, K.; Kumar, P.; Verma, K. Food adulteration detection using artificial intelligence: A systematic review. Arch. Comput. Methods Eng. 2021, 29, 397–426.
- Kotsanopoulos, K.V.; Exadactylos, A.; Gkafas, G.A.; Martsikalis, P.V.; Parlapani, F.F.; Boziaris, I.S.; Arvanitoyannis, I.S. The use of molecular markers in the verification of fish and seafood authenticity and the detection of adulteration. Compr. Rev. Food Sci. Food Saf. 2021, 20, 1584–1654.
- Rasmussen, R.S.; Morrissey, M.T. DNA-based methods for the identification of commercial fish and seafood species. Compr. Rev. Food Sci. Food Saf. 2008, 7, 280–295.
- Civera, T. Species identification and safety of fish products. Vet. Res. Commun. 2003, 27, 481.
- Pauly, D.; Christensen, V.; Dalsgaard, J.; Froese, R.; Torres, F., Jr. Fishing down marine food webs. Science 1998, 279, 860–863.
- Food and Agriculture Organization of the United Nations. Report of the Fourteenth Session of the Sub-Committee on Fish Trade; COFI:FT/XIV/2014/5; FAO: Rome, Italy, 2014; 86p.
- Buyuktiryaki, B.; Masini, M.; Mori, F.; Barni, S.; Liccioli, G.; Sarti, L.; Lodi, L.; Giovannini, M.; du Toit, G.; Lopata, A.L. IgE-mediated fish allergy in children. Medicina 2021, 57, 76.
- Moonesinghe, H.; Mackenzie, H.; Venter, C.; Kilburn, S.; Turner, P.; Weir, K.; Dean, T. Prevalence of fish and shellfish allergy: A systematic review. Ann. Allergy Asthma Immunol. 2016, 117, 264–272.e264.
- Kuehn, A.; Swoboda, I.; Arumugam, K.; Hilger, C.; Hentges, F. Fish allergens at a glance: Variable allergenicity of parvalbumins, the major fish allergens. Front. Immunol. 2014, 5, 179.
- Griesmeier, U.; Vázquez-Cortés, S.; Bublin, M.; Radauer, C.; Ma, Y.; Briza, P.; Fernández-Rivas, M.; Breiteneder, H. Expression levels of parvalbumins determine allergenicity of fish species. Allergy 2010, 65, 191–198.
- Lee, P.-W.; Nordlee, J.A.; Koppelman, S.J.; Baumert, J.L.; Taylor, S.L. Measuring parvalbumin levels in fish muscle tissue: Relevance of muscle locations and storage conditions. Food Chem. 2012, 135, 502–507.
- Taylor, S.; Kabourek, J.; Hefle, S. Fish allergy: Fish and products thereof. J. Food Sci. 2004, 69, R175–R180.
- Sharp, M.F.; Lopata, A.L. Fish allergy: In review. Clin. Rev. Allergy Immunol. 2014, 46, 258–271.
- Mukherjee, S.; Bartoš, O.; Zdeňková, K.; Hanák, P.; Horká, P.; Musilova, Z. Evolution of the Parvalbumin Genes in Teleost Fishes after the Whole-Genome Duplication. Fishes 2021, 6, 70.
- Saptarshi, S.R.; Sharp, M.F.; Kamath, S.D.; Lopata, A.L. Antibody reactivity to the major fish allergen parvalbumin is determined by isoforms and impact of thermal processing. Food Chem. 2014, 148, 321–328.
- Sletten, G.; Van Do, T.; Lindvik, H.; Egaas, E.; Florvaag, E. Effects of industrial processing on the immunogenicity of commonly ingested fish species. Int. Arch. Allergy Immunol. 2010, 151, 223–236.
- Aiello, D.; Materazzi, S.; Risoluti, R.; Thangavel, H.; Di Donna, L.; Mazzotti, F.; Casadonte, F.; Siciliano, C.; Sindona, G.; Napoli, A. A major allergen in rainbow trout (Oncorhynchus mykiss): Complete sequences of parvalbumin by MALDI tandem mass spectrometry. Mol. Biosyst. 2015, 11, 2373–2382.
- Hubalkova, Z.; Kralik, P.; Tremlova, B.; Rencova, E. Methods of gadoid fish species identification in food and their economic impact in the Czech Republic: A review. Vet. Med. 2007, 52, 273.
- Leduc, F.; Krzewinski, F.; Le Fur, B.; N’Guessan, A.; Malle, P.; Kol, O.; Duflos, G. Differentiation of fresh and frozen/thawed fish, European sea bass (Dicentrarchus labrax), gilthead seabream (Sparus aurata), cod (Gadus morhua) and salmon (Salmo salar), using volatile compounds by SPME/GC/MS. J. Sci. Food Agric. 2012, 92, 2560–2568.
- Edirisinghe, R.K.; Graffham, A.J.; Taylor, S.J. Characterisation of the volatiles of yellowfin tuna (Thunnus albacares) during storage by solid phase microextraction and GC–MS and their relationship to fish quality parameters. Int. J. Food Sci. Technol. 2007, 42, 1139–1147.
- Carrera, M.; Cañas, B.; Gallardo, J.M. Rapid direct detection of the major fish allergen, parvalbumin, by selected MS/MS ion monitoring mass spectrometry. J. Proteom. 2012, 75, 3211–3220.
- Carrera, M.; Canas, B.; López-Ferrer, D.; Pineiro, C.; Vázquez, J.; Gallardo, J.M. Fast monitoring of species-specific peptide biomarkers using high-intensity-focused-ultrasound-assisted tryptic digestion and selected MS/MS ion monitoring. Anal. Chem. 2011, 83, 5688–5695.
- Alamprese, C.; Casiraghi, E. Application of FT-NIR and FT-IR spectroscopy to fish fillet authentication. LWT-Food Sci. Technol. 2015, 63, 720–725.
- Aursand, M.; Standal, I.B.; Praël, A.; McEvoy, L.; Irvine, J.; Axelson, D.E. 13C NMR pattern recognition techniques for the classification of Atlantic salmon (Salmo salar L.) according to their wild, farmed, and geographical origin. J. Agric. Food Chem. 2009, 57, 3444–3451.
- Standal, I.B.; Axelson, D.E.; Aursand, M. 13C NMR as a tool for authentication of different gadoid fish species with emphasis on phospholipid profiles. Food Chem. 2010, 121, 608–615.
- Asensio, L.; González, I.; García, T.; Martín, R. Determination of food authenticity by enzyme-linked immunosorbent assay (ELISA). Food Control 2008, 19, 1–8.
- Martinez, I.; Jakobsen Friis, T. Application of proteome analysis to seafood authentication. Proteomics 2004, 4, 347–354.
- Hoffman, J. Tracing the origins, migrations, and other movements of fishes using stable isotopes. In An Introduction to Fish Migration; CRC Press: Boca Raton, FL, USA, 2016; pp. 169–196.
- Molkentin, J.; Lehmann, I.; Ostermeyer, U.; Rehbein, H. Traceability of organic fish—Authenticating the production origin of salmonids by chemical and isotopic analyses. Food Control 2015, 53, 55–66.
- Akasaki, T.; Yanagimoto, T.; Yamakami, K.; Tomonaga, H.; Sato, S. Species identification and PCR-RFLP analysis of cytochrome b gene in cod fish (order Gadiformes) products. J. Food Sci. 2006, 71, C190–C195.
- Moretti, V.; Turchini, G.; Bellagamba, F.; Caprino, F. Traceability issues in fishery and aquaculture products. Vet. Res. Commun. 2003, 27, 497–505.
- Mazzeo, M.F.; Siciliano, R.A. Proteomics for the authentication of fish species. J. Proteom. 2016, 147, 119–124.
- Hubalkova, Z.; Kralik, P.; Kasalova, J.; Rencova, E. Identification of gadoid species in fish meat by polymerase chain reaction (PCR) on genomic DNA. J. Agric. Food Chem. 2008, 56, 3454–3459.
- Hanák, P.; Laknerová, I.; Švátora, M. Second intron in the protein-coding region of the fish parvalbumin gene—A promising platform for polymerase chain reaction-based discrimination of fish meat of various species. J. Food Nutr. Res. 2012, 51, 81–88.
- Griffiths, A.M.; Sotelo, C.G.; Mendes, R.; Pérez-Martín, R.I.; Schröder, U.; Shorten, M.; Silva, H.A.; Verrez-Bagnis, V.; Mariani, S. Current methods for seafood authenticity testing in Europe: Is there a need for harmonisation? Food Control 2014, 45, 95–100.
- Gil, L.A. PCR-based methods for fish and fishery products authentication. Trends Food Sci. Technol. 2007, 18, 558–566.
- Clark, L.F. The current status of DNA barcoding technology for species identification in fish value chains. Food Policy 2015, 54, 85–94.
- Fernandes, T.J.; Costa, J.; Carrapatoso, I.; Oliveira, M.B.P.; Mafra, I. Advances on the molecular characterization, clinical relevance, and detection methods of Gadiform parvalbumin allergens. Crit. Rev. Food Sci. Nutr. 2017, 57, 3281–3296.
- Tomás, C.; Ferreira, I.; Faria, M. Codfish authentication by a fast short amplicon high resolution melting analysis (SA-HRMA) method. Food Control 2017, 71, 255–263.
- Cai, X.; Xu, M.; Wang, Y.; Qiu, D.; Liu, G.; Lin, A.; Tang, J.; Zhang, R.; Zhu, X. Sensitive and rapid detection of Clonorchis sinensis infection in fish by loop-mediated isothermal amplification (LAMP). Parasitol. Res. 2010, 106, 1379–1383.
- Caipang, C.M.A.; Haraguchi, I.; Ohira, T.; Hirono, I.; Aoki, T. Rapid detection of a fish iridovirus using loop-mediated isothermal amplification (LAMP). J. Virol. Methods 2004, 121, 155–161.
- Lin, T.C.; Hsiao, W.V.; Han, S.J.; Joung, S.J.; Shiao, J.C. A direct multiplex loop-mediated isothermal amplification method to detect three CITES-listed shark species. Aquat. Conserv. Mar. Freshw. Ecosyst. 2021, 31, 2193–2203.
- But, G.W.-C.; Wu, H.-Y.; Shao, K.-T.; Shaw, P.-C. Rapid detection of CITES-listed shark fin species by loop-mediated isothermal amplification assay with potential for field use. Sci. Rep. 2020, 10, 4455.
- Wang, Y.; Feng, J.; Tian, X. Application of loop-mediated isothermal amplification (LAMP) for rapid detection of Atlantic cod (Gadus morhua), Pacific cod (Gadus macrocephalus) and haddock (Melanogrammus aeglefinus). Mol. Cell. Probes 2019, 47, 101420.
- Li, Q.; Cheng, Y.; Xu, W.; Cui, X.; Cao, M.; Xiong, X.; Wang, L.; Xiong, X. Rapid identification of Atlantic salmon (Salmo salar) based on loop-mediated isothermal amplification (LAMP) using self-quenching fluorogenic approach. J. Food Compos. Anal. 2022, 105, 104224.
- Hanner, R.; Desalle, R.; Ward, R.D.; Kolokotronis, S.-O. The fish Barcode of Life (FISH-BOL) special issue. Mitochondrial DNA 2011, 22, 1–2.
- Böhme, K.; Calo-Mata, P.; Barros-Velázquez, J.; Ortea, I. Review of Recent DNA-Based Methods for Main Food-Authentication Topics. J. Agric. Food Chem. 2019, 67, 3854–3864.
- Aparicio, S.; Chapman, J.; Stupka, E.; Putnam, N.; Chia, J.-m.; Dehal, P.; Christoffels, A.; Rash, S.; Hoon, S.; Smit, A. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 2002, 297, 1301–1310.
- Randhawa, S.S.; Pawar, R. Fish genomes: Sequencing trends, taxonomy and influence of taxonomy on genome attributes. J. Appl. Ichthyol. 2021, 37, 553–562.
- Nelson, J.S.; Grande, T.C.; Wilson, M.V. Fishes of the World; John Wiley & Sons: Hoboken, NJ, USA, 2016.
- Rehbein, H. Differentiation of fish species by PCR-based DNA analysis of nuclear genes. Eur. Food Res. Technol. 2013, 236, 979–990.
- Paracchini, V.; Petrillo, M.; Lievens, A.; Kagkli, D.-M.; Angers-Loustau, A. Nuclear DNA barcodes for cod identification in mildly-treated and processed food products. Food Addit. Contam. Part A 2019, 36, 1–14.
- Cline, S.D. Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2012, 1819, 979–991.
- Pamilo, P.; Nei, M. Relationships between gene trees and species trees. Mol. Biol. Evol. 1988, 5, 568–583.
- Bermingham, E.; Forbes, S.H.; Friedland, K.; Pla, C. Discrimination between Atlantic salmon (Salmo salar) of North American and European origin using restriction analyses of mitochondrial DNA. Can. J. Fish. Aquat. Sci. 1991, 48, 884–893.
- Lencová, S.; Zdeňková, K.; Akhatova, D.; Demnerová, K. Aktuální trendy druhové identifikace rybího masa pomocí molekulárně-biologických metod. Chem. Listy 2019, 113, 292–300.
- Hebert, P.D.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B Biol. Sci. 2003, 270, 313–321.
- Ward, R.D.; Zemlak, T.S.; Innes, B.H.; Last, P.R.; Hebert, P.D. DNA barcoding Australia’s fish species. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 1847–1857.
- Hisar, O.; Erdogan, O.; Aksakal, E.; Hisar, S.A. Authentication of fish species using a simple PCR-RFLP method. Isr. J. Aquac.-Bamigdeh 2006, 58, 62–65.
- Calo-Mata, P.; Sotelo, C.G.; Pérez-Martín, R.I.; Rehbein, H.; Hold, G.L.; Russell, V.J.; Pryde, S.; Quinteiro, J.; Rey-Méndez, M.; Rosa, C. Identification of gadoid fish species using DNA-based techniques. Eur. Food Res. Technol. 2003, 217, 259–264.
- Sevilla, R.G.; Diez, A.; Norén, M.; Mouchel, O.; Jérôme, M.; Verrez-Bagnis, V.; Van Pelt, H.; Favre-Krey, L.; Krey, G.; The Fishtrace Consortium. Primers and polymerase chain reaction conditions for DNA barcoding teleost fish based on the mitochondrial cytochrome b and nuclear rhodopsin genes. Mol. Ecol. Notes 2007, 7, 730–734.
- Teletchea, F. Molecular identification methods of fish species: Reassessment and possible applications. Rev. Fish Biol. Fish. 2009, 19, 265–293.
- Bossier, P. Authentication of Seafood Products by DNA Patterns. J. Food Sci. 1999, 64, 189–193.
- Ballin, N.Z.; Vogensen, F.K.; Karlsson, A.H. Species determination—Can we detect and quantify meat adulteration? Meat Sci. 2009, 83, 165–174.
- Mariani, S.; Bekkevold, D. The nuclear genome: Neutral and adaptive markers in fisheries science. In Stock Identification Methods; Elsevier: Amsterdam, The Netherlands, 2014; pp. 297–327.
- Akhatova, D.; Laknerova, I.; Zdenkova, K.; Ólafsdo Ttir, G.; Magnúsdó, S.; Piknova, Ľ.; Kyrova, V.; Lerch, Z.; Hanak, P. International interlaboratory study on TaqMan real-time polymerase chain reaction authentication of black seabream (Spondyliosoma cantharus). J. Food Nutr. Res. 2018, 57, 27–37.
- Lee, J.-S. The internally self-fertilizing hermaphroditic teleost Rivulus marmoratus (Cyprinodontiformes, Rivulidae) β-actin gene: Amplification and sequence analysis with conserved primers. Mar. Biotechnol. 2000, 2, 161–166.
- Sohn, Y.C.; Kobayashi, M.; Aida, K. Regulation of gonadotropin β subunit gene expression by testosterone and gonadotropin-releasing hormones in the goldfish, Carassius auratus. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2001, 129, 419–426.
- Wang, Y.; Ge, W. Gonadotropin regulation of follistatin expression in the cultured ovarian follicle cells of zebrafish, Danio rerio. Gen. Comp. Endocrinol. 2003, 134, 308–315.
- Prado, M.; Ortea, I.; Vial, S.; Rivas, J.; Calo-Mata, P.; Barros-Velázquez, J. Advanced DNA-and protein-based methods for the detection and investigation of food allergens. Crit. Rev. Food Sci. Nutr. 2016, 56, 2511–2542.
- Paracchini, V.; Petrillo, M.; Lievens, A.; Gallardo, A.P.; Martinsohn, J.T.; Hofherr, J.; Maquet, A.; Silva, A.P.B.; Kagkli, D.M.; Querci, M. Novel nuclear barcode regions for the identification of flatfish species. Food Control 2017, 79, 297–308.
- Edwards, A.; Civitello, A.; Hammond, H.A.; Caskey, C.T. DNA typing and genetic mapping with trimeric and tetrameric tandem repeats. Am. J. Hum. Genet. 1991, 49, 746.
- Tautz, D. Hypervariability of simple sequences as a general source for polymorphic DNA markers. Nucleic Acids Res. 1989, 17, 6463–6471.
- Tóth, G.; Gáspári, Z.; Jurka, J. Microsatellites in different eukaryotic genomes: Survey and analysis. Genome Res. 2000, 10, 967–981.
- Litt, M.; Luty, J.A. A hypervariable microsatellite revealed by in vitro amplification of a dinucleotide repeat within the cardiac muscle actin gene. Am. J. Hum. Genet. 1989, 44, 397.
- Lauerman, L.H. Advances in PCR technology. Anim. Health Res. Rev. 2004, 5, 247–248.
- Rahman, M.T.; Uddin, M.S.; Sultana, R.; Moue, A.; Setu, M. Polymerase chain reaction (PCR): A short review. Anwer Khan Mod. Med. Coll. J. 2013, 4, 30–36.
- Kubista, M.; Andrade, J.M.; Bengtsson, M.; Forootan, A.; Jonák, J.; Lind, K.; Sindelka, R.; Sjöback, R.; Sjögreen, B.; Strömbom, L. The real-time polymerase chain reaction. Mol. Asp. Med. 2006, 27, 95–125.
- Dingle, T.C.; Sedlak, R.H.; Cook, L.; Jerome, K.R. Tolerance of Droplet-Digital PCR vs Real-Time Quantitative PCR to Inhibitory Substances. Clin. Chem. 2013, 59, 1670–1672.
- Fernandes, T.J.; Costa, J.; Oliveira, M.B.P.; Mafra, I. COI barcode-HRM as a novel approach for the discrimination of hake species. Fish. Res. 2018, 197, 50–59.
- Fernandes, T.J.; Costa, J.; Oliveira, M.B.P.; Mafra, I. DNA barcoding coupled to HRM analysis as a new and simple tool for the authentication of Gadidae fish species. Food Chem. 2017, 230, 49–57.
- Silva, A.J.; Hellberg, R.S. DNA-based techniques for seafood species authentication. In Advances in Food and Nutrition Research; Elsevier: Amsterdam, The Netherlands, 2021; Volume 95, pp. 207–255.
- Fernandes, T.J.; Amaral, J.S.; Mafra, I. DNA barcode markers applied to seafood authentication: An updated review. Crit. Rev. Food Sci. Nutr. 2021, 61, 3904–3935.
- Druml, B.; Cichna-Markl, M. High resolution melting (HRM) analysis of DNA—Its role and potential in food analysis. Food Chem. 2014, 158, 245–254.
- Fernandes, T.J.; Silva, C.R.; Costa, J.; Oliveira, M.B.P.; Mafra, I. High resolution melting analysis of a COI mini-barcode as a new approach for Penaeidae shrimp species discrimination. Food Control 2017, 82, 8–17.
- Verrez-Bagnis, V.; Sotelo, C.G.; Mendes, R.; Silva, H.; Kappel, K.; Schröder, U. Methods for seafood authenticity testing in Europe. In Bioactive Molecules in Food; Springer: Berlin/Heidelberg, Germany, 2019; pp. 2063–2117.
- Whitford, W.; Hawkins, V.; Moodley, K.S.; Grant, M.J.; Lehnert, K.; Snell, R.G.; Jacobsen, J.C. Proof of concept for multiplex amplicon sequencing for mutation identification using the MinION nanopore sequencer. Sci. Rep. 2022, 12, 8572.
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59.
- Fakruddin, M.; Chowdhury, A. Pyrosequencing an alternative to traditional Sanger sequencing. Am. J. Biochem. Biotechnol. 2012, 8, 14–20.
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 251364.
- Illumina. Sequencing and Array-Based Solutions for Genetic Research. Available online: https://www.illumina.com (accessed on 21 May 2022).
- Canadian Agency for Drugs Technologies in Health. Next Generation DNA Sequencing: A Review of the Cost Effectiveness and Guidelines; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2014; 19p.
- Young, A.D.; Gillung, J.P. Phylogenomics—Principles, opportunities and pitfalls of big-data phylogenetics. Syst. Entomol. 2020, 45, 225–247.
- Xie, Z.; Yu, C.; Guo, L.; Li, M.; Yong, Z.; Liu, X.; Meng, Z.; Lin, H. Ion Torrent next-generation sequencing reveals the complete mitochondrial genome of black and reddish morphs of the Coral Trout Plectropomus leopardus. Mitochondrial DNA Part A 2016, 27, 609–612.
- Adelyna, M.N.; Jung, H.; Chand, V.; Mather, P.; Azizah, M.S. A genome survey sequence (GSS) analysis and microsatellite marker development for Indian mackerel, Rastrelliger kanagurta, using Ion Torrent technology. Meta Gene 2016, 10, 67–72.
- De Battisti, C.; Marciano, S.; Magnabosco, C.; Busato, S.; Arcangeli, G.; Cattoli, G. Pyrosequencing as a tool for rapid fish species identification and commercial fraud detection. J. Agric. Food Chem. 2014, 62, 198–205.
- Keller, I.; Taverna, A.; Seehausen, O. A pyrosequencing assay for the rapid discrimination of mitochondrial lineages in the Salmo trutta species complex. Mol. Ecol. Resour. 2011, 11, 196–199.
- Levsky, J.M.; Singer, R.H. Fluorescence in situ hybridization: Past, present and future. J. Cell Sci. 2003, 116, 2833–2838.
- Bayani, J.; Squire, J.A. Fluorescence in situ Hybridization (FISH). Curr. Protoc. Cell Biol. 2004, 23, 22.4.1–22.4.52.
- Zhang, J.; Huang, H.; Cai, Z.; Huang, L. Species identification in salted products of red snappers by semi-nested PCR-RFLP based on the mitochondrial 12S rRNA gene sequence. Food Control 2006, 17, 557–563.
- Pardo, M.A.; Pérez-Villareal, B. Identification of commercial canned tuna species by restriction site analysis of mitochondrial DNA products obtained by nested primer PCR. Food Chem. 2004, 86, 143–150.
- Hayashi, K. PCR-SSCP: A method for detection of mutations. Genet. Anal. Biomol. Eng. 1992, 9, 73–79.
- Earley, M.C.; Vogt, R.F., Jr.; Shapiro, H.M.; Mandy, F.F.; Kellar, K.L.; Bellisario, R.; Pass, K.A.; Marti, G.E.; Stewart, C.C.; Hannon, W.H. Report from a workshop on multianalyte microsphere assays. Cytom. (Clin. Cytom.) 2002, 50, 239–242.
- Bartlett, S.; Davidson, W. FINS (forensically informative nucleotide sequencing): A procedure for identifying the animal origin of biological specimens. Biotechniques 1992, 12, 408–411.
- Espiñeira, M.; Vieites, J.M.; Santaclara, F.J. Development of a genetic method for the identification of salmon, trout, and bream in seafood products by means of PCR–RFLP and FINS methodologies. Eur. Food Res. Technol. 2009, 229, 785–793.
- Jérôme, M.; Martinsohn, J.T.; Ortega, D.; Carreau, P.; Verrez-Bagnis, V.R.; Mouchel, O. Toward fish and seafood traceability: Anchovy species determination in fish products by molecular markers and support through a public domain database. J. Agric. Food Chem. 2008, 56, 3460–3469.
- Hebert, P.D.; Ratnasingham, S.; De Waard, J.R. Barcoding animal life: Cytochrome c oxidase subunit 1 divergences among closely related species. Proc. R. Soc. Lond. Ser. B Biol. Sci. 2003, 270, S96–S99.
- Mueller, S.; Handy, S.M.; Deeds, J.R.; George, G.O.; Broadhead, W.J.; Pugh, S.E.; Garrett, S.D. Development of a COX1 based PCR-RFLP method for fish species identification. Food Control 2015, 55, 39–42.
- Hellberg, R.S.; Kawalek, M.D.; Van, K.T.; Shen, Y.; Williams-Hill, D.M. Comparison of DNA extraction and PCR setup methods for use in high-throughput DNA barcoding of fish species. Food Anal. Methods 2014, 7, 1950–1959.
- Delrieu-Trottin, E.; Williams, J.T.; Pitassy, D.; Driskell, A.; Hubert, N.; Viviani, J.; Cribb, T.H.; Espiau, B.; Galzin, R.; Kulbicki, M. A DNA barcode reference library of French Polynesian shore fishes. Sci. Data 2019, 6, 114.
- Sarmiento-Camacho, S.; Valdez-Moreno, M. DNA barcode identification of commercial fish sold in Mexican markets. Genome 2018, 61, 457–466.
- Jin, L.; Yu, J.; Yuan, X.; Du, X. Fish classification using DNA barcode sequences through deep learning method. Symmetry 2021, 13, 1599.
- Ward, R.D.; Hanner, R.; Hebert, P.D. The campaign to DNA barcode all fishes, FISH-BOL. J. Fish Biol. 2009, 74, 329–356.
- Steinke, D.; Hanner, R. The FISH-BOL collaborators’ protocol. Mitochondrial DNA 2011, 22, 10–14.
- Ivanova, N.V.; Zemlak, T.S.; Hanner, R.H.; Hebert, P.D. Universal primer cocktails for fish DNA barcoding. Mol. Ecol. Notes 2007, 7, 544–548.
- Li, M.; Zhang, K.Y.-B.; But, P.P.-H.; Shaw, P.-C. Forensically informative nucleotide sequencing (FINS) for the authentication of Chinese medicinal materials. Chin. Med. 2011, 6, 42.
- Santaclara, F.J.; Espiñeira, M.; Vieites, J.M. Genetic identification of squids (families Ommastrephidae and Loliginidae) by PCR–RFLP and FINS methodologies. J. Agric. Food Chem. 2007, 55, 9913–9920.
- Hellberg, R.S.R.; Morrissey, M.T. Advances in DNA-based techniques for the detection of seafood species substitution on the commercial market. JALA J. Assoc. Lab. Autom. 2011, 16, 308–321.
- Sotelo, C.G.; Calo-Mata, P.; Chapela, M.J.; Pérez-Martín, R.I.; Rehbein, H.; Hold, G.L.; Russell, V.J.; Pryde, S.; Quinteiro, J.; Izquierdo, M. Identification of flatfish (Pleuronectiforme) species using DNA-based techniques. J. Agric. Food Chem. 2001, 49, 4562–4569.
- Notomi, T.; Mori, Y.; Tomita, N.; Kanda, H. Loop-mediated isothermal amplification (LAMP): Principle, features, and future prospects. J. Microbiol. 2015, 53, 1–5.
- Liu, Z.J.; Cordes, J. DNA marker technologies and their applications in aquaculture genetics. Aquaculture 2004, 238, 1–37.
- Rehbein, H.; Kress, G.; Schmidt, T. Application of PCR-SSCP to species identification of fishery products. J. Sci. Food Agric. 1997, 74, 35–41.
- Lockley, A.; Bardsley, R. DNA-based methods for food authentication. Trends Food Sci. Technol. 2000, 11, 67–77.
- Mackie, I.; Pryde, S.E.; Gonzales-Sotelo, C.; Medina, I.; Pérez-Martın, R.; Quinteiro, J.; Rey-Mendez, M.; Rehbein, H. Challenges in the identification of species of canned fish. Trends Food Sci. Technol. 1999, 10, 9–14.
- Čížková, H. Falšování Potravin; Verlag Dashöfer: Hamburg, Germany, 2019.
- Chiesa, L.M.; Pavlovic, R.; Nobile, M.; Di Cesare, F.; Malandra, R.; Pessina, D.; Panseri, S. Discrimination between Fresh and Frozen-Thawed Fish Involved in Food Safety and Fraud Protection. Foods 2020, 9, 1896.
- Monteiro, C.S.; Deconinck, D.; Eljasik, P.; Sobczak, M.; Derycke, S.; Panicz, R.; Kane, N.; Mazloomrezaei, M.; Devlin, R.; Faria, M.A. A fast HRMA tool to authenticate eight salmonid species in commercial food products. Food Chem. Toxicol. 2021, 156, 112440.
- Chen, C.-H.; Hsieh, C.-H.; Hwang, D.-F. Species identification of Cyprinidae fish in Taiwan by FINS and PCR–RFLP analysis. Food Control 2012, 28, 240–245.
- Wang, Y.; Hamid, N.; Jia, P.P.; Pei, D.S. A comprehensive review on genetically modified fish: Key techniques, applications and future prospects. Rev. Aquac. 2021, 13, 1635–1660.
- Maclean, N.; Talwar, S. Injection of cloned genes into rainbow trout eggs. J. Embryol. Exp. Morphol. 1984, 82, 187–200.
- Debode, F.; Marien, A.; Ledoux, Q.; Janssen, E.; Ancion, C.; Berben, G. Detection of ornamental transgenic fish by real-time PCR and fluorescence microscopy. Transgenic Res. 2020, 29, 283–294.
- Gong, Z.; Ju, B.; Wan, H. Green fluorescent protein (GFP) transgenic fish and their applications. Genetica 2001, 111, 213–225.
- Tonelli, F.M.; Lacerda, S.M.; Tonelli, F.C.; Costa, G.M.; de Franca, L.R.; Resende, R.R. Progress and biotechnological prospects in fish transgenesis. Biotechnol. Adv. 2017, 35, 832–844.
- Green, D.P. Genetically Engineered Salmon Approved for Food by US FDA. J. Aquat. Food Prod. Technol. 2016, 25, 145–146.
- Debode, F.; Janssen, E.; Marien, A.; Devlin, R.H.; Lieske, K.; Mankertz, J.; Berben, G. Detection of transgenic Atlantic and Coho salmon by real-time PCR. Food Anal. Methods 2018, 11, 2396–2406.
- Masri, S.; Rast, H.; Ripley, T.; James, D.; Green, M.; Jia, X.; Devlin, R.H. Detection of genetically modified coho salmon using polymerase chain reaction (PCR) amplification. J. Agric. Food Chem. 2002, 50, 3161–3164.
- Hafsa, A.B.; Nabi, N.; Zellama, M.S.; Said, K.; Chaouachi, M. A new specific reference gene based on growth hormone gene (GH1) used for detection and relative quantification of Aquadvantage® GM salmon (Salmo salar L.) in food products. Food Chem. 2016, 190, 1040–1045.
- Kishimoto, K.; Washio, Y.; Yoshiura, Y.; Toyoda, A.; Ueno, T.; Fukuyama, H.; Kato, K.; Kinoshita, M. Production of a breed of red sea bream Pagrus major with an increase of skeletal muscle mass and reduced body length by genome editing with CRISPR/Cas9. Aquaculture 2018, 495, 415–427.
- Hallerman, E. Genome editing in cultured fishes. CABI Agric. Biosci. 2021, 2, 46.


