Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | An Su | -- | 1799 | 2023-02-14 11:55:24 | | | |
| 2 | Peter Tang | Meta information modification | 1799 | 2023-02-15 02:37:35 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
He, C.; Zhang, C.; Bian, T.; Jiao, K.; Su, W.; Wu, K.; Su, A. Artificial Intelligence Enabled Chemical Process Intensification. Encyclopedia. Available online: https://encyclopedia.pub/entry/41208 (accessed on 21 July 2026).
He C, Zhang C, Bian T, Jiao K, Su W, Wu K, et al. Artificial Intelligence Enabled Chemical Process Intensification. Encyclopedia. Available at: https://encyclopedia.pub/entry/41208. Accessed July 21, 2026.
He, Chasheng, Chengwei Zhang, Tengfei Bian, Kaixuan Jiao, Weike Su, Ke-Jun Wu, An Su. "Artificial Intelligence Enabled Chemical Process Intensification" Encyclopedia, https://encyclopedia.pub/entry/41208 (accessed July 21, 2026).
He, C., Zhang, C., Bian, T., Jiao, K., Su, W., Wu, K., & Su, A. (2023, February 14). Artificial Intelligence Enabled Chemical Process Intensification. In Encyclopedia. https://encyclopedia.pub/entry/41208
He, Chasheng, et al. "Artificial Intelligence Enabled Chemical Process Intensification." Encyclopedia. Web. 14 February, 2023.
Copy Citation
An overview of the application of AI techniques is provided, in particular machine learning, in chemical design, synthesis, and process optimization over the past years. The application of AI for structure-function relationship analysis, synthetic route planning, and automated synthesis is highly highlighted.
artificial intelligence
machine learning
automated synthesis
synthetic route planning
structure-function relationship
1. AI for Structure-Function Relationship Analysis
1.1. Molecular Property Prediction
Molecular property prediction is an important problem in computer-aided molecular design, and excellent deep-learning models for molecular property prediction can greatly accelerate the progress of experimental studies. Two main types of models are prominent in molecular property prediction—graphical neural networks and sequence-based neural networks, which differ in their representation of different molecules, with the former requiring molecular graphical information and the latter requiring string representations of molecular structures (Figure 1) [1].
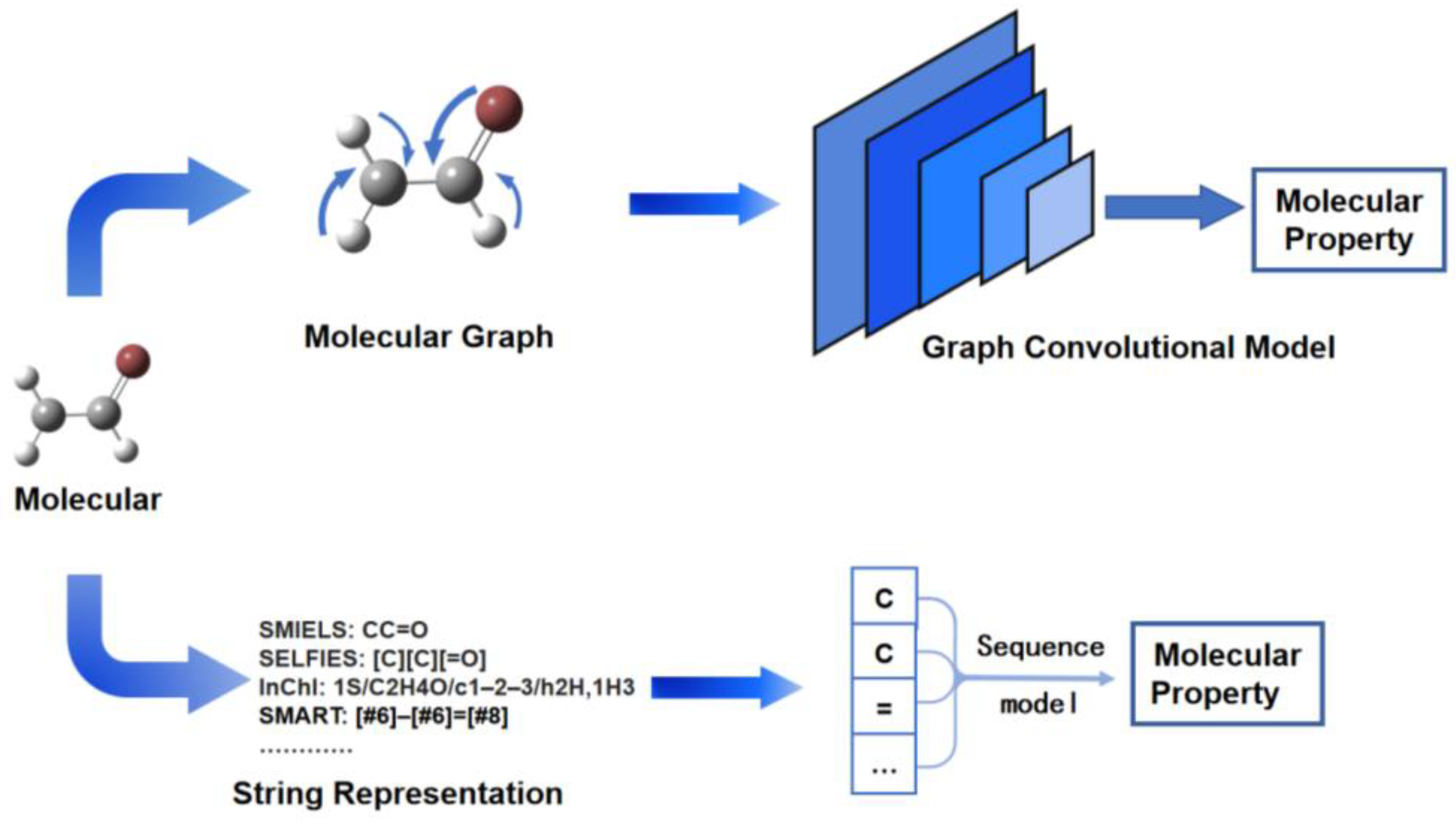
Figure 1. Molecular graph-based and sequence-based models in molecular property prediction.
The direct use of matrices to record molecular structure information is a widely used method of molecular representation known as molecular graphs. Molecular graphs can be trained using graph neural networks. Lu et al. reported the prediction of molecular properties using multilevel Graph Convolutional Neural Networks (MGCN). Different layers of convolutional layers learn the atomic feature information and chemical bond feature information of the molecule and then process the information to predict the molecular properties [2]. In QM9, the MGCN model gains a mean absolute error (MAE) of 0.0642 eV in the HOMO-LUMO gap. The model has excellent predictive performance with generalization capability. Gilmer et al. used a Message Passing Neural Network (MPNN) to predict the QM9 public data set and obtained better performance than any previous model [3][4]. The ratio of the MAE of the MPNN models to the provided chemical accuracy estimate was reported, with a HOMO-LUMO gap of 1.60 eV in QM9. In the framework of the MPNN model, the design of appropriate functions can effectively improve the prediction effect. The directed-MPNN model was used by Yang et al. for the extraction of molecular graph features and predicting the properties of molecules, and the model was tested on 19 public datasets and 16 industry datasets, and the model performance was better than previous models on most tasks [5]. Compared to other papers, the paper gives an MAE of 2.766 ± 0.022 for multi-task prediction of the QM9 database and provides more comparison of model performance.
The recording of molecules using strings is another mainstream molecular representation method, of which the most widely used is SMILES [6]. Deep learning models for natural language processing are well suited to process these sequences, which record molecular information. There is no more effective model for string processing in recent years than the Transformer [7]. Honda et al. reported the use of the Transformer for the prediction of molecular properties in 2019 [8]. Schwaller et al., on the other hand, applied the Transformer model to the prediction of reaction yields [9]. Chithrananda et al. then built several pre-trained models for chemical molecules using the BERT model, which allowed for a significant reduction in training time for later Transformer-based models [10]. Su et al. used these pre-trained models for a transfer learning study to predict the energy gap of metalloporphyrin, spending only one-third of the training time that would have been spent if transfer learning had not been used [11]. Jo et al., on the other hand, used MPNN for processing SMILES information, and the model obtained better results when performing classification tasks on multiple datasets [12]. The molecular graph-based models and sequence-based models, though both perform well in molecular property prediction tasks, have their own advantages. The molecular structure information recorded in molecular graphs is significantly richer than that of sequence methods, and the prediction of molecular properties will be more accurate. The use of sequences to record molecular information has high freedom and can reduce the training cost more easily using transfer learning methods. The two families of models should be selected according to the research content in the next study, or multimodal models can be used to combine their advantages.
1.2. Molecular Design
Computer-aided molecular design is another important research direction in cheminformatics, and the design of suitable molecules according to requirements has been a dream function for chemists [13]. Similar to molecular property prediction, both graph generation models and text generation models in the field of deep learning can be used for the molecular design (Figure 2).
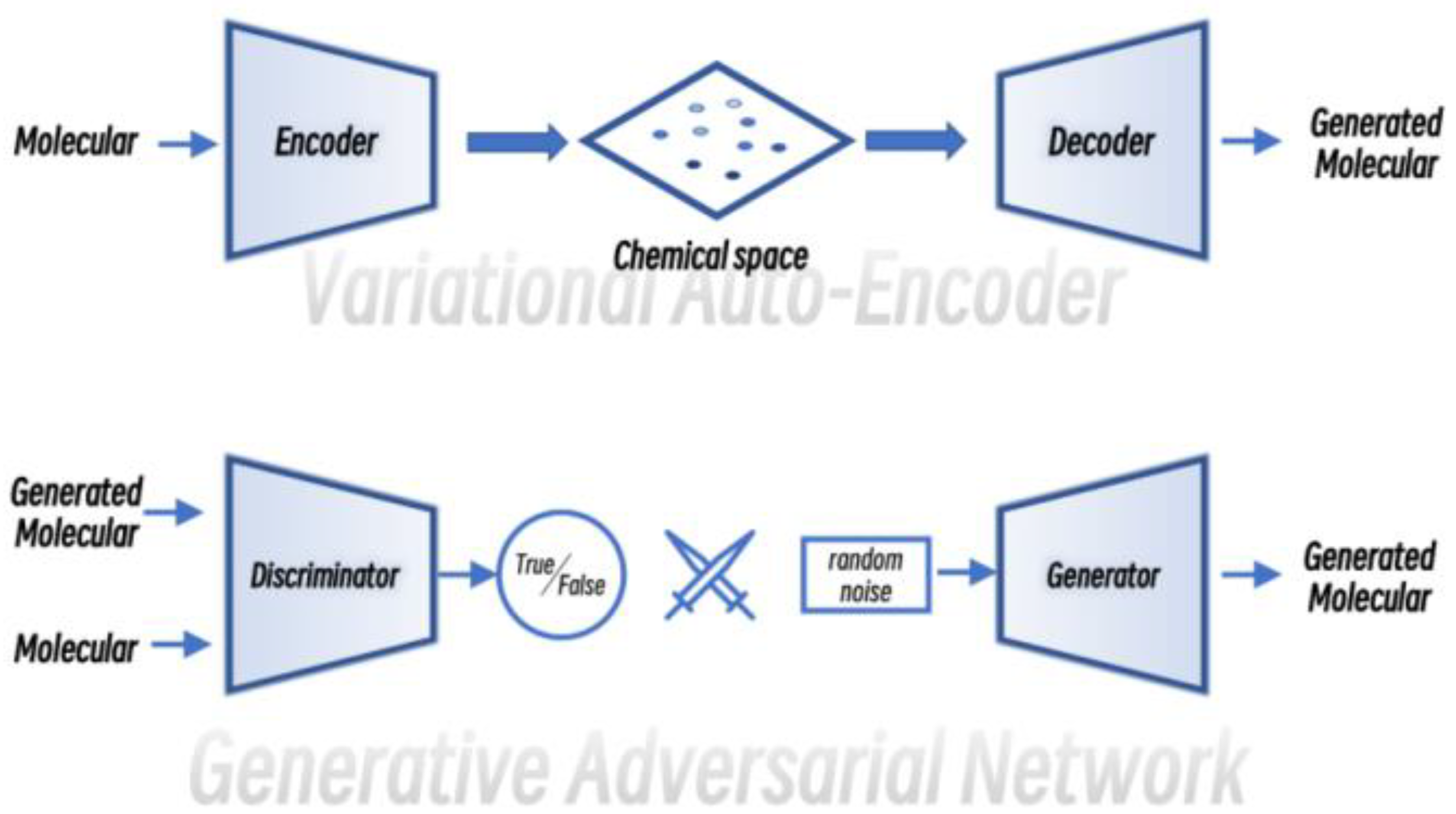
Figure 2. The schematics of Variational Auto-Encoder (VAE) and Generative Adversarial Network (GAN).
In 2018, Gómez-Bombarelli et al. reported the design of new molecules using Variational Auto-Encoder (VAE), a study that will perform molecule generation while mapping the encoded potential chemical space to the corresponding molecular properties, allowing the model to explore the chemical space more efficiently and purposefully [14]. Segler et al., on the other hand, applied recurrent neural networks based on Long Short-Term Memory (LSTM) for ab initio drug design [15]. In this model, transfer learning and reinforcement learning are introduced to improve the validity of the designed new molecules. In the same year, Cao et al. applied Generative Adversarial Network (GAN) to chemical molecule generation, and reinforcement learning also was introduced in the model to score the generated molecules in order to be able to generate molecules that meet the desired target [16]. Flam-Shepherd et al. added MPNN to the decoder and encoder of the VAE model, which greatly improved performance of the VAE model [17].
The two most difficult problems to overcome in computer-aided molecular design are the generation of legitimate chemical molecules and the generation of molecules with target properties or target characteristics, in other words, distribution learning for molecular design and goal-directed molecular optimization [13]. Comparing the performance of molecular design models is not a trivial task. Brown et al. 2019 proposed the GuacaMol platform, which gives different evaluation criteria for the two task models [18]. From current approaches, the use of transfer learning in a separate generative model can improve the chance of generating valid molecules. On the other hand, the development of novel molecular representation methods with greater robustness, such as SELFIES, can also be effective for the task of distribution-learning of molecular design [19]. In addition, in goal-directed molecular optimization with targets, when the design targets can be quickly computed by computer (e.g., LogP, TPSA, etc.), reinforcement learning can help the model to find the target molecules faster. Furthermore, when the desired property cannot be obtained by simple computation, the potential chemical space in the model can be mapped to the corresponding property before the molecule is designed.
For the design of new molecules, one of the important application areas of AI is interpretable machine learning [20]. For example, Verkhivker et al. developed and implemented interpretable machine learning models for the molecular design of Tyrosine Kinase Inhibitors by combining ChemVAE embedding architecture and cluster decomposition [21]. Recently, a computer-aided molecular design (CAMD) framework for molecular design has been reported. Hatamleh et al. developed a CAMD framework for mosquito repellents to mitigate the drawbacks of currently used repellents [22]. In this framework, a data-driven Hyperbox-based machine learning approach was used to predict the mosquito rejection properties of molecules in the absence of a mechanistic prediction model. Ooi et al. proposed a CAMD-based approach to design fragrance molecules and used a Hyperbox classifier to predict fragrance properties [23]. The resulting model can be interpreted as a parsing decision support rule that establishes a quantitative relationship between the structural parameters of a molecule and its odor characteristics. In addition, a novel data-driven rough set-based machine learning (RSML) model was used as a predictive or diagnostic modeling tool for odor properties to design fragrance molecules [24]. The RSML generates deterministic rules based on the relationship between the topology of fragrant molecules and the odor characteristics from existing odor databases. The generated rules are then integrated into CAMD problems as constraints. The results show that the new method is capable of identifying non-intuitive and promising fragrant molecules that can be used for various applications.
Moreover, in addition to molecular design, several fields are beginning to take advantage of the integration of ML and systems biology, including pathways identification and analysis, modeling of metabolisms and growth, and 3D protein modeling [25]. For example, AI is being used for the dynamic modeling of signaling networks, which helps to understand cellular pathways and facilitate drug discovery. It allows cataloging the changes in gene expression and signaling that occur when cells are exposed to various perturbations, building a network-based understanding of biology [26][27][28]. For example, in metabolic engineering, ML models, including naive Bayes, decision trees, and logistic regression trained on the pathway information of many organisms, were used in MetaCyc to predict the presence of a novel metabolic pathway in a newly-sequenced organism [25]. In general, the ML models used for pathway prediction showed better performance than standard mathematical and statistical methods. Nevertheless, pathway discovery still relies heavily on traditional approaches such as gene sequence similarity and network analysis. Therefore, better ML algorithms/methods for improving Dynamic and Constraint-based Metabolic Modelling, such as FBA modeling, are needed [25].
2. AI for Synthetic Route Planning
AI has been successful in planning synthetic routes performed in the laboratory or evaluated by chemists, including (1) retrosynthetic planning, (2) forward reaction prediction, and (3) condition recommendation. In chemistry, the origins of Computer-assisted synthesis planning (CASP) can be traced back to the translation of retrosynthetic logic into computer code by Corey in the 1960s [29]. Nevertheless, early synthetic route planning relied entirely on the expertise of chemists and did not use statistical learning based on large amounts of data [30][31][32]. Given the limitation of computational resources, complex algorithms cannot be widely used in synthetic planning. Fortunately, with the growing availability of molecular property datasets, reaction datasets, and increased computational power, AI for synthetic planning is once again gaining widespread attention [33][34][35][36][37]. In the last 20 years, patterns of reactivity inferred from published response data by AI have become viable alternatives to algorithms based on “expert” rules. It can automate the extraction and training of data, making it easily scalable to merge new responses, which eases the burden on scientists. Today, the retrosynthesis of complex molecules, high-fidelity prediction of reaction outcomes, and automation of chemical reactions are still major research fields.
3. AI for Automated Synthesis
Applications of AI in chemical reactions include not only synthetic route planning but also automated synthesis. Traditionally, scientists have been exposed to hazardous, repetitive chemical manipulations for long periods, resulting in a significant waste of resources and time [38][39]. Additionally, cost and condition constraints prevent scientists from conducting too many experiments to obtain desired results. Most importantly, traditional chemical synthesis relies heavily on labor-intensive practices such as scientific training, planning, experience, observation, and interpretation. Fortunately, AI is changing the productivity of modern manufacturing, and modern automation of organic chemistry operations is gradually freeing the hands and minds of organic chemists [40][41][42]. For example, with an auto mated platform, the anti-arrhythmic drug lidocaine, the anti-epileptic drug rufinamide, and the anti-cardiovascular drug sildenafil have been synthesized automatically without human intervention [43][44]. Exactly, AI alleviates the operator from tedious work and manual intervention.
References
- Venkatasubramanian, V.; Mann, V. Artificial intelligence in reaction prediction and chemical synthesis. Curr. Opin. Chem. Eng. 2022, 36, 100749.
- Lu, C.; Liu, Q.; Wang, C.; Huang, Z.; Lin, P.; He, L. Molecular property prediction: A multilevel quantum interactions modeling perspective. In Proceedings of the AAAI Conference on Artificial Intelligence, Honolulu, HI, USA, 27 January–1 February 2019; pp. 1052–1060.
- Gilmer, J.; Schoenholz, S.S.; Riley, P.F.; Vinyals, O.; Dahl, G.E. Neural Message Passing for Quantum Chemistry. In Proceedings of the 34th International Conference on Machine Learning, Proceedings of Machine Learning Research, Sydney, NSW, Australia, 6–11 August 2017; pp. 1263–1272.
- Blomberg, M.R.A.; Borowski, T.; Himo, F.; Liao, R.-Z.; Siegbahn, P.E.M. Quantum Chemical Studies of Mechanisms for Metalloenzymes. Chem. Rev. 2014, 114, 3601–3658.
- Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M.; et al. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388.
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 1988, 28, 31–36.
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. arXiv 2017.
- Honda, S.; Shi, S.; Hiroki, R. Ueda. SMILES Transformer: Pre-trained Molecular Fingerprint for Low Data Drug Discovery. arXiv 2019, arXiv:1911.04738.
- Schwaller, P.; Vaucher, A.C.; Laino, T.; Reymond, J.-L. Prediction of chemical reaction yields using deep learning. Mach. Learn. Sci. Technol. 2021, 2, 015016.
- Chithrananda, S.; Grand, G.; Ramsundar, B. Chemberta: Large-scale self- supervised pretraining for molecular property prediction. arXiv 2020, arXiv:2010.09885.
- Su, A.; Zhang, C.; She, Y.-B.; Yang, Y.-F. Exploring Deep Learning for Metalloporphyrins: Databases, Molecular Representations, and Model Architectures. Catalysts 2022, 12, 1485.
- Jo, J.; Kwak, B.; Choi, H.-S.; Yoon, S. The message passing neural networks for chemical property prediction on SMILES. Methods 2020, 179, 65–72.
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Ad-vances in De Novo Drug Design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci. 2021, 22, 1676.
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.K.; Hernandez-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Central Sci. 2018, 4, 268–276.
- Segler, M.H.S.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Central Sci. 2017, 4, 120–131.
- De Cao, N.; Kipf, T. MolGAN: An implicit generative model for small molecular graphs. arXiv 2018, arXiv:1805.11973.
- Flam-Shepherd, D.; Wu, T.; Aspuru-Guzik, A. Graph deconvolutional generation. arXiv 2020, arXiv:2002.07087.
- Brown, N.; Fiscato, M.; Segler, M.H.; Vaucher, A.C. GuacaMol: Benchmarking Models for de Novo Mo-lecular Design. J. Chem. Inf. Model. 2019, 59, 1096–1108.
- Krenn, M.; Hase, F.; Nigam, A.K.; Friederich, P.; Aspuru-Guzik, A. Self- referencing embedded strings (SELFIES): A 100% robust molecular string representation. Mach. Learn. Sci. Technol. 2020, 1, 045024.
- Dybowski, R. Interpretable machine learning as a tool for scientific discovery in chemistry. New J. Chem. 2020, 44, 20914–20920.
- Krishnan, K.; Kassab, R.; Agajanian, S.; Verkhivker, G. Interpretable Machine Learning Models for Molecular Design of Ty-rosine Kinase Inhibitors Using Variational Autoencoders and Perturbation-Based Approach of Chemical Space Exploration. Int. J. Mol. Sci. 2022, 23, 11262.
- Hatamleh, M.; Chong, J.W.; Tan, R.R.; Aviso, K.B.; Janairo, J.I.B.; Chemmangattuvalappil, N.G. Design of mosquito repellent molecules via the integration of hyperbox machine learning and computer aided molecular design. Digit. Chem. Eng. 2022, 3, 100018.
- Ooi, Y.J.; Aung, K.N.G.; Chong, J.W.; Tan, R.R.; Aviso, K.B.; Chemmangattuvalappil, N.G. Design of fragrance molecules using computer-aided molecular design with machine learning. Comput. Chem. Eng. 2021, 157, 107585.
- Radhakrishnapany, K.T.; Wong, C.Y.; Tan, F.K.; Chong, J.W.; Tan, R.R.; Aviso, K.B.; Janairo, J.I.B.; Chemmangattuvalappil, N.G. Design of fragrant molecules through the incorporation of rough sets into computer-aided molecular design. Mol. Syst. Des. Eng. 2020, 5, 1391–1416.
- Helmy, M.; Smith, D.; Selvarajoo, K. Systems biology approaches integrated with artificial intelligence for optimized food-focused metabolic engineering. Metab. Eng. Commun. 2020, 11, e00149.
- Ji, Z.; Su, J.; Liu, C.; Wang, H.; Huang, D.; Zhou, X. Integrating Genomics and Proteomics Data to Predict Drug Effects Using Binary Linear Programming. PLoS ONE 2014, 9, e102798.
- Ji, Z.; Wu, D.; Zhao, W.; Peng, H.; Zhao, S.; Huang, D.; Zhou, X. Systemic modeling myeloma-osteoclast interactions under normoxic/hypoxic condition using a novel computational approach. Sci. Rep. 2015, 5, 13291.
- Peng, H.; Zhao, W.; Tan, H.; Ji, Z.; Li, J.; Li, K.; Zhou, X. Prediction of treatment efficacy for prostate cancer using a mathematical model. Sci. Rep. 2016, 6, 21599.
- Corey, E.J.; Wipke, W.T. Computer-Assisted Design of Complex Organic Syntheses. Science 1969, 166, 178–192.
- Cook, A.; Johnson, A.P.; Law, J.; Mirzazadeh, M.; Ravitz, O.; Simon, A. Computer-aided synthesis design: 40 years on. WIREs Comput. Mol. Sci. 2011, 2, 79–107.
- Ihlenfeldt, W.-D.; Gasteiger, J. Computer-Assisted Planning of Organic Syntheses: The Second Generation of Programs. Angew. Chem. Int. Ed. 1996, 34, 2613–2633.
- Todd, M.H. Computer-aided organic synthesis. Chem. Soc. Rev. 2005, 34, 247–266.
- Ruddigkeit, L.; van Deursen, R.; Blum, L.C.; Reymond, J.-L. Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875.
- Davies, I.W. The digitization of organic synthesis. Nature 2019, 570, 175–181.
- Coley, C.W.; Eyke, N.S.; Jensen, K.F. Autonomous Discovery in the Chemical Sciences Part I: Progress. Angew. Chem. Int. Ed. 2020, 59, 22858–22893.
- Coley, C.W.; Eyke, N.S.; Jensen, K.F. Autonomous Discovery in the Chemical Sciences Part II: Outlook. Angew. Chem. Int. Ed. 2019, 59, 23414–23436.
- Shen, Y.; Borowski, J.E.; Hardy, M.A.; Sarpong, R.; Doyle, A.G.; Cernak, T. Automation and computer-assisted planning for chemical synthesis. Nat. Rev. Methods Prim. 2021, 1, 23.
- Ley, S.V.; Fitzpatrick, D.E.; Ingham, R.J.; Myers, R.M. Organic Synthesis: March of the Machines. Angew. Chem. Int. Ed. 2015, 54, 3449–3464.
- Ley, S.V.; Fitzpatrick, D.E.; Myers, R.M.; Battilocchio, C.; Ingham, R.J. Machine-Assisted Organic Synthesis. Angew. Chem. Int. Ed. 2015, 54, 10122–10136.
- Ahneman, D.T.; Estrada, J.G.; Lin, S.; Dreher, S.D.; Doyle, A.G. Predicting reaction performance in C–N cross-coupling using machine learning. Science 2018, 360, 186–190.
- Li, J.; Ballmer, S.G.; Gillis, E.P.; Fujii, S.; Schmidt, M.J.; Palazzolo, A.M.E.; Lehmann, J.W.; Morehouse, G.F.; Burke, M.D. Synthesis of many different types of organic small molecules using one automated process. Science 2015, 347, 1221–1226.
- Chatterjee, S.; Guidi, M.; Seeberger, P.H.; Gilmore, K. Automated radial synthesis of organic molecules. Nature 2020, 579, 379–384.
- Mehr, S.H.M.; Craven, M.; Leonov, A.I.; Keenan, G.; Cronin, L. A universal system for digitization and automatic execution of the chemical synthesis literature. Science 2020, 370, 101–108.
- Steiner, S.; Wolf, J.; Glatzel, S.; Andreou, A.; Granda, J.M.; Keenan, G.; Hinkley, T.; Aragon-Camarasa, G.; Kitson, P.J.; Angelone, D.; et al. Organic synthesis in a modular robotic system driven by a chemical programming language. Science 2019, 363, eaav2211.
More
Information
Subjects:
Engineering, Chemical
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.8K
Revisions:
2 times
(View History)
Update Date:
15 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No