Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yusuke Nakamura | -- | 5614 | 2023-02-16 04:21:21 | | | |
| 2 | Camila Xu | Meta information modification | 5614 | 2023-02-16 07:39:51 | | | | |
| 3 | Camila Xu | Meta information modification | 5614 | 2023-02-20 08:22:11 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nakamura, Y.; Shimizu, Y. Idiopathic Pulmonary Fibrosis and Lipid Metabolism. Encyclopedia. Available online: https://encyclopedia.pub/entry/41269 (accessed on 08 August 2026).
Nakamura Y, Shimizu Y. Idiopathic Pulmonary Fibrosis and Lipid Metabolism. Encyclopedia. Available at: https://encyclopedia.pub/entry/41269. Accessed August 08, 2026.
Nakamura, Yusuke, Yasuo Shimizu. "Idiopathic Pulmonary Fibrosis and Lipid Metabolism" Encyclopedia, https://encyclopedia.pub/entry/41269 (accessed August 08, 2026).
Nakamura, Y., & Shimizu, Y. (2023, February 16). Idiopathic Pulmonary Fibrosis and Lipid Metabolism. In Encyclopedia. https://encyclopedia.pub/entry/41269
Nakamura, Yusuke and Yasuo Shimizu. "Idiopathic Pulmonary Fibrosis and Lipid Metabolism." Encyclopedia. Web. 16 February, 2023.
Copy Citation
It has become evident that lipid metabolism is involved in the fibrosis of lungs, and various reports have been made at the cellular level as well as at the organic level. The balance among eicosanoids, sphingolipids, and lipid composition has been reported to be involved in fibrosis, with particularly close attention being paid to a bioactive lipid “lysophosphatidic acid (LPA)” and its pathway.
idiopathic pulmonary fibrosis
lipid metabolism
lysophosphatidic acid
induced pluripotent stem cells
S1P
pirfenidone
nintedanib
1. Eicosanoids
Eicosanoids, which are metabolites of arachidonic acid, are also reported to be involved in fibrosis. Arachidonic acid esterified phospholipid (Ara-PL) undergoes cleavage of arachidonic acid (AA) by phospholipase A2 (PLA2) induced by various stimuli. Thus, the freed AA is converted into various eicosanoids by cyclooxygenase-1 or -2 (COX1 or COX2) or by 5-lipoxygenase (5-LOX) (Figure 1) [1]. This section will describe the mechanism for pulmonary fibrosis involving various eicosanoids.
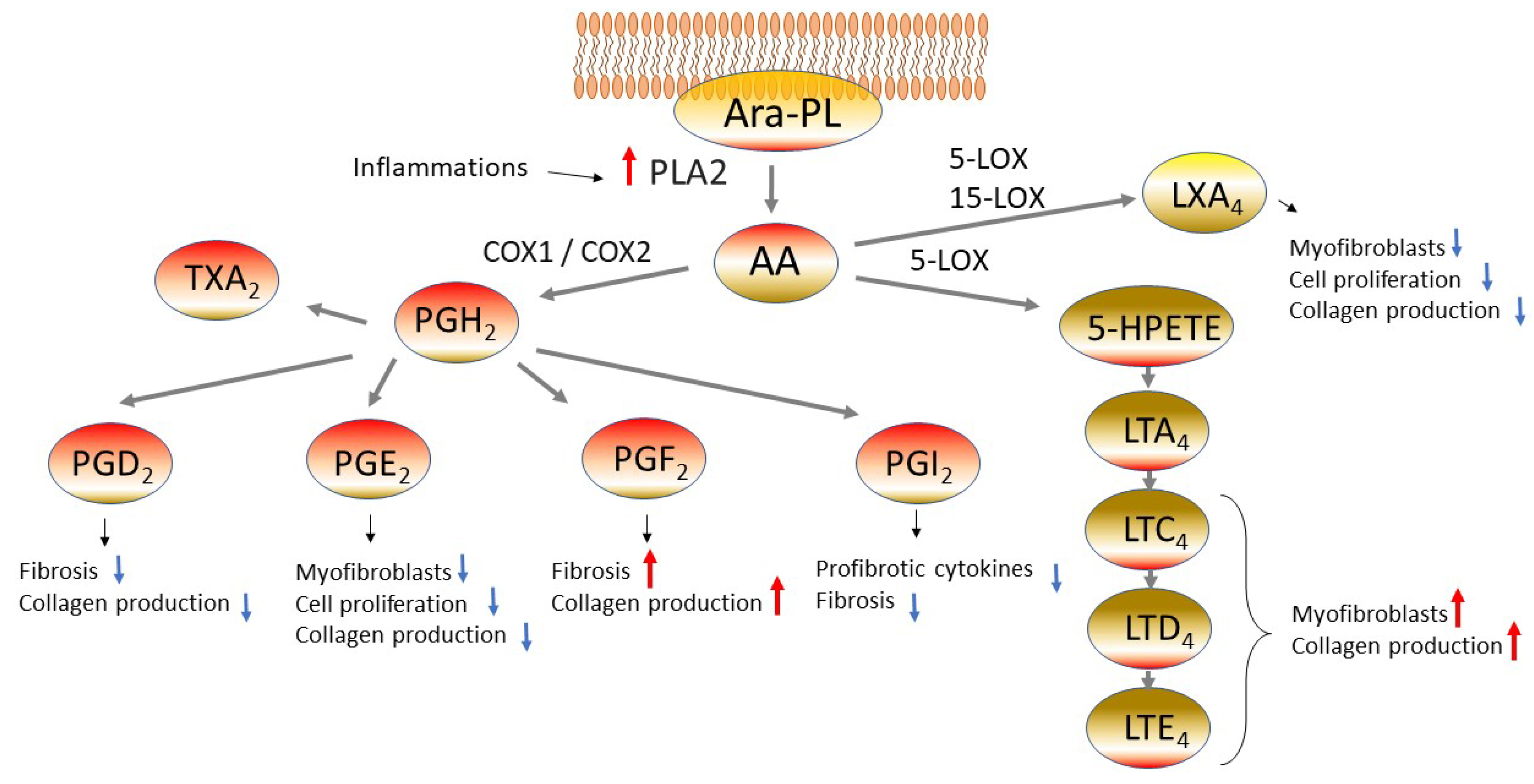
Figure 1. Eicosanoids pathway in fibrotic lung diseases. Arachidonic acid (AA) is freed from arachidonic acid esterified phospholipid (Ara-PL) by phospholipaseA2 (PLA2). AA is metabolized by cyclooxygenase-1 or -2 (COX1 or COX2) or by 5-lipoxygenase (5-LOX), yielding various eicosanoids. The eicosanoids manifest diverse physiological actions related to the pathogenesis of pulmonary fibrosis. Abbreviations: prostaglandin D2 (PGD2), prostaglandin H2 (PGH2), prostaglandin F2 (PGF2), prostaglandin I2 (PGI2, prostacyclin), thromboxane A2 (TXA2), 5-hydroxyeicosatetraenoic acids (5-HETE), leukotriene A4 (LTA4), leukotriene C4 (LTC4), leukotriene D4 (LTD4), leukotriene E4 (LTE4), lipoxin A4 (LXA4).
1.1. Prostaglandin E2
The cells derived from IPF patients have been reported to show reduced levels of cyclooxygenase-2 (COX-2) and prostaglandin E2 (PGE2), leading to the long-lasting view that eicosanoids are involved in the pathogenesis of IPF [2][3]. It has also been reported that the expression of prostaglandin E synthetase (PTGES), an enzyme involved in PGE2 synthesis, is low in the lungs of IPF patients [4]. PGE2 is one of the lipids having been actively studied as a lipid with antifibrotic activity. PGE2 has been reported to suppress the proliferation of fibroblasts and their transformation into myofibroblasts and to reduce collagen production [3][5]. In a mouse model of bleomycin-induced fibrosis, PGE2 was suggested to work protectively against fibrosis [6]. The bronchoalveolar lavage fluid (BALF) after a bleomycin dose showed elevation of the PGE2 level, suggesting the possibility that PGE2 works in physiological defensive reactions. When PGE2 was administered in advance to bleomycin in an animal model of bleomycin-induced interstitial pneumonia, the collagen level and the histological fibrosis score (Ashcroft score) on 21 days after the dose showed improvement, while administration of PGE2 after the bleomycin dose failed to manifest therapeutic efficacy [6]. These results suggest that PGE2 has physiological antifibrotic activity. In the lungs, PGE2 is produced by epithelial cells and fibroblasts [7], and a study using IPF-derived fibroblasts showed less suppression of collagen synthesis and cell proliferation by PGE2 [8]. There is also a report that the stiffened matrix arising from fibrosis reduces PGE2 synthesis [4]. Thus, PGE2 is considered to manifest protective activity during the early phase of fibrosis and can serve as a critical target of IPF treatment.
1.2. Cysteinyl Leukotrienes
The senescent cells, which are unable to proliferate as a result of aging, are considered one of the significant etiological factors for IPF, and it has been reported that IPF can be characterized by an increase of alveolar epithelial type II cells (AECII) which are p53 and p21WAF (senescent cell markers) positive [9]. Lipid composition in senescent cells has also been studied, revealing elevated levels of 5-lipoxygenase (5-LOX), LTC4S, and LTA4H in human fibroblasts and elevation of cysteinyl leukotrienes (cysLTs) in conditioned medium (CM) [10]. The senescent fibroblasts prepared from IPF patient-derived cells showed increased expression of arachidonate 5-lipoxygenase (ALOX5) but no elevation of the enzymes involved in the synthesis of prostaglandins such as PTGS2 (COX-2), PTGDS, and PTGES [10]. It is probable that the lipoxygenase-inducing cascade becomes dominant during the processes of arachidonic acid metabolism in the presence of IPF. When the cysLTs-rich CM collected from senescent fibroblasts was administered to non-senescent fibroblasts, the expression of COL1A2 and αSMA increased, and this change was inhibited by a 5-LOX inhibitor (zileuton) but not by a COX-2 inhibitor (NS-398) [10]. These results suggest the involvement of cysLTs in the pathogenesis of pulmonary fibrosis. Furthermore, the conditioned medium derived from senescent cells induced COL1A2 expression in fibroblasts, while montelukast, known to inhibit cysteinyl leukotriene receptors 1 (CysLT1), suppressed COL1A2 [10][11]. There are multiple other reports suggesting that montelukast can serve as a target of fibroblast treatment [12][13], providing a perspective for its clinical application in IPF management in the future.
1.3. Lipoxin A4
Similar to LTs, lipoxin A4 (LXA4) is an eicosanoid metabolized by 5-LOX and 15-lipoxygenase (15-LOX). LXA4 has been shown to suppress TGFβ1-dependent collagen secretion, αSMA expression, and cell proliferation in fibroblasts derived from IPF patients. It is desirable that LXA4 is further studied from now on [14].
1.4. Prostaglandin I2
Of the other eicosanoids, prostaglandin I2 (PGI2; prostacyclin) has also been reported to be antifibrogenic. Iloprost, a prostacyclin analogue, has been reported to have suppressed profibrotic cytokines (TNFα, IL-6, and TGFβ1) and fibrosis in a mouse model of bleomycin-induced fibrosis [15].
1.5. Prostaglandin F2α
Prostaglandin F2α (PGF2α) is considered to be profibrogenic. The level of PGF2α in the BALF was higher in IPF patients than in the disease control group [16], and this prostaglandin was shown to stimulate fibroblast proliferation and collagen production in a manner independent from TGFβ [16]. In mice lacking the PGF2α receptor “prostaglandin F (PGF) receptor (FP)” (Ptgfr−/−), bleomycin-induced pulmonary fibrosis was suppressed [16]. Thus, PGF2α is involved in the pathogenesis of pulmonary fibrosis and can serve as a target for treatment of pulmonary fibrosis.
1.6. Prostaglandin D2
Prostaglandin D2 (PGD2) is catalyzed by hematopoietic prostaglandin D synthase in the presence of glutathione and produced primarily in activated mast cells and T cells, and has a suppressive effect on fibrosis [17][18]. The treatment of human fibroblasts with PGD2 resulted in c-AMP-mediated suppression of TGFβ-induced type I collagen secretion. A similar suppressive activity has been observed also with BW245C, a PGD2 receptor (DP-receptor: DP1) agonist, thus suggesting the involvement of PGD2-DP1 signals in suppression of fibrosis [19]. Another PGD2 receptor “chemoattractant receptor homologous with T-helper cell type 2 cells (CRTH2)” is also considered to be involved in the suppression of fibrosis. CRTH2 is expressed in type 2 cytokine-producing T cells and in group 2 innate lymphoid cells (ILC2). CRTH2 deficient mice (CRTH2−/−) showed aggravation of bleomycin-indued fibrosis as well as elevation of inflammatory cells in BALF and elevation of lung tissue Col1a1. The transplant of splenic δγT cells into CRTH2−/− mice resulted in the suppression of fibrosis, suggesting that CRTH2 in these cells affects fibrosis [20].
2. Sphingolipids
The cell membrane is composed of lipids such as phospholipids and sphingolipids. These lipids work not only as cell membrane components but also as factors with physiological activity of transducing diverse signals in response to various stimuli [21]. Glycerophospholipid will be discussed later, and this paragraph focuses on sphingolipids. Sphingomyelin (SM) is a representative of sphingolipids found in cell membranes, assuming the form of sphingophospholipid composed of phosphocholine (PC) or phosphoethanolamine (PE) bound to the ceramide (Cer). Sphingomyelinase (SMase) is formed by various external factors, and Cer is formed as a result of hydrolysis. Cer is converted into sphingosine (Sph) by ceramidase. Then, under the catalytic activity of sphingosine kinase 1 (Sphk1) distributed in the cell membrane, sphingosine-1-phosphate (S1P) is formed [22]. S1P is released from cells mediated by ATP-binding cassette (ABC) transporters [23] or spinster homolog 2 (Spns2) [24]. Five subtypes of S1P receptor (S1P1–S1P5) are known, all assuming the form of seven-transmembrane trimer type G protein-coupled receptor (Figure 2). Niemann–Pick disease types A and B are caused by SM accumulation due to a shortage of acid sphingomyelinase (ASM), and these diseases occasionally induce pulmonary fibrosis [25]. Sphk1 is induced by TGFβ, a cytokine playing a central role in fibrosis [26].

Figure 2. Sphingolipids pathway in fibrotic lung diseases. Sphingomyelin (SM) is metabolized by sphingomyelinase (SMase), yielding ceramide (Cer). It is later converted by ceramidase into sphingosine (Sph) and subsequently converted by sphingosine kinase 1 (Sphk1) into sphingosine-1-phosphate (S1P). S1P communicates with the inside and outside of cells via ATP-binding cassette (ABC) transporters and spinster homolog 2 (Spns2) and binds to S1P receptors to manifest physiological activity. Abbreviations: Bleomycin (BLM), Hippo/yes-associated protein (YAP), mitochondrial reactive oxygen species (mtROS).
When studied in mice, administration of bleomycin elevated the lung tissue ASM activity, accompanied by elevation in Cer and acid ceramidase (AC) activity. In ASM−/− mice, collagen production was suppressed, accompanied by a reduction of the apoptosis rate and suppression of transformation into myofibroblasts. When ASM in NIH3T3 fibroblasts was blocked with small interfering (si) RNA, a reduction in sphingosine-1-phosphate (S1P) levels was induced and collagen production was suppressed [27]. This result suggests the possible influence of S1P on fibrosis and indicates that activation of each synthetase in the presence of fibrosis-inducing stimuli will lead to S1P production.
When human fibroblasts were stimulated with TGFβ or bleomycin, the levels of mitochondrial reactive oxygen species (mtROS) and transcription factors, such as Hippo/yes-associated protein (YAP), fibronectin (FN), and αSMA, were elevated. Inhibition of Sphk1 or YAP resulted in a decrease of mtROS and less expression of FN and αSMA. Furthermore, inhibition of each of S1P and YAP resulted in a similar decrease of mtROS and less expression of FN and αSMA [28]. On the basis of these findings, S1P is considered to possibly induce mitochondrial damage and YAP-mediated fibrosis.
There is a report demonstrating a significant elevation of the S1P level in the serum and BALF of IPF patients. S1P has additionally been reported to have roles in epithelial-mesenchymal transition (EMT) in stimulated-AECII, cell proliferation, and collagen production [29].
Regarding the activity of S1P on vascular endothelial cells, the effects in reducing vascular permeability and the protective activity are known [30]. The elevation of vascular permeability is involved in the pathophysiology of pulmonary fibrosis at the acute stage [31]. Mice with endothelial cell-(EC) specific deletion of the S1P receptor 1 (S1PR1) (EC-S1pr1−/−) showed enhanced permeability across the pulmonary vessels. Intratracheal administration of bleomycin to EC-S1pr1−/− mice resulted in enhanced pulmonary vascular permeability and aggravation of fibrosis compared to wild-type mice [31]. It seems possible that the S1P-S1PR1 pathway in vascular endothelial cells induced immune cell infiltration and activation of the coagulation via enhanced pulmonary vascular permeability, leading to aggravation of fibrosis [31].
A sphingosine analogue, FTY720 (Fingolimod) is a drug known to be phosphorylated by sphingosine kinase 2 and to function as an S1P analogue [32]. It has already been introduced clinically as a drug for the treatment of multiple sclerosis. In an animal model of bleomycin-induced interstitial pneumonia, low doses of FTY720 during inflammatory phases could suppress fibrosis, while such doses during remodeling phases had the potential of aggravating the fibrosis [33]. S1P may work protectively if used during the inflammatory phases of acute vascular permeability reduction. On the other hand, if used during the fibrotic phases, it can stimulate fibroblast proliferation and collagen production, leading to aggravation of fibrosis. More detailed fundamental evaluation is desirable before clinical application of S1P to the management of pulmonary fibrosis.
3. Lipid Balance and Fibrosis
Elongation of long-chain fatty acid family member 6 (Elovl6), a kind of fatty acid elongation, has been reported to aggravate bleomycin-induced pulmonary fibrosis through disturbing the balance in fatty acid composition [34]. Elovl6 is a rate-limiting enzyme for the elongation of saturated and monounsaturated long chain fatty acids and is known to catalyze the elongation of palmitate (PA: C16:0) to stearate (C18:0). Stearate is converted by stearoyl CoA desaturase (SCD) into oleic acid (OA; C18:1 n-9). If the expression of Elovl6 is reduced by bleomycin, the PA level rises, leading to elevation of reactive oxygen species (ROS), eventually resulting in the progression of fibrosis mediated by TGFβ elevation and apoptosis [34]. This has additionally been shown to induce apoptosis in a study of PA at the cellular level [35]. That is a significant report indicating that changes in fatty acids are involved in fibrosis. Therefore, measurement and adjustment of lipid profiles in the living body may serve as a therapeutic strategy.
Phospholipid profiles in the serum of IPF patients have also been measured. Analysis of the serum lipids in IPF patients by ultra-high performance liquid chromatography coupled to high-resolution mass spectrometry (UHPLCHRMS) revealed elevated levels of lysophosphatidylcholine (LPC) [36]. That report referred also to the usefulness of LPA (lysophosphatidic acid) as a biomarker, and there are also reports demonstrating elevated LPA levels in the BALF and exhaled breath condensate from IPF patients [37][38]. Therefore, the above-mentioned elevation in the LPC level may reflect the event during the course of LPC metabolism into LPA. Elevation in the LPA level has also been reported for patients with other diseases such as bronchial asthma [39], hyperoxic lung injury [40], and COPD [41]. Another study measured 507 individual serum circulating lipid profile of IPF by quadrupole time of flight mass spectrometry (UPLC-QTOF/MS) with lipidomics, and six discriminating lipids were selected, such as stigmasteryl ester, dodecatrienyl dodecanoate, deoxyvitamin D3, triacylglycerol, and diacylglycerol, as the potential biomarkers in diagnosing IPF [42].
Lipid composition has also been studied in the IPF-affected lung tissue, revealing the elevation of palmitic acid (16:0) and stearic acid (18:0) [43]. In a study of a mouse model of bleomycin-induced interstitial pneumonia, the mice fed with a palmitic acid-rich high-fat diet showed exacerbation of fibrosis and decreased survival rate, accompanied by elevation in the TGFβ level [43]. In the epithelial cells, treatment with palmitic acid (16:0) induced increases in endoplasmic reticulum (ER) stress and apoptosis [43]. In mouse lung epithelial cells (MLE12 cells), inhibition of CD36 (a fatty acid transporter) reduced the effect of palmitic acid (16:0) on cell viability and caused decreases in profibrotic cytokines (MCP-1 and IL-6), ER stress markers, and apoptosis markers [43]. That study indicates the possibility that oral intake of palmitic acid serves as a factor responsible for the pathogenesis of IPF, and the data from that study are valuable. If the details of ingested foods are analyzed in relation to pulmonary fibrosis in clinical cases, a new approach for the treatment of pulmonary fibrosis may be cultivated.
The role of other lipids has also been investigated in preclinical studies [44]. Exposure to high-fat diet can be a trigger of lung fibrosis by neutrophilic inflammation or TGFβ stimulation [45][46]. Docosahexaenoic acid (DHA) is a ω-3 polyunsaturated fatty acid (PUFA), and this lipid has reported to attenuate lung fibrosis by upregulation of Smad7 [47]. Another PUFA of gamma-linolenic acid (GLA) has also attenuated fibrosis. In hamster bleomycin-induced fibrosis model, GLA dietary intake leads to elevation of prostaglandin E1 (PGE1) which is metabolite of PGE2 and has anti-inflammatory effect, then showed attenuate lung inflammation and fibrosis [47].
4. Statins
Statins are extensively used for the treatment of hypercholesterolemia during clinical practice. They have recently been shown to have anti-inflammatory activity as well. In a mouse model of bleomycin-induced fibrosis, treatment with pravastatin suppressed the expression of TGFβ, CTGF, and RhoA as well as the expression of a fibrosis marker, hydroxyproline [48]. In recent studies, the ACEII and fibroblasts of patients with pulmonary fibrosis were shown to have markedly low levels of low-density lipoprotein receptor (LDLR). In Ldlr deficient (Ldlr−/−) mice, the level of low-density lipoprotein (LDL) was markedly high, and the administration of bleomycin resulted in the rapid onset of intense fibrosis. LDL was thus estimated as a factor responsible for the induction of apoptosis and TGFβ [49]. Also, clinically, there is a report that the use of statins (drugs for the treatment of hypercholesterolemia) in IPF patients reduced the hospitalization rate and the death rate due to IPF [50].
5. Involvement of Mitochondria
There is a report that mitochondrial dysfunction in type II alveolar epithelial cells exacerbated fibrosis [51]. In that report, increased mitochondrial injury was noted in IPF patients. When airway epithelial cells were studied in vitro, stimulation with TGFβ resulted in signs of mitochondrial depolarization and mitochondrial ROS (reactive oxygens species), thus indicating that mitochondrial injury was caused. Mitochondrial injury is known to stimulate apoptosis and profibrotic cytokines, leading to pulmonary fibrosis [51][52]. As stated above, the appearance of mitochondrial ROS (mtROS) possibly leading to mitochondrial injury can also be induced from S1P signals [28].
In fibroblasts, knocking down the PISD resulted in the elevation of αSMA and collagen production, suggesting that PISD may be contributing to the mechanism for fibrosis through the adjustment of phospholipids [53].
6. Lipofibroblast
Myofibroblasts are an important source for collagen production. Metformin, a drug for the treatment of diabetes mellitus, has been reported to have the potential of inducing the transformation of myofibroblasts into lipid droplet-containing interstitial fibroblasts (lipofibroblasts) mediated by AMP-activated protein kinase (AMPK). COL1A1 expression is lower in lipofibroblasts than in myofibroblasts, and the effect in maintaining AECII (known to be involved in lung regeneration) is expected of lipofibroblasts. Thus, lipofibroblasts are promising as a target for the treatment of pulmonary fibrosis from now on [54][55].
7. Points for Consideration in Connection with Regenerative Medicine
As illustrated above, lipid metabolism is involved in the pathogenesis of fibrosis. Novel treatment methods are being developed as mentioned above, including the evaluation of cell-based therapy. Cell-based therapy is an approach of regenerative medicine expected to manifest anti-inflammatory activity through the administration of stem cells. When regenerative medicine is considered, it is also essential to evaluate the lipid composition in each product used for regenerative medicine [56]. Suppression of fibrosis by means of stem cell administration has been attempted with mesenchymal stem cells (MSCs) [57][58], adipose-derived MSCs [59][60], induced pluripotent stem cells (iPSC) [61][62], and AECII [63][64]. Stem cell-based treatment is still challenging; however, MSC therapy in IPF has shown beneficial results [65]. Because the lipid composition in stem cells is altered by the differentiation of stem cells [66], evaluation of lipid composition is essential in evaluation of the quality of regenerative medicine products. It also needs to be taken into consideration that the altered lipid profile can give rise to profibrotic lipid mediators.
The relationship between vascular meshwork and fibrosis during IPF pathogenesis has been actively analyzed, yielding reports that lung tissue fibrosis is affected by the stimulation of fibroblasts mediated by capillary endothelial cells [67] and that myofibroblasts produce neovascularization suppressive factors, leading to suppressed neovascularization [68]. There is also a view that the reduction in vascular meshwork causes abnormal repair of tissues, possibly leading to fibrosis [69]. It is important to analyze the lipid profile of vascular endothelial cells which constitute blood vessels and to obtain basic data. Researchers revealed elevation of plasmalogen phosphatidylethanolamines (38:5) and (38:4) during the course of differentiation from iPSC to vascular endothelial cells [70]. It was additionally shown that lysophosphatidylcholine (LPC) (22:5) was predominantly detected during the course of network formation by iPSC-derived vascular endothelial cells and that their distribution was not uniform [71].
When regenerative medicine is considered, it needs to be borne in mind that the lipid composition is likely to change depending on the conditions and that attention is needed to be paid not only to the composition but also to the distribution of lipids.
8. LPA Involvement in IPF and Its Pathogenesis
8.1. Outline
The LPA cascade has been attracting close attention in recent years because it has the potential of explaining the pathogenesis of IPF and being utilized for therapeutic strategy. The BALF of IPF patients is known to be significantly rich in LPA [37], and LPA has been studied for analyzing the pathogenesis of IPF and as a target of IPF treatment. LPA receptors assume the form of G protein-coupled receptors (GPCRs). Six subtypes are known, and LPA1 and LPA2 are considered to be primarily involved in IPF [72]. This section will outline LPA and describe the clinical studies conducted to date.
8.2. Pathways for LPA Metabolism
The pathway for LPA metabolism is diverse [73], and a representative pathway is shown below. The formation of LPA begins with the metabolism of glycerophospholipids (GPs) such as phosphatidylcholine (PC) and phosphatidylethanolamine (PE) abundantly found in the cell membranes. Dipalmitoylphosphatidylcholine (DPPC) is a major phospholipid found in the alveoli, and here PC is taken as an example for further discussion. PC is degraded by phospholipase A2 or phospholipase A1 (PLA2 or PLA1) into lysophosphatidylcholine (LPC) [74] and LPC is converted by lysophospholipase D (lysoPLD: Autotaxin/ATX) into lysophosphatidic acid (LPA) [75]. In addition, phosphatidic acid (PA) is formed by metabolism with lysoPLD/ATX (Figure 3).

Figure 3. LPA pathway in fibrotic lung diseases. A representative pathway for lysophosphatidic acid (LPA) production is shown here. Glycerophospholipids, such as phosphatidylcholine (PC) and phosphatidylethanolamine (PE), are converted by phospholipase A2 (PLA2) into lysophosphatidylcholine (LPC). LPC is then converted by lysophospholipase D (lysoPLD: Autotaxin: ATX) into LPA. Another pathway involves the formation of lysoPLD/ATX from phosphatidic acid (PA). Later, the signals are transduced to the LPA receptors (LPA1/LPA2), resulting in the manifestation of physiological activity related to pulmonary fibrosis.
Using an animal model of bleomycin-induced interstitial pneumonia, the profiles of PC, PE, and LPC in the serum and BALF was analyzed. The levels of PC, PE, and LPC in the BALF were found to be significantly elevated for many fatty-acid-binding patterns (unpublished data). These results suggest the possibility that phospholipids, such as PC and PE, were released locally in the alveolar epithelium and underwent metabolism into lysophospholipids. If this finding is combined with the past report that the lung tissue’s chromatic response to ATX was elevated in IPF patients [76], it seems likely that the metabolism of lysophospholipids into LPA occurs locally, contributing to the pathogenesis of interstitial lung disease.
8.3. LPA Signal Transduction and Mechanism for Fibrosis
Resembling the migration of fibroblasts to a certain site of the fibrin matrix to form granuloma there during the course of skin injury, it is known that fibroblasts accumulate in the fibrin-rich exudate from airspaces during the course of IPF [77]. The BALF from IPF patients has been reported to increase the chemotaxis of pulmonary fibroblasts significantly [78][79], and this activity seems to be involved in the pathogenesis of IPF. Data are available concerning the correlation between such chemotactic response of fibroblasts and lung function [78], allowing an expectation to use it as an important target of treatment. Subsequent studies revealed that LPA is a substance involved in this response, thus triggering research into the involvement of LPA in IPF.
8.3.1. LPA1
Stimulation of fibroblast’s chemotaxis by LPA was inhibited by an LPA1 inhibitor, Ki16425 [37], indicating that the LPA/LPA1 signals are important for chemotaxis. In LPA1 knockout (KO) mice, bleomycin-induced fibrosis was reduced and the hydroxyproline content in the lungs decreased significantly, accompanied by a significant improvement of the 21-day survival rate [37]. There are data indicating that LPA1 receptors are significantly predominant among the LPA receptors in IPF patients [36], suggesting the possibility for LPA1 to serve as the target for treatment. There is also a report that LPA/LPA1 signals affected the vascular endothelial cells, resulting in exacerbation of the vascular permeability in LPA1 KO mice [37].
The LPA/LPA1 cascade also affects the epithelial cells. Apoptosis of epithelial cells is considered to be involved in the onset of IPF [80]. LPA1 KO mice showed significantly less apoptosis of alveolar epithelial cells. Apoptosis of bronchial epithelial cells was also suppressed in these mice. Of the LPA species, LPA (16:0) and LPA (18:0) increased soon (Day 1) after bleomycin treatment, followed 3 days later by an increase of LPA(18:1), LPA(18:2), and LPA(20:4) [81]. In kidney epithelial cells, stimulation of EMT (epithelial-mesenchymal transition) via the LPA1/MAPK-AKT/KLF5 signaling pathway has been reported, and a similar event may be occurring also in the lungs [82]. The LPA1/3 antagonist VPC12249 has been reported to have attenuated the pulmonary fibrosis in a mouse model of radiation lung fibrosis. Following the finding of enhanced LPA1/3 expression in mice induced by 16 Gy radiation, VPC12249 was administered to these mice, resulting in the suppression of survival shortening and pulmonary fibrosis and decrease of profibrotic cytokines such as TGFβ and CTGF. That study is interesting in that the LPA1/3 signals stimulated the expression of CTGF. An anti-CTGF antibody (pamrevlumab) is now under clinical trial (currently at phase 3) as a novel drug for IPF treatment [83].
8.3.2. LPA2
Evaluation has also been made on LPA2. LPA2 KO (Lpar2−/−) mice, studied as a model of bleomycin-induced interstitial pneumonia, showed suppressed expression of fibronectin and αSMA, accompanied by decreases in lung tissue collagen and pulmonary vascular leakage and improvement of the survival rate [84]. Furthermore, the levels of profibrotic cytokines (IL-6, TGFβ) in BALF decreased. Also, when human fibroblasts were inhibited with small interfering siRNA for LPA2, the expression of fibronectin, Col1A2, αSMA, and TGFβ was decreased [84]. A possible mechanism for this change is the influence of LPA2 signals on the extracellular regulated kinase (ERK)1/2, Akt, Smad3, and p38 mitogen-activated protein kinase (p38) cascade [84]. Interesting enough, LPA2 was shown to have the potential of enhancing the TGFβ/Smad2/3 signals [84].
There is also a report showing that in epithelial cells, LPA activates TGFβ mediated by αvβ6 integrin in the LPA2-RhoA/Rho kinase cascade [85]. In that study, data showing increases of LPA2 and αvβ6 integrin in IPF patients were also collected, suggesting the possibility of clinically applying LPA2 as a target of treatment [85].
8.3.3. ATX
ATX (autotaxin), i.e., ectonucleotide pyrophosphatase/phosphodiesterase family member 2 (Enpp2), is an enzyme involved in LPA formation and has been reported to be expressed abundantly in the alveolar epithelium, fibroblastic foci, and alveolar macrophages of IPF patients [76] (Figure 3). It has been studied also in an animal model of bleomycin-induced interstitial pneumonia, revealing significant elevation of the ATX level in BALF. ATX−/− animals died at embryonic day 10.5 because of vascular hypoplasia [86]. For this reason, conditionally knockout animals are used for the evaluation of ATX after maturation. Mice with conditionally knockout bronchiolar epithelial cells or alveolar macrophages were created to evaluate ATX deficiency. Although some parameters did not differ significantly between these two groups of knockout mice, ATX conditionally knockout mice showed a reduction in lung collagen, total cell counts in BALFs, BALF soluble collagen concentrations, BALF ATX concentrations, and ATX activity. These results suggest that bronchiolar epithelial cells and alveolar macrophages are contributing at least to the formation of ATX in lungs [76]. In the same study, it was additionally shown that treatment with the ATX inhibitor GWJ-A-23 alleviated fibrosis in the mouse model of bleomycin-induced interstitial pneumonia [76].
8.4. Therapeutic Strategy Focusing on the LPA Cascade
As described above, LPA1 antagonists and ATX inhibitors deserve evaluation as targets of IPF treatment. Table 1 lists the drugs under clinical studies revealed by the literature search via U.S. National Library of Medicine, ClinicalTrials.gov (Table 1). This chapter will outline the candidate drugs currently evaluated for use in treatment.
Table 1. Clinical trials on the LPA pathway for IPF. Clinical studies on LPA pathways related to IPF (revealed by search via the U.S. National Library of Medicine, ClinicalTrials.gov).
| Drug Name | Mechanism | Trial Number | Phase | Disease | Status at Nov 2022 |
|---|---|---|---|---|---|
| BMS-986020 | LPA1 antagonist | NCT01766817 | Phase II | IPF | Completed |
| BMS-986278 | LPA1 antagonist | NCT04308681 | Phase II | IPF and PF-ILD | Active, not recruiting |
| GLPG1690 | ATX inhibitor | NCT02738801 | Phase II | IPF | Completed |
| NCT03711162 | Phase III | IPF | Terminated | ||
| NCT03733444 | Phase III | IPF | Terminated | ||
| BBT-877 | ATX inhibitor | NCT03830125 | Phase I | Completed |
8.4.1. LPA1 Antagonist
Clinical studies on LPA1 antagonists are now under way on BMS-986020 and BMS-986278.
BMS-986020
IM136003 (NCT01766817) is a phase 2 clinical trial on the use of BMS-986020 for IPF. Between April 2013 and February 2016, 373 participants were enrolled to the trial and 143 of them were randomized to three groups (placebo group, n = 47; BMS-986020 600 mg quaque die (qd) group, n = 48; BMS-986020 600 mg bis in die (bid) group, n = 48) for this phase 2, multicenter, randomized, double-blind trial involving patients with IPF having FVC 45–90% and diffusing capacity for carbon monoxide (DLCO) 30–80%. The primary endpoint was the change from baseline in FVC rate to Week 26. FVC reduction was significantly smaller in the BMS-986020 600 mg bid group than in the control group (−0.134 L vs. −0.042 L, p = 0.049) [87]. In the BMS-986020 600 mg bid group, three patients prematurely quit the study because of treatment-related cholecystitis that resulted in cholecystectomy [87]. No case of such hepatic impairment was noted in the phase 1 trial in which the subjects were confined to healthy individuals and the dosing period was short. Furthermore, alanine aminotransferase (ALT) elevation (≥3× upper limit of normal) arose at an incidence of 7% (n = 3/48) in the BMS-986020 600 mg qd group, 21% (n = 10/48) in the 600 mg bid group, and 0% (0/47) in the placebo group. The incidence of elevation in other enzymes was as follows: aspartate aminotransferase (AST) placebo 0% vs. 600 mg qd 4% vs. 600 mg bid 6%, alkaline phosphatase (ALP) placebo 0% vs. 600 mg qd 0% vs. 600 mg bid 4%, and total bilirubin placebo 0% vs. 600 mg qd 4% vs. 600 mg bid 0%. The 600 mg bid regimen for this drug therapy seems to be useful as a therapeutic option because it significantly suppressed FVC reduction, but how to alleviate the hepatic impairment caused by this therapy is an open issue.
The Hepatobiliary Toxicity of BMS-986020
A paper by Gill et al. dealing with the mechanism for the hepatobiliary toxicity of BMS-986020 was published recently [88]. BMS-986020 was shown to inhibit bile acid (BA) efflux transporters BSEP, MRP3, and MRP4 as well as BA canalicular efflux. In human liver cells and bile duct cells, this drug inhibited the mitochondrial function. BMS-986278, another LAP1 antagonist, did not inhibit BA canalicular efflux. Therefore, the hepatobiliary toxicity of BMS-986020 does not seem to be associated with the LPA1-antagonizing activity.
BMS-986278
BMS-986278 is a second-generation LPA1 antagonist differing structurally from BMS-986020 [88]. In the structure-activity relationship (SAR) studies on LPA1, the discovery of BMS-986020 and subsequent modifications led to development of BMS-986278 as the final compound [89]. BMS-986278 is free of the efflux transporter inhibitory activity observed with BMS-986020 [88][90], and it did not cause hepatic impairment in the phase 1 study [91]. A phase 2 clinical trial on BMS-986278 (NCT04308681) was conducted in patients with IPF or PF-ILD [92]. The trial started on July 29, 2020 and was completed on August 4, 2022 (Actual Primary Completion Date). The trial was planned to enroll IPF patients (n = 240) and PF-ILD patients (n = 120), to be allocated at random to the BMS-986278 30 mg PO bid group, the BMS-986278 60 mg PO bid group, and the placebo PO bid group at a ratio of 1:1:1. The inclusion criteria were patients satisfying the diagnostic criteria for IPF or PF-ILD, with predicted %FVC ≥ 40%, a forced expiratory volume in 1 s (FEV1)/FVC ≥ 0.7 and DLCO ≥ 25%. The primary endpoint was the reduction rate in predicted %FVC [92]. Although the results of analysis from this trial have not yet been reported, effectiveness and tolerability in terms of adverse events are expected of this drug which manifests LPA1 inhibitory activity akin to BMS-986020 but is free of efflux transporter inhibitory activity which can lead to hepatic impairment (a problem with BMS-986020).
8.4.2. ATX Inhibitors
Clinical studies on ATX inhibitors have been conducted on GLPG1690 and BBT-877.
Phase 2 FLORA Trial
GLPG1690 is the first ATX inhibitor used for IPF. Clinical trials on this drug have proceeded to the phase 3. Here, the phase 2 FLORA trial (NCT02738801) conducted previously is presented first. During the period from March 24, 2016, to May 2, 2017, screening was conducted on 72 patients and 23 of them were enrolled to the trial. Six patients were allocated to the placebo group and 17 patients to the GLPG1690 600 mg qd group, each planned to receive 12-week oral treatment according to the protocol [93]. The primary outcomes were safety (adverse events), tolerability, pharmacokinetics, and pharmacodynamics. Spirometry was assessed as a secondary outcome. The inclusion criteria were patients confirmed to have IPF by the central review, with FVC ≥ 50%, DLCO ≥ 30%, FEV1/FVC ≥ 0.70, and life expectancy ≥ 12 months. One patient in each group quit the trial prematurely because of adverse events and another patient in the GLPG1690 group cancelled consent to the trial. As a result, the data were analyzed on 5 patients in the placebo group and 15 patients in the GLPG1690 group [93]. Treatment-related adverse events, such as infection, developed in 4 patients (67%) from the placebo group and 11 patients (65%) from the GLPG1690 group, but most events were mild to moderate. Severe adverse events were recorded in 3 cases (2 cases from the placebo group and 1 case from the GLPG1690 group), including a case of urinary tract infection, acute nephropathy and lower airway infection, and a case of second-degree atrioventricular block from the placebo group as well as a case of cholangiocarcinoma from the GLPG1690 group. ATX activity was evaluated on the basis of changes in the serum LPA (18:2) level, revealing a reduction of the LPA level in the GLPG1690 group and improvement to the baseline level after discontinuation of treatment [93]. This result was close to the result of phase 1 clinical trials involving healthy volunteers (NCT02179502, NCT03143712) [94]. The mean FVC at Week 12, evaluated as a secondary outcome, was −70 mL in the placebo group and +25 mL in the GLPG1690 group [93].
Phase 3 ISABELA1 and ISABELA2
In the phase 2 trial, the safety of GLPG1690 comparable to a placebo was demonstrated, and the reduction in respiratory function at Week 12 (a secondary outcome) was also suppressed. On the basis of these results, two phase 3 clinical trials, i.e., ISABELA1 (NCT03711162) and ISABELA2 (NCT03733444), were started in November 2018 [95]. Both trials had an identical design, involving IPF patients (international, randomized, double-blind, placebo-controlled, parallel-group, multicenter studies). In each trial, 750 patients with IPF were allocated at a ratio of 1:1:1 to three groups (GLPG1690 600 mg po qd, GLPG1690 200 mg po qd, and placebo) in which GLPG1690 or a placebo was administered as an add-on to the standard therapy). The primary endpoint was the rate of decline of forced vital capacity (FVC) over 52 weeks. The trial was discontinued prematurely on the grounds that the benefit-risk profile no longer supported continuing the study based on recommendations of the Independent Data Monitoring Committee. The exact reason for its discontinuation is unknown, but the study data can be accessed at the U.S. National Library of Medicine, ClinicalTrials.gov. It seems that the all-cause mortality was probably related to the discontinuation of this trial.
BBT-877
BBT-877 is an ATX inhibitor on which antifibrotic activity and toxicity have been checked in animals [96]. On February 13, 2019, a phase 1 clinical trial (NCT03830125) on this drug in healthy adults was started. It was designed to allocate the subjects to a placebo group, a BBT-877 single dose group, and a BBT-877 multiple dose group. The trial was completed on November 24, 2019. The publication of its results is now anticipated.
References
- Taketomi, Y.; Murakami, M. Regulatory Roles of Phospholipase A(2) Enzymes and Bioactive Lipids in Mast Cell Biology. Front. Immunol. 2022, 13, 923265.
- Wilborn, J.; Crofford, L.J.; Burdick, M.D.; Kunkel, S.L.; Strieter, R.M.; Peters-Golden, M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J. Clin. Investig. 1995, 95, 1861–1868.
- Clark, J.G.; Kostal, K.M.; Marino, B.A. Modulation of collagen production following bleomycin-induced pulmonary fibrosis in hamsters. Presence of a factor in lung that increases fibroblast prostaglandin E2 and cAMP and suppresses fibroblast proliferation and collagen production. J. Biol. Chem. 1982, 257, 8098–8105.
- Berhan, A.; Harris, T.; Jaffar, J.; Jativa, F.; Langenbach, S.; Lönnstedt, I.; Alhamdoosh, M.; Ng, M.; Lee, P.; Westall, G.; et al. Cellular Microenvironment Stiffness Regulates Eicosanoid Production and Signaling Pathways. Am. J. Respir. Cell Mol. Biol. 2020, 63, 819–830.
- Kolodsick, J.E.; Peters-Golden, M.; Larios, J.; Toews, G.B.; Thannickal, V.J.; Moore, B.B. Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am. J. Respir. Cell Mol. Biol. 2003, 29, 537–544.
- Dackor, R.T.; Cheng, J.; Voltz, J.W.; Card, J.W.; Ferguson, C.D.; Garrett, R.C.; Bradbury, J.A.; DeGraff, L.M.; Lih, F.B.; Tomer, K.B.; et al. Prostaglandin E₂ protects murine lungs from bleomycin-induced pulmonary fibrosis and lung dysfunction. Am. J. Physiol. Lung. Cell Mol. Physiol. 2011, 301, L645–L655.
- Hodges, R.J.; Jenkins, R.G.; Wheeler-Jones, C.P.; Copeman, D.M.; Bottoms, S.E.; Bellingan, G.J.; Nanthakumar, C.B.; Laurent, G.J.; Hart, S.L.; Foster, M.L.; et al. Severity of lung injury in cyclooxygenase-2-deficient mice is dependent on reduced prostaglandin E(2) production. Am. J. Pathol. 2004, 165, 1663–1676.
- Huang, S.K.; Wettlaufer, S.H.; Hogaboam, C.M.; Flaherty, K.R.; Martinez, F.J.; Myers, J.L.; Colby, T.V.; Travis, W.D.; Toews, G.B.; Peters-Golden, M. Variable prostaglandin E2 resistance in fibroblasts from patients with usual interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2008, 177, 66–74.
- Akram, K.M.; Lomas, N.J.; Forsyth, N.R.; Spiteri, M.A. Alveolar epithelial cells in idiopathic pulmonary fibrosis display upregulation of TRAIL, DR4 and DR5 expression with simultaneous preferential over-expression of pro-apoptotic marker p53. Int. J. Clin. Exp. Pathol. 2014, 7, 552–564.
- Wiley, C.D.; Brumwell, A.N.; Davis, S.S.; Jackson, J.R.; Valdovinos, A.; Calhoun, C.; Alimirah, F.; Castellanos, C.A.; Ruan, R.; Wei, Y.; et al. Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis. JCI Insight 2019, 4, e130056.
- Castelino, F.V. Lipids and eicosanoids in fibrosis: Emerging targets for therapy. Curr. Opin. Rheumatol. 2012, 24, 649–655.
- Shaker, O.G.; Sourour, D.A. Effect of leukotriene receptor antagonists on lung fibrosis in rats. J. Appl. Toxicol. 2011, 31, 678–684.
- Shimbori, C.; Shiota, N.; Okunishi, H. Effects of montelukast, a cysteinyl-leukotriene type 1 receptor antagonist, on the pathogenesis of bleomycin-induced pulmonary fibrosis in mice. Eur. J. Pharmacol. 2011, 650, 424–430.
- Roach, K.M.; Feghali-Bostwick, C.A.; Amrani, Y.; Bradding, P. Lipoxin A4 Attenuates Constitutive and TGF-β1-Dependent Profibrotic Activity in Human Lung Myofibroblasts. J. Immunol. 2015, 195, 2852–2860.
- Zhu, Y.; Liu, Y.; Zhou, W.; Xiang, R.; Jiang, L.; Huang, K.; Xiao, Y.; Guo, Z.; Gao, J. A prostacyclin analogue, iloprost, protects from bleomycin-induced pulmonary fibrosis in mice. Respir. Res. 2010, 11, 34.
- Oga, T.; Matsuoka, T.; Yao, C.; Nonomura, K.; Kitaoka, S.; Sakata, D.; Kita, Y.; Tanizawa, K.; Taguchi, Y.; Chin, K.; et al. Prostaglandin F(2alpha) receptor signaling facilitates bleomycin-induced pulmonary fibrosis independently of transforming growth factor-beta. Nat. Med. 2009, 15, 1426–1430.
- Murakami, M.; Matsumoto, R.; Urade, Y.; Austen, K.F.; Arm, J.P. c-kit ligand mediates increased expression of cytosolic phospholipase A2, prostaglandin endoperoxide synthase-1, and hematopoietic prostaglandin D2 synthase and increased IgE-dependent prostaglandin D2 generation in immature mouse mast cells. J. Biol. Chem. 1995, 270, 3239–3246.
- Tanaka, K.; Ogawa, K.; Sugamura, K.; Nakamura, M.; Takano, S.; Nagata, K. Cutting edge: Differential production of prostaglandin D2 by human helper T cell subsets. J. Immunol. 2000, 164, 2277–2280.
- Ayabe, S.; Kida, T.; Hori, M.; Ozaki, H.; Murata, T. Prostaglandin D2 inhibits collagen secretion from lung fibroblasts by activating the DP receptor. J. Pharmacol. Sci. 2013, 121, 312–317.
- Ueda, S.; Fukunaga, K.; Takihara, T.; Shiraishi, Y.; Oguma, T.; Shiomi, T.; Suzuki, Y.; Ishii, M.; Sayama, K.; Kagawa, S.; et al. Deficiency of CRTH2, a Prostaglandin D(2) Receptor, Aggravates Bleomycin-induced Pulmonary Inflammation and Fibrosis. Am. J. Respir. Cell Mol. Biol. 2019, 60, 289–298.
- Suryadevara, V.; Ramchandran, R.; Kamp, D.W.; Natarajan, V. Lipid Mediators Regulate Pulmonary Fibrosis: Potential Mechanisms and Signaling Pathways. Int. J. Mol. Sci. 2020, 21, 4257.
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191.
- van der Deen, M.; de Vries, E.G.; Timens, W.; Scheper, R.J.; Timmer-Bosscha, H.; Postma, D.S. ATP-binding cassette (ABC) transporters in normal and pathological lung. Respir. Res. 2005, 6, 59.
- Fukuhara, S.; Simmons, S.; Kawamura, S.; Inoue, A.; Orba, Y.; Tokudome, T.; Sunden, Y.; Arai, Y.; Moriwaki, K.; Ishida, J.; et al. The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J. Clin. Investig. 2012, 122, 1416–1426.
- O’Neill, R.S.; Belousova, N.; Malouf, M.A. Pulmonary Type B Niemann-Pick Disease Successfully Treated with Lung Transplantation. Case Rep. Transplant. 2019, 2019, 9431751.
- Yamanaka, M.; Shegogue, D.; Pei, H.; Bu, S.; Bielawska, A.; Bielawski, J.; Pettus, B.; Hannun, Y.A.; Obeid, L.; Trojanowska, M. Sphingosine kinase 1 (SPHK1) is induced by transforming growth factor-beta and mediates TIMP-1 up-regulation. J. Biol. Chem. 2004, 279, 53994–54001.
- Dhami, R.; He, X.; Schuchman, E.H. Acid sphingomyelinase deficiency attenuates bleomycin-induced lung inflammation and fibrosis in mice. Cell Physiol. Biochem. 2010, 26, 749–760.
- Huang, L.S.; Sudhadevi, T.; Fu, P.; Punathil-Kannan, P.K.; Ebenezer, D.L.; Ramchandran, R.; Putherickal, V.; Cheresh, P.; Zhou, G.; Ha, A.W.; et al. Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts. Int. J. Mol. Sci 2020, 21, 2064.
- Milara, J.; Navarro, R.; Juan, G.; Peiró, T.; Serrano, A.; Ramón, M.; Morcillo, E.; Cortijo, J. Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax 2012, 67, 147–156.
- Garcia, J.G.; Liu, F.; Verin, A.D.; Birukova, A.; Dechert, M.A.; Gerthoffer, W.T.; Bamberg, J.R.; English, D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Investig. 2001, 108, 689–701.
- Knipe, R.S.; Spinney, J.J.; Abe, E.A.; Probst, C.K.; Franklin, A.; Logue, A.; Giacona, F.; Drummond, M.; Griffith, J.; Brazee, P.L.; et al. Endothelial-Specific Loss of Sphingosine-1-Phosphate Receptor 1 Increases Vascular Permeability and Exacerbates Bleomycin-induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2022, 66, 38–52.
- Paugh, S.W.; Payne, S.G.; Barbour, S.E.; Milstien, S.; Spiegel, S. The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. FEBS Lett. 2003, 554, 189–193.
- Gendron, D.R.; Lemay, A.M.; Lecours, P.B.; Perreault-Vallières, V.; Huppé, C.A.; Bossé, Y.; Blanchet, M.R.; Dion, G.; Marsolais, D. FTY720 promotes pulmonary fibrosis when administered during the remodelling phase following a bleomycin-induced lung injury. Pulm. Pharmacol. Ther. 2017, 44, 50–56.
- Sunaga, H.; Matsui, H.; Ueno, M.; Maeno, T.; Iso, T.; Syamsunarno, M.R.; Anjo, S.; Matsuzaka, T.; Shimano, H.; Yokoyama, T.; et al. Deranged fatty acid composition causes pulmonary fibrosis in Elovl6-deficient mice. Nat. Commun. 2013, 4, 2563.
- Listenberger, L.L.; Ory, D.S.; Schaffer, J.E. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J. Biol. Chem. 2001, 276, 14890–14895.
- Rindlisbacher, B.; Schmid, C.; Geiser, T.; Bovet, C.; Funke-Chambour, M. Serum metabolic profiling identified a distinct metabolic signature in patients with idiopathic pulmonary fibrosis—A potential biomarker role for LysoPC. Respir. Res. 2018, 19, 7.
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; Karimi-Shah, B.A.; Kim, N.D.; et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54.
- Montesi, S.B.; Mathai, S.K.; Brenner, L.N.; Gorshkova, I.A.; Berdyshev, E.V.; Tager, A.M.; Shea, B.S. Docosatetraenoyl LPA is elevated in exhaled breath condensate in idiopathic pulmonary fibrosis. BMC Pulm. Med. 2014, 14, 5.
- Ackerman, S.J.; Park, G.Y.; Christman, J.W.; Nyenhuis, S.; Berdyshev, E.; Natarajan, V. Polyunsaturated lysophosphatidic acid as a potential asthma biomarker. Biomark Med. 2016, 10, 123–135.
- Nowak-Machen, M.; Lange, M.; Exley, M.; Wu, S.; Usheva, A.; Robson, S.C. Lysophosphatidic acid generation by pulmonary NKT cell ENPP-2/autotaxin exacerbates hyperoxic lung injury. Purinergic Signal. 2015, 11, 455–461.
- Naz, S.; Kolmert, J.; Yang, M.; Reinke, S.N.; Kamleh, M.A.; Snowden, S.; Heyder, T.; Levänen, B.; Erle, D.J.; Sköld, C.M.; et al. Metabolomics analysis identifies sex-associated metabotypes of oxidative stress and the autotaxin-lysoPA axis in COPD. Eur. Respir. J. 2017, 49, 1602322.
- Yan, F.; Wen, Z.; Wang, R.; Luo, W.; Du, Y.; Wang, W.; Chen, X. Identification of the lipid biomarkers from plasma in idiopathic pulmonary fibrosis by Lipidomics. BMC Pulm. Med. 2017, 17, 174.
- Chu, S.G.; Villalba, J.A.; Liang, X.; Xiong, K.; Tsoyi, K.; Ith, B.; Ayaub, E.A.; Tatituri, R.V.; Byers, D.E.; Hsu, F.F.; et al. Palmitic Acid-Rich High-Fat Diet Exacerbates Experimental Pulmonary Fibrosis by Modulating Endoplasmic Reticulum Stress. Am. J. Respir. Cell Mol. Biol. 2019, 61, 737–746.
- Mercader-Barceló, J.; Truyols-Vives, J.; Río, C.; López-Safont, N.; Sala-Llinàs, E.; Chaplin, A. Insights into the Role of Bioactive Food Ingredients and the Microbiome in Idiopathic Pulmonary Fibrosis. Int J. Mol. Sci 2020, 21, 6051.
- Vedova, M.C.D.; Soler Garcia, F.M.; Muñoz, M.D.; Fornes, M.W.; Gomez Mejiba, S.E.; Gómez, N.N.; Ramirez, D.C. Diet-Induced Pulmonary Inflammation and Incipient Fibrosis in Mice: A Possible Role of Neutrophilic Inflammation. Inflammation 2019, 42, 1886–1900.
- Ge, X.N.; Greenberg, Y.; Hosseinkhani, M.R.; Long, E.K.; Bahaie, N.S.; Rao, A.; Ha, S.G.; Rao, S.P.; Bernlohr, D.A.; Sriramarao, P. High-fat diet promotes lung fibrosis and attenuates airway eosinophilia after exposure to cockroach allergen in mice. Exp. Lung. Res. 2013, 39, 365–378.
- Chen, J.; Zeng, T.; Zhao, X.; Xiea, K.; Bi, Y.; Zhong, Z.; Zhao, X. Docosahexaenoic acid (DHA) ameliorates paraquat-induced pulmonary fibrosis in rats possibly through up-regulation of Smad 7 and SnoN. Food Chem. Toxicol. 2013, 57, 330–337.
- Kim, J.W.; Rhee, C.K.; Kim, T.J.; Kim, Y.H.; Lee, S.H.; Yoon, H.K.; Kim, S.C.; Lee, S.Y.; Kwon, S.S.; Kim, K.H.; et al. Effect of pravastatin on bleomycin-induced acute lung injury and pulmonary fibrosis. Clin. Exp. Pharmacol. Physiol. 2010, 37, 1055–1063.
- Shi, X.; Chen, Y.; Liu, Q.; Mei, X.; Liu, J.; Tang, Y.; Luo, R.; Sun, D.; Ma, Y.; Wu, W.; et al. LDLR dysfunction induces LDL accumulation and promotes pulmonary fibrosis. Clin. Transl. Med. 2022, 12, e711.
- Kreuter, M.; Bonella, F.; Maher, T.M.; Costabel, U.; Spagnolo, P.; Weycker, D.; Kirchgaessler, K.U.; Kolb, M. Effect of statins on disease-related outcomes in patients with idiopathic pulmonary fibrosis. Thorax 2017, 72, 148–153.
- Patel, A.S.; Song, J.W.; Chu, S.G.; Mizumura, K.; Osorio, J.C.; Shi, Y.; El-Chemaly, S.; Lee, C.G.; Rosas, I.O.; Elias, J.A.; et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS ONE 2015, 10, e0121246.
- Bueno, M.; Lai, Y.C.; Romero, Y.; Brands, J.; St Croix, C.M.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538.
- Uchida, N.; Shimizu, Y.; Fujimaki, M.; Horibata, Y.; Nakamura, Y.; Horigane, Y.; Chibana, K.; Takemasa, A.; Sugimoto, H.; Niho, S. Metabolic changes induced by TGF-β1 via reduced expression of phosphatidylserine decarboxylase during myofibroblast transition. J. Clin. Biochem. Nutr. 2022, 70, 108–116.
- El Agha, E.; Moiseenko, A.; Kheirollahi, V.; De Langhe, S.; Crnkovic, S.; Kwapiszewska, G.; Szibor, M.; Kosanovic, D.; Schwind, F.; Schermuly, R.T.; et al. Two-Way Conversion between Lipogenic and Myogenic Fibroblastic Phenotypes Marks the Progression and Resolution of Lung Fibrosis. Cell Stem Cell 2017, 20, 261–273.e3.
- Kheirollahi, V.; Wasnick, R.M.; Biasin, V.; Vazquez-Armendariz, A.I.; Chu, X.; Moiseenko, A.; Weiss, A.; Wilhelm, J.; Zhang, J.S.; Kwapiszewska, G.; et al. Metformin induces lipogenic differentiation in myofibroblasts to reverse lung fibrosis. Nat. Commun. 2019, 10, 2987.
- Nakamura, Y.; Shimizu, Y. Induced Pluripotent Stem Cells for Regenerative Medicine: Quality Control Based on Evaluation of Lipid Composition. Adv. Exp. Med. Biol. 2020, 1212, 49–56.
- Chambers, D.C.; Enever, D.; Ilic, N.; Sparks, L.; Whitelaw, K.; Ayres, J.; Yerkovich, S.T.; Khalil, D.; Atkinson, K.M.; Hopkins, P.M. A phase 1b study of placenta-derived mesenchymal stromal cells in patients with idiopathic pulmonary fibrosis. Respirology 2014, 19, 1013–1018.
- Glassberg, M.K.; Minkiewicz, J.; Toonkel, R.L.; Simonet, E.S.; Rubio, G.A.; DiFede, D.; Shafazand, S.; Khan, A.; Pujol, M.V.; LaRussa, V.F.; et al. Allogeneic Human Mesenchymal Stem Cells in Patients with Idiopathic Pulmonary Fibrosis via Intravenous Delivery (AETHER): A Phase I Safety Clinical Trial. Chest 2017, 151, 971–981.
- Ntolios, P.; Manoloudi, E.; Tzouvelekis, A.; Bouros, E.; Steiropoulos, P.; Anevlavis, S.; Bouros, D.; Froudarakis, M.E. Longitudinal outcomes of patients enrolled in a phase Ib clinical trial of the adipose-derived stromal cells-stromal vascular fraction in idiopathic pulmonary fibrosis. Clin. Respir. J. 2018, 12, 2084–2089.
- Tzouvelekis, A.; Paspaliaris, V.; Koliakos, G.; Ntolios, P.; Bouros, E.; Oikonomou, A.; Zissimopoulos, A.; Boussios, N.; Dardzinski, B.; Gritzalis, D.; et al. A prospective, non-randomized, no placebo-controlled, phase Ib clinical trial to study the safety of the adipose derived stromal cells-stromal vascular fraction in idiopathic pulmonary fibrosis. J. Transl. Med. 2013, 11, 171.
- Zhou, Y.; He, Z.; Gao, Y.; Zheng, R.; Zhang, X.; Zhao, L.; Tan, M. Induced Pluripotent Stem Cells Inhibit Bleomycin-Induced Pulmonary Fibrosis in Mice through Suppressing TGF-β1/Smad-Mediated Epithelial to Mesenchymal Transition. Front. Pharmacol. 2016, 7, 430.
- How, C.K.; Chien, Y.; Yang, K.Y.; Shih, H.C.; Juan, C.C.; Yang, Y.P.; Chiou, G.Y.; Huang, P.I.; Chang, Y.L.; Chen, L.K.; et al. Induced pluripotent stem cells mediate the release of interferon gamma-induced protein 10 and alleviate bleomycin-induced lung inflammation and fibrosis. Shock 2013, 39, 261–270.
- Serrano-Mollar, A.; Gay-Jordi, G.; Guillamat-Prats, R.; Closa, D.; Hernandez-Gonzalez, F.; Marin, P.; Burgos, F.; Martorell, J.; Sánchez, M.; Arguis, P.; et al. Safety and Tolerability of Alveolar Type II Cell Transplantation in Idiopathic Pulmonary Fibrosis. Chest 2016, 150, 533–543.
- Zhou, Q.; Ye, X.; Sun, R.; Matsumoto, Y.; Moriyama, M.; Asano, Y.; Ajioka, Y.; Saijo, Y. Differentiation of mouse induced pluripotent stem cells into alveolar epithelial cells in vitro for use in vivo. Stem Cells Transl. Med. 2014, 3, 675–685.
- Averyanov, A.; Koroleva, I.; Konoplyannikov, M.; Revkova, V.; Lesnyak, V.; Kalsin, V.; Danilevskaya, O.; Nikitin, A.; Sotnikova, A.; Kotova, S.; et al. First-in-human high-cumulative-dose stem cell therapy in idiopathic pulmonary fibrosis with rapid lung function decline. Stem Cells Transl. Med. 2020, 9, 6–16.
- Shimizu, Y.; Satou, M.; Hayashi, K.; Nakamura, Y.; Fujimaki, M.; Horibata, Y.; Ando, H.; Watanabe, T.; Shiobara, T.; Chibana, K.; et al. Matrix-assisted laser desorption/ionization imaging mass spectrometry reveals changes of phospholipid distribution in induced pluripotent stem cell colony differentiation. Anal. Bioanal. Chem. 2017, 409, 1007–1016.
- Cao, Z.; Lis, R.; Ginsberg, M.; Chavez, D.; Shido, K.; Rabbany, S.Y.; Fong, G.H.; Sakmar, T.P.; Rafii, S.; Ding, B.S. Targeting of the pulmonary capillary vascular niche promotes lung alveolar repair and ameliorates fibrosis. Nat. Med. 2016, 22, 154–162.
- Ebina, M. Pathognomonic remodeling of blood and lymphatic capillaries in idiopathic pulmonary fibrosis. Respir. Investig. 2017, 55, 2–9.
- Hanumegowda, C.; Farkas, L.; Kolb, M. Angiogenesis in pulmonary fibrosis: Too much or not enough? Chest 2012, 142, 200–207.
- Nakamura, Y.; Shimizu, Y.; Horibata, Y.; Tei, R.; Koike, R.; Masawa, M.; Watanabe, T.; Shiobara, T.; Arai, R.; Chibana, K.; et al. Changes of plasmalogen phospholipid levels during differentiation of induced pluripotent stem cells 409B2 to endothelial phenotype cells. Sci. Rep. 2017, 7, 9377.
- Shimizu, Y.; Nakamura, Y.; Horibata, Y.; Fujimaki, M.; Hayashi, K.; Uchida, N.; Morita, H.; Arai, R.; Chibana, K.; Takemasa, A.; et al. Imaging of lysophosphatidylcholine in an induced pluripotent stem cell-derived endothelial cell network. Regen Ther. 2020, 14, 299–305.
- Tager, A.M. Autotaxin emerges as a therapeutic target for idiopathic pulmonary fibrosis: Limiting fibrosis by limiting lysophosphatidic acid synthesis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 563–565.
- Arisz, S.A.; Munnik, T. The salt stress-induced LPA response in Chlamydomonas is produced via PLA₂ hydrolysis of DGK-generated phosphatidic acid. J. Lipid Res. 2011, 52, 2012–2020.
- Law, S.H.; Chan, M.L.; Marathe, G.K.; Parveen, F.; Chen, C.H.; Ke, L.Y. An Updated Review of Lysophosphatidylcholine Metabolism in Human Diseases. Int. J. Mol. Sci. 2019, 20, 1149.
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442.
- Oikonomou, N.; Mouratis, M.A.; Tzouvelekis, A.; Kaffe, E.; Valavanis, C.; Vilaras, G.; Karameris, A.; Prestwich, G.D.; Bouros, D.; Aidinis, V. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol 2012, 47, 566–574.
- Kuhn, C., 3rd; Boldt, J.; King, T.E., Jr.; Crouch, E.; Vartio, T.; McDonald, J.A. An immunohistochemical study of architectural remodeling and connective tissue synthesis in pulmonary fibrosis. Am. Rev. Respir. Dis 1989, 140, 1693–1703.
- Behr, J.; Adelmann-Grill, B.C.; Krombach, F.; Beinert, T.; Schwaiblmair, M.; Fruhmann, G. Fibroblast chemotactic response elicited by native bronchoalveolar lavage fluid from patients with fibrosing alveolitis. Thorax 1993, 48, 736–742.
- Selman, M.; Carrillo, G.; Estrada, A.; Mejia, M.; Becerril, C.; Cisneros, J.; Gaxiola, M.; Pérez-Padilla, R.; Navarro, C.; Richards, T.; et al. Accelerated variant of idiopathic pulmonary fibrosis: Clinical behavior and gene expression pattern. PLoS ONE 2007, 2, e482.
- Plataki, M.; Koutsopoulos, A.V.; Darivianaki, K.; Delides, G.; Siafakas, N.M.; Bouros, D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest 2005, 127, 266–274.
- Funke, M.; Zhao, Z.; Xu, Y.; Chun, J.; Tager, A.M. The lysophosphatidic acid receptor LPA1 promotes epithelial cell apoptosis after lung injury. Am. J. Respir. Cell Mol. Biol 2012, 46, 355–364.
- Lee, G.H.; Cheon, J.; Kim, D.; Jun, H.S. Lysophosphatidic Acid Promotes Epithelial-Mesenchymal Transition in Kidney Epithelial Cells via the LPAR1/MAPK-AKT/KLF5 Signaling Pathway in Diabetic Nephropathy. Int. J. Mol. Sci. 2022, 23, 10497.
- Richeldi, L.; Fernández Pérez, E.R.; Costabel, U.; Albera, C.; Lederer, D.J.; Flaherty, K.R.; Ettinger, N.; Perez, R.; Scholand, M.B.; Goldin, J.; et al. Pamrevlumab, an anti-connective tissue growth factor therapy, for idiopathic pulmonary fibrosis (PRAISE): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Respir. Med. 2020, 8, 25–33.
- Huang, L.S.; Fu, P.; Patel, P.; Harijith, A.; Sun, T.; Zhao, Y.; Garcia, J.G.; Chun, J.; Natarajan, V. Lysophosphatidic acid receptor-2 deficiency confers protection against bleomycin-induced lung injury and fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 2013, 49, 912–922.
- Xu, M.Y.; Porte, J.; Knox, A.J.; Weinreb, P.H.; Maher, T.M.; Violette, S.M.; McAnulty, R.J.; Sheppard, D.; Jenkins, G. Lysophosphatidic acid induces alphavbeta6 integrin-mediated TGF-beta activation via the LPA2 receptor and the small G protein G alpha(q). Am. J. Pathol. 2009, 174, 1264–1279.
- Tanaka, M.; Okudaira, S.; Kishi, Y.; Ohkawa, R.; Iseki, S.; Ota, M.; Noji, S.; Yatomi, Y.; Aoki, J.; Arai, H. Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J. Biol. Chem. 2006, 281, 25822–25830.
- Palmer, S.M.; Snyder, L.; Todd, J.L.; Soule, B.; Christian, R.; Anstrom, K.; Luo, Y.; Gagnon, R.; Rosen, G. Randomized, Double-Blind, Placebo-Controlled, Phase 2 Trial of BMS-986020, a Lysophosphatidic Acid Receptor Antagonist for the Treatment of Idiopathic Pulmonary Fibrosis. Chest 2018, 154, 1061–1069.
- Gill, M.W.; Murphy, B.J.; Cheng, P.T.W.; Sivaraman, L.; Davis, M.; Lehman-McKeeman, L. Mechanism of hepatobiliary toxicity of the LPA(1) antagonist BMS-986020 developed to treat idiopathic pulmonary fibrosis: Contrasts with BMS-986234 and BMS-986278. Toxicol. Appl. Pharmacol. 2022, 438, 115885.
- Cheng, P.T.W.; Kaltenbach, R.F., 3rd; Zhang, H.; Shi, J.; Tao, S.; Li, J.; Kennedy, L.J.; Walker, S.J.; Shi, Y.; Wang, Y.; et al. Discovery of an Oxycyclohexyl Acid Lysophosphatidic Acid Receptor 1 (LPA(1)) Antagonist BMS-986278 for the Treatment of Pulmonary Fibrotic Diseases. J. Med. Chem. 2021, 64, 15549–15581.
- Gill, M.W.; Lakshmi, S.; Cheng, P.T.W.; Murphy, B.J.; Chadwick, K.; Lehman-McKeeman, L.; Graziano, M. An LPA1 receptor antagonist for idiopathic pulmonary fibrosis: Preclinical assessments of potential hepatobiliary toxicity. Am. J. Respir. Crit. Care Med. 2019, 199, A5882.
- Tirucherai, G.S.; Revankar, D.Y.R.; Klinger, G.; van Lier, J.J.; Taubel, J.; Charles, E.D. BMS-986278, A Lysophosphatidic Acid 1 (LPA1) Receptor Antagonist, in Healthy Participants: A Single/Multiple Ascending Dose (SAD/MAD) and Japanese MAD (JMAD) Phase 1 Study. Am. J. Respir. Crit. Care Med. 2020, 201, A1492.
- Corte, T.J.; Lancaster, L.; Swigris, J.J.; Maher, T.M.; Goldin, J.G.; Palmer, S.M.; Suda, T.; Ogura, T.; Minnich, A.; Zhan, X.; et al. Phase 2 trial design of BMS-986278, a lysophosphatidic acid receptor 1 (LPA(1)) antagonist, in patients with idiopathic pulmonary fibrosis (IPF) or progressive fibrotic interstitial lung disease (PF-ILD). BMJ Open Respir. Res. 2021, 8, e001026.
- Maher, T.M.; van der Aar, E.M.; Van de Steen, O.; Allamassey, L.; Desrivot, J.; Dupont, S.; Fagard, L.; Ford, P.; Fieuw, A.; Wuyts, W. Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): A phase 2a randomised placebo-controlled trial. Lancet Respir. Med. 2018, 6, 627–635.
- van der Aar, E.; Desrivot, J.; Dupont, S.; Heckmann, B.; Fieuw, A.; Stutvoet, S.; Fagard, L.; Van de Wal, K.; Helmer, E. Safety, Pharmacokinetics, and Pharmacodynamics of the Autotaxin Inhibitor GLPG1690 in Healthy Subjects: Phase 1 Randomized Trials. J. Clin. Pharmaco.l 2019, 59, 1366–1378.
- Maher, T.M.; Kreuter, M.; Lederer, D.J.; Brown, K.K.; Wuyts, W.; Verbruggen, N.; Stutvoet, S.; Fieuw, A.; Ford, P.; Abi-Saab, W.; et al. Rationale, design and objectives of two phase III, randomised, placebo-controlled studies of GLPG1690, a novel autotaxin inhibitor, in idiopathic pulmonary fibrosis (ISABELA 1 and 2). BMJ Open Respir. Res. 2019, 6, e000422.
- Lee, G.; Kang, S.U.; Ryou, J.-H.; Lim, J.-J.; Lee, D.-Y.; Kwon, H.-J.; Ha, G.-H.; Lee, Y.-H. BBT-877, a Potent Autotaxin Inhibitor in Clinical Development to Treat Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, A2577.
More
Information
Subjects:
Respiratory System
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
3 times
(View History)
Update Date:
20 Feb 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No