Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Charles Lefèvre | -- | 1933 | 2023-02-14 10:59:13 | | | |
| 2 | Rita Xu | Meta information modification | 1933 | 2023-02-15 02:38:36 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lefèvre, C.R.; Labarthe, F.; Dufour, D.; Moreau, C.; Faoucher, M.; Rollier, P.; Arnoux, J.; Tardieu, M.; Damaj, L.; Bendavid, C.; et al. Primary Carnitine Deficiency Newborn Screening. Encyclopedia. Available online: https://encyclopedia.pub/entry/41203 (accessed on 08 August 2026).
Lefèvre CR, Labarthe F, Dufour D, Moreau C, Faoucher M, Rollier P, et al. Primary Carnitine Deficiency Newborn Screening. Encyclopedia. Available at: https://encyclopedia.pub/entry/41203. Accessed August 08, 2026.
Lefèvre, Charles R., François Labarthe, Diane Dufour, Caroline Moreau, Marie Faoucher, Paul Rollier, Jean-Baptiste Arnoux, Marine Tardieu, Léna Damaj, Claude Bendavid, et al. "Primary Carnitine Deficiency Newborn Screening" Encyclopedia, https://encyclopedia.pub/entry/41203 (accessed August 08, 2026).
Lefèvre, C.R., Labarthe, F., Dufour, D., Moreau, C., Faoucher, M., Rollier, P., Arnoux, J., Tardieu, M., Damaj, L., Bendavid, C., Dessein, A., Acquaviva-Bourdain, C., & Cheillan, D. (2023, February 14). Primary Carnitine Deficiency Newborn Screening. In Encyclopedia. https://encyclopedia.pub/entry/41203
Lefèvre, Charles R., et al. "Primary Carnitine Deficiency Newborn Screening." Encyclopedia. Web. 14 February, 2023.
Copy Citation
Primary Carnitine Deficiency (PCD) is a fatty acid oxidation disorder that will be included in the expansion of the French newborn screening (NBS) program at the beginning of 2023.
primary carnitine deficiency
CDSP
PCD
newborn screening
1. Introduction
Primary Carnitine Deficiency (PCD) (OMIM #212140)—also referred to as systemic primary carnitine deficiency (CDSP), carnitine transporter defect (CTD), or carnitine uptake deficiency (CUD)—is an autosomal recessive inborn error of metabolism involving a disorder of the carnitine cycle. It is a part of fatty acid oxidation (FAO) disorders and is caused by a partial or complete loss of function of the membrane transporter organic cation/carnitine transporter novel 2 (OCTN2). This solute carrier is coded by the SLC22A5 gene, comprising 10 exons, located approximately on a 26 kb region on chromosome 5q31.1 (chr5:132,369,710–132,395,612) [1][2]. This sodium-dependent carnitine symporter is the main carnitine (3-hydroxy-4-(trimethylazaniumyl)butanoate) transporter in mammals, displaying a high affinity for carnitine (KM = 4.3 µmol·L−1) [3]. OCTN2 is ubiquitous, with expression predominantly in kidney and intestinal cells, to ensure absorption and reabsorption of L-carnitine, and in skeletal muscles, to allow the shuttling of long chain fatty acids across inner mitochondrial membranes toward the fatty acid oxidation process [4]. Carnitine is almost exclusively intracellular (>99% of the total pool), with high tissue concentrations [5]. Carnitine homeostasis is balanced by dietary intake, endogenous biosynthesis, and especially by renal reabsorption. There are compensatory mechanisms, thus even a poor carnitine diet or a defect in carnitine biosynthesis does not affect FAO [6][7]. However, when OCTN2 function is impaired, a major urinary leak of free carnitine leads to a progressively significant decrease in both intracellular and circulating carnitine concentrations, resulting in PCD. Clinical characteristics of PCD encompass a broad clinical spectrum and have been widely assessed in high quality reviews [8][9][10][11]. In absence of newborn screening (NBS), patients usually present in their infancy: acute metabolic decompensation with hypoketotic hypoglycemia; dilated cardiomyopathy; and hepatic cytolysis. Without L-carnitine treatment, death can occur due to heart failure. Fortunately, PCD has an excellent prognosis upon L-carnitine supplementation and almost all patients remain asymptomatic [11]. Incidence of primary carnitine deficiency was quite variable depending on the studied population, ranging from 1:300 in the Faroe Islands [12] where there was a founding mutation, to 1:30-142,000 in Japan, Australia, or USA [13][14][15]. Regarding the incidence, the knowledge of this disease’s natural history, and the availability of a safe and efficient treatment, PCD follows consolidated principles for newborn screening [16], especially as free carnitine (C0) represents an easily measurable biomarker on dried blood spot (DBS) [17]. New South Wales (Australia) was the first state to evaluate PCD newborn screening in the late 1990s [18], and this was usually conducted by expanded newborn screening programs deployed since then [19][20][21]. Nevertheless, screening of primary carnitine deficiency is not simple, due to various secondary carnitine deficiencies that may generate false-positives (e.g., maternal carnitine deficiency, organic acidurias, pivalic acid-based antibiotherapy, pre-term birth, etc.) which represent pitfalls for the diagnosis and management of newborn PCD. Consequently, several algorithms for screening have been proposed, which include: different thresholds for C0 and other biomarkers; molecular sequencing of SLC22A5; and functional confirmation by carnitine uptake assay on skin fibroblasts.
2. Worldwide Overview of Primary Carnitine Deficiency Newborn Screening
2.1. Countries/Regions Screening PCD
To evaluate the extent of PCD NBS worldwide, researchers have screened national NBS programs and the literature for countries/regions that have implemented this condition. Actual NBS programs including PCD and excluding PCD are represented in Figure 1.
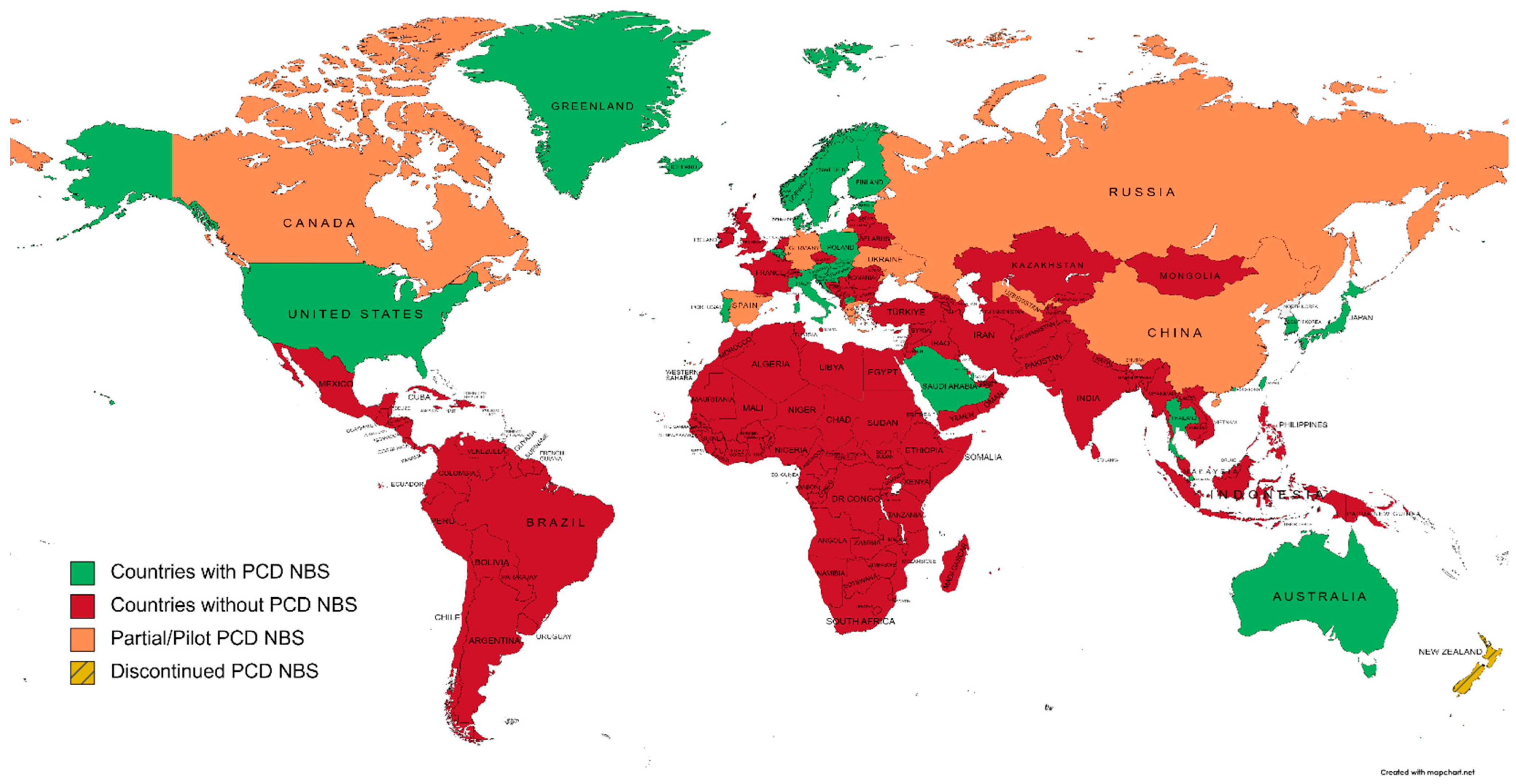
Figure 1. A world map of primary carnitine deficiency newborn screening programs, according to national NBS societies and the literature (created on mapchart.net, accessed on 27 October 2022).
2.1.1. Australia and New Zealand
Australia was the first country to include PCD in NBS [18], in 1998. The cut-off for low free carnitine to trigger a retest during screening was set to 10 µmol·L−1. A confirmed level of C0 < 5 µmol·L−1 generated a second sample, and PCD diagnosis was confirmed through OCTN2 activity on fibroblasts. To date, PCD NBS is performed nationwide [22].
New Zealand implemented PCD to ENBS in 2006, with a screen-positive level of C0 of 5 µmol·L−1 and molecular confirmation [23]. However, due to the low incidence (two cases in ten years; 1:300,000 births); the prevalence of asymptomatic patients; and the impact of diagnosing more mothers with PCD than newborns, PCD screening was considered unsuitable for NBS and was therefore discontinued [24].
2.1.2. North America
North America has almost a full coverage of PCD screening. Newborn screening has been nationally organized since mid-1980s under the aegis of the Council of Regional Networks for Genetic Services (CORN) [25]. In 2006, the American College of Medical Genetics (ACMG) provided guidelines to promote a standardized and uniform newborn screening program [26]. These guidelines had a substantial impact on perinatal healthcare through early identification and treatment of inborn errors of metabolism, including PCD, reducing morbidity and mortality [20][27][28]. Consequently, the USA has a solid nationwide background on NBS, its follow-up and outcomes [29], enhanced by standardization tools and programs [30].
English-speaking Canadian regions follow the ACMG’s guidelines [31][32][33], whereas French-Canadian provinces do not screen DBS for PCD even though they provide an interesting urinary NBS for organic acidurias, urea cycle disorders, cystinuria, homocystinuria, and creatine synthesis and transport disorders [33].
2.1.3. Central and South America
Mexico and South America started NBS in mid-1970s with PKU and struggled to achieve a whole coverage of population since then, with the exception of Cuba, Costa Rica, Chile, and Uruguay, where around 100% coverage was reached [34][35]. Difficulties encountered were mostly due to the lack of financial resources and fundings for free national NBS programs (especially regarding the high cost of tandem MS equipment), and the high prevalence of major, priority health issues, such as malnutrition and infectious diseases. As a result, most Central and South American countries have NBS programs, but to date, none of them include PCD, and only Costa Rica and Uruguay have set up a tandem MS NBS program [36]. It is to be noted that French Guyana and French Polynesia follow French guidelines for NBS.
2.1.4. Europe
PCD NBS in Europe is heterogeneous. Germany was the first country to include PCD to their program in a pilot study between 1998 and 2001 [37]. Biomarkers used for the screening were C0 < 10 µmol·L−1 and total acylcarnitines (C3 to C18) of <10 µmol·L−1. Confirmation was biochemically deduced by determining OCTN2 activity on fibroblasts. In 2007, only Austria, Belgium, Denmark, and Poland had PCD on their ENBS panel [19]. NBS for Greenland and Faroe Islands are managed by Denmark. In Faroese population, there was a high incidence (~1:300) of PCD caused by a founder pathogenic variant on SLC22A5: c.95A>G, p.(Asn32Ser) [38]. Consequently, to minimize false negatives on NBS and to ensure diagnosis of all PCD patients, a nationwide second screening performed at two months of age had been introduced [39]. In addition, all Scandinavian countries have included PCD to NBS, and few other countries have conducted pilot studies or have already began to screen PCD, such as Portugal or Italy [21][40]. It is to be noted that the UK does not screen PCD, to date, even though in 2009, England had conducted a pilot study of amino acid and acylcarnitines analysis on cord blood samples to identify inborn errors of metabolism [41]. The Netherlands have listed PCD, along with seven other diseases, in the upcoming schedule of NBS expansion [42], and France will be including PCD to NBS at the beginning of 2023.
2.1.5. Africa
Middle East and North Africa (MENA) is a large region consisting of 21 countries; from Morocco in northwestern Africa, to Iran in southwestern Asia. Genetic disorders are relatively common in this area due to the high rate of consanguinity [43]. Efforts have allowed NBS programs to emerge, resulting from pilot programs and successful studies [44][45][46]. However, to date, only Qatar and Saudi Arabia have included PCD in their programs [47][48][49].
Regarding Sub-Saharan Africa, NBS implementation is still at its beginning. However, much efforts are being made by a Pan-African Workshop on Newborn Screening [50]. Sickle Cell Disease (SCD) is the disease with the highest prevalence in this region and is, therefore, of priority and collaborations will be needed to expand NBS toward a larger panel in the future.
2.1.6. Asia
Teams in Asia were the first to elude that pathogenic variations in the SLC22A5 gene was the molecular basis of primary carnitine deficiency [14][51][52]. Incidence of PCD appears to be more frequent in Asian populations than in those from western countries, even being one of the most prevalent inherited metabolic diseases in the Chinese population [53]. However, the first pilot study of ESI-MS/MS-based NBS in Japan, led by Schigematsu et al., did not report any cases of PCD. First Asian studies and experience on PCD NBS started in late 2000s in China (province-based program), Taiwan, South Korea, and Japan. More recently, Thailand and Philippines included PCD to the ENBS program as well [40][54][55]. To the knowledge, India still struggles to initiate a nationwide NBS program, and other Asian Pacific or Central Asia countries have not included PCD to their program to date [56]. The high incidence of PCD in Asian populations, along with the development of Next Generation Sequencing, have led to the emergence of a systematic study of the SLC22A5 gene as a second-tier testing after phenotypic screening [57][58][59].
2.1.7. Russia
As it is part of both Europe and Asia, Russia is addressed as a separate entity. To date, the nationwide NBS program in Russia includes: phenylketonuria, congenital hypothyroidism, congenital adrenal hyperplasia, galactosemia, and cystic fibrosis. However, Primorsky and Moscow regions are currently performing tandem MS ENBS to identify 39 and 11 diseases, respectively, and the national expansion of NBS is being discussed [60].
3. Pitfalls of Newborn Screening for PCD
Including primary carnitine deficiency to NBS is not as simple as it is for other inborn errors of metabolism. Indeed, PCD is eligible based on the Wilson and Jungner criteria, as it is an easily treatable and very serious condition. Nonetheless, there are substantial obstacles. It is one of the rare diseases for which the screening biomarker is not expected to be detectable above a cut-off value, but below. This is a major problem as there are common causes of decreased free carnitine levels in a newborn, such as: preterm birth [49], maternal PCD, inborn errors of metabolism or vegetarian/vegan diet [61][62], and pivalic acid-based therapeutics in the mother (e.g., pivmecillinam, cephalosporin antibiotics, sivelestat, etc.) [63][64]. These situations are causes of false positive screening test results.
In addition, preanalytical issues can impact the DBS test. For example, the extraction method, using derivatization or not, will lead to different levels of C0, which are higher with derivatized methods [65]. Therefore, there is a need for standardization, at least for screening centers within the same country. The timing of blood collection is crucial as well, as C0 seems to decrease during the first 48 h of life before increasing until 120 h after [66][67]. Ethnicity can be a variating factor as well, as Asian populations seem to have higher C0 levels [68].
Particularly, the high incidence of identification of asymptomatic mothers with PCD from their child’s NBS, raises the question of the limit of the screening approach. Associated with the low sensitivity and positive predictive value of PCD NBS, it would lead to the diagnosis of more mothers than children. For this reason, and the possible side effects of a long term supplementation with L-carnitine, such as trimethyl-N-oxide (TMAO) accumulation and repression of compensation mechanisms observed in PCD, New-Zealand decided to discontinue PCD from their NBS program [24].
In summary, newborn screening for PCD is undoubtedly useful, but only if the following requirements are fulfilled: an algorithm to reduce false positive results, and to increase the positive predictive value by utilizing 1st, 2nd, and even 3rd tier analyses comprising C0 test and retest, along with 2nd tier biomarkers, and molecular and/or functional studies.
References
- Wu, X.; Prasad, P.D.; Leibach, F.H.; Ganapathy, V. CDNA Sequence, Transport Function, and Genomic Organization of Human OCTN2, a New Member of the Organic Cation Transporter Family. Biochem. Biophys. Res. Commun. 1998, 246, 589–595.
- Nezu, J.; Tamai, I.; Oku, A.; Ohashi, R.; Yabuuchi, H.; Hashimoto, N.; Nikaido, H.; Sai, Y.; Koizumi, A.; Shoji, Y.; et al. Primary Systemic Carnitine Deficiency Is Caused by Mutations in a Gene Encoding Sodium Ion-Dependent Carnitine Transporter. Nat. Genet. 1999, 21, 91–94.
- Juraszek, B.; Nałęcz, K.A. SLC22A5 (OCTN2) Carnitine Transporter—Indispensable for Cell Metabolism, a Jekyll and Hyde of Human Cancer. Molecules 2020, 25, 14.
- Tamai, I.; Ohashi, R.; Nezu, J.; Yabuuchi, H.; Oku, A.; Shimane, M.; Sai, Y.; Tsuji, A. Molecular and Functional Identification of Sodium Ion-Dependent, High Affinity Human Carnitine Transporter OCTN2*. J. Biol. Chem. 1998, 273, 20378–20382.
- Stanley, C.A. Carnitine Deficiency Disorders in Children. Ann. N. Y. Acad. Sci. 2004, 1033, 42–51.
- Longo, N. Primary Carnitine Deficiency and Newborn Screening for Disorders of the Carnitine Cycle. Ann. Nutr. Metab. 2016, 68, 5–9.
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine Transport and Fatty Acid Oxidation. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2016, 1863, 2422–2435.
- Stanley, C.A.; DeLeeuw, S.; Coates, P.M.; Vianey-Liaud, C.; Divry, P.; Bonnefont, J.P.; Saudubray, J.M.; Haymond, M.; Trefz, F.K.; Breningstall, G.N. Chronic Cardiomyopathy and Weakness or Acute Coma in Children with a Defect in Carnitine Uptake. Ann. Neurol. 1991, 30, 709–716.
- El-Hattab, A.W.; Scaglia, F. Disorders of Carnitine Biosynthesis and Transport. Mol. Genet. Metab. 2015, 116, 107–112.
- El-Hattab, A.W. Systemic Primary Carnitine Deficiency. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 1993.
- Crefcoeur, L.L.; Visser, G.; Ferdinandusse, S.; Wijburg, F.A.; Langeveld, M.; Sjouke, B. Clinical Characteristics of Primary Carnitine Deficiency—A Structured Review Using a Case by Case Approach. J. Inherit. Metab. Dis. 2022, 45, 386–405.
- Rasmussen, J.; Nielsen, O.W.; Janzen, N.; Duno, M.; Køber, L.; Steuerwald, U.; Lund, A.M. Carnitine Levels in 26,462 Individuals from the Nationwide Screening Program for Primary Carnitine Deficiency in the Faroe Islands. J. Inherit. Metab. Dis. 2014, 37, 215–222.
- Rose, E.C.; di San Filippo, C.A.; Ndukwe Erlingsson, U.C.; Ardon, O.; Pasquali, M.; Longo, N. Genotype-Phenotype Correlation in Primary Carnitine Deficiency. Hum. Mutat. 2012, 33, 118–123.
- Koizumi, A.; Nozaki, J.; Ohura, T.; Kayo, T.; Wada, Y.; Nezu, J.; Ohashi, R.; Tamai, I.; Shoji, Y.; Takada, G.; et al. Genetic Epidemiology of the Carnitine Transporter OCTN2 Gene in a Japanese Population and Phenotypic Characterization in Japanese Pedigrees with Primary Systemic Carnitine Deficiency. Hum. Mol. Genet. 1999, 8, 2247–2254.
- Wilcken, B.; ChB, M. Disorders of the Carnitine Cycle and Detection by Newborn Screening. Ann. Acad. Med. Singap. 2008, 37, 3.
- Dobrow, M.J.; Hagens, V.; Chafe, R.; Sullivan, T.; Rabeneck, L. Consolidated Principles for Screening Based on a Systematic Review and Consensus Process. CMAJ Can. Med. Assoc. J. 2018, 190, E422–E429.
- Barns, R.J.; Bowling, F.G.; Brown, G.; Clague, A.E.; Thompson, A. Carnitine in Dried Blood Spots: A Method Suitable for Neonatal Screening. Clin. Chim. Acta 1991, 197, 27–33.
- Wilcken, B.; Wiley, V.; Sim, K.G.; Carpenter, K. Carnitine Transporter Defect Diagnosed by Newborn Screening with Electrospray Tandem Mass Spectrometry. J. Pediatr. 2001, 138, 581–584.
- Bodamer, O.A.; Hoffmann, G.F.; Lindner, M. Expanded Newborn Screening in Europe 2007. J. Inherit. Metab. Dis. 2007, 30, 439–444.
- Sontag, M.K.; Yusuf, C.; Grosse, S.D.; Edelman, S.; Miller, J.I.; McKasson, S.; Kellar-Guenther, Y.; Gaffney, M.; Hinton, C.F.; Cuthbert, C.; et al. Infants with Congenital Disorders Identified Through Newborn Screening—United States, 2015–2017. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1265–1268.
- Koracin, V.; Mlinaric, M.; Baric, I.; Brincat, I.; Djordjevic, M.; Drole Torkar, A.; Fumic, K.; Kocova, M.; Milenkovic, T.; Moldovanu, F.; et al. Current Status of Newborn Screening in Southeastern Europe. Front. Pediatr. 2021, 9, 648939.
- Care, A.G.D. List of Conditions Screened by Jurisdiction—September 2022. Available online: https://www.health.gov.au/resources/publications/list-of-conditions-screened-by-jurisdiction-september-2022?language=en (accessed on 30 July 2022).
- Wilson, C.; Kerruish, N.J.; Wilcken, B.; Wiltshire, E.; Webster, D. Diagnosis of Disorders of Intermediary Metabolism in New Zealand before and after Expanded Newborn Screening: 2004–2009. N. Z. Med. J. 2012, 125, 42–50.
- Wilson, C.; Knoll, D.; de Hora, M.; Kyle, C.; Glamuzina, E.; Webster, D. The Decision to Discontinue Screening for Carnitine Uptake Disorder in New Zealand. J. Inherit. Metab. Dis. 2019, 42, 86–92.
- Therrell, B.L.; Hannon, W.H. National Evaluation of US Newborn Screening System Components. Ment. Retard. Dev. Disabil. Res. Rev. 2006, 12, 236–245.
- Watson, M.S.; Mann, M.Y.; Lloyd-Puryear, M.A.; Rinaldo, P.; Howell, R.R. American College of Medical Genetics Newborn Screening Expert Group Newborn Screening: Toward a Uniform Screening Panel and System—Executive Summary. Pediatrics 2006, 117, S296–S307.
- Centers for Disease Control and Prevention (CDC) Impact of Expanded Newborn Screening--United States, 2006. MMWR Morb. Mortal. Wkly. Rep. 2008, 57, 1012–1015.
- Centers for Disease Control and Prevention (CDC) CDC Grand Rounds: Newborn Screening and Improved Outcomes. MMWR Morb. Mortal. Wkly. Rep. 2012, 61, 390–393.
- Magoulas, P.L.; El-Hattab, A.W. Systemic Primary Carnitine Deficiency: An Overview of Clinical Manifestations, Diagnosis, and Management. Orphanet J. Rare Dis. 2012, 7, 68.
- Ojodu, J.; Singh, S.; Kellar-Guenther, Y.; Yusuf, C.; Jones, E.; Wood, T.; Baker, M.; Sontag, M. NewSTEPs: The Establishment of a National Newborn Screening Technical Assistance Resource Center. Int. J. Neonatal Screen. 2017, 4, 1.
- Hanley, W.B. Newborn Screening in Canada—Are We out of Step? Paediatr. Child Health 2005, 10, 203–207.
- Dyack, S. Expanded Newborn Screening: Lessons Learned from MCAD Deficiency. Paediatr. Child Health 2004, 9, 241–243.
- Auray-Blais, C.; Boutin, M.; Lavoie, P.; Maranda, B. Neonatal Urine Screening Program in the Province of Quebec: Technological Upgrade from Thin Layer Chromatography to Tandem Mass Spectrometry. Int. J. Neonatal Screen. 2021, 7, 18.
- Borrajo, G.J.C. Newborn Screening in Latin America at the Beginning of the 21st Century. J. Inherit. Metab. Dis. 2007, 30, 466–481.
- Cabello, J.F.; Novoa, F.; Huff, H.V.; Colombo, M. Expanded Newborn Screening and Genomic Sequencing in Latin America and the Resulting Social Justice and Ethical Considerations. Int. J. Neonatal Screen. 2021, 7, 6.
- Borrajo, G.J.C. Newborn Screening in Latin America: A Brief Overview of the State of the Art. Am. J. Med. Genet. C Semin. Med. Genet. 2021, 187, 322–328.
- Schulze, A.; Lindner, M.; Kohlmüller, D.; Olgemöller, K.; Mayatepek, E.; Hoffmann, G.F. Expanded Newborn Screening for Inborn Errors of Metabolism by Electrospray Ionization-Tandem Mass Spectrometry: Results, Outcome, and Implications. Pediatrics 2003, 111, 1399–1406.
- Lund, A.M.; Joensen, F.; Hougaard, D.M.; Jensen, L.K.; Christensen, E.; Christensen, M.; Nørgaard-Petersen, B.; Schwartz, M.; Skovby, F. Carnitine Transporter and Holocarboxylase Synthetase Deficiencies in The Faroe Islands. J. Inherit. Metab. Dis. 2007, 30, 341–349.
- Steuerwald, U.; Lund, A.M.; Rasmussen, J.; Janzen, N.; Hougaard, D.M.; Longo, N. Neonatal Screening for Primary Carnitine Deficiency: Lessons Learned from the Faroe Islands. Int. J. Neonatal Screen. 2017, 3, 1.
- Loeber, J.G.; Platis, D.; Zetterström, R.H.; Almashanu, S.; Boemer, F.; Bonham, J.R.; Borde, P.; Brincat, I.; Cheillan, D.; Dekkers, E.; et al. Neonatal Screening in Europe Revisited: An ISNS Perspective on the Current State and Developments Since 2010. Int. J. Neonatal Screen. 2021, 7, 15.
- Walter, J.H.; Patterson, A.; Till, J.; Besley, G.T.N.; Fleming, G.; Henderson, M.J. Bloodspot Acylcarnitine and Amino Acid Analysis in Cord Blood Samples: Efficacy and Reference Data from a Large Cohort Study. J. Inherit. Metab. Dis. 2009, 32, 95–101.
- Jansen, M.E.; Klein, A.W.; Buitenhuis, E.C.; Rodenburg, W.; Cornel, M.C. Expanded Neonatal Bloodspot Screening Programmes: An Evaluation Framework to Discuss New Conditions With Stakeholders. Front. Pediatr. 2021, 9, 635353.
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.C.; Adams, J. Current Status of Newborn Screening Worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187.
- Al Hosani, H.; Salah, M.; Osman, H.M.; Farag, H.M.; El Assiouty, L.; Saade, D.; Hertecant, J. Expanding the Comprehensive National Neonatal Screening Programme in the United Arab Emirates from 1995 to 2011. East. Mediterr. Health J. 2014, 20, 17–23.
- Golbahar, J.; Al-Jishi, E.A.; Altayab, D.D.; Carreon, E.; Bakhiet, M.; Alkhayyat, H. Selective Newborn Screening of Inborn Errors of Amino Acids, Organic Acids and Fatty Acids Metabolism in the Kingdom of Bahrain. Mol. Genet. Metab. 2013, 110, 98–101.
- Hassan, F.A.; El-Mougy, F.; Sharaf, S.A.; Mandour, I.; Morgan, M.F.; Selim, L.A.; Hassan, S.A.; Salem, F.; Oraby, A.; Girgis, M.Y.; et al. Inborn Errors of Metabolism Detectable by Tandem Mass Spectrometry in Egypt: The First Newborn Screening Pilot Study. J. Med. Screen. 2016, 23, 124–129.
- Mohamed, S.; Elsheikh, W.; Al-Aqeel, A.I.; Alhashem, A.M.; Alodaib, A.; Alahaideb, L.; Almashary, M.; Alharbi, F.; AlMalawi, H.; Ammari, A.; et al. Incidence of Newborn Screening Disorders among 56632 Infants in Central Saudi Arabia: A 6-Year Study. Saudi Med. J. 2020, 41, 703–708.
- Lindner, M.; Abdoh, G.; Fang-Hoffmann, J.; Shabeck, N.; Al Sayrafi, M.; Al Janahi, M.; Ho, S.; Abdelrahman, M.O.; Ben-Omran, T.; Bener, A.; et al. Implementation of Extended Neonatal Screening and a Metabolic Unit in the State of Qatar: Developing and Optimizing Strategies in Cooperation with the Neonatal Screening Center in Heidelberg. J. Inherit. Metab. Dis. 2007, 30, 522–529.
- Ramaswamy, M.; Anthony Skrinska, V.; Fayez Mitri, R.; Abdoh, G. Diagnosis of Carnitine Deficiency in Extremely Preterm Neonates Related to Parenteral Nutrition: Two Step Newborn Screening Approach. Int. J. Neonatal Screen. 2019, 5, 29.
- Therrell, B.L.; Lloyd-Puryear, M.A.; Ohene-Frempong, K.; Ware, R.E.; Padilla, C.D.; Ambrose, E.E.; Barkat, A.; Ghazal, H.; Kiyaga, C.; Mvalo, T.; et al. Empowering Newborn Screening Programs in African Countries through Establishment of an International Collaborative Effort. J. Community Genet. 2020, 11, 253–268.
- Lamhonwah, A.M.; Tein, I. Carnitine Uptake Defect: Frameshift Mutations in the Human Plasmalemmal Carnitine Transporter Gene. Biochem. Biophys. Res. Commun. 1998, 252, 396–401.
- Tang, N.L.S.; Hwu, W.L.; Chan, R.T.; Law, L.K.; Fung, L.M.; Zhang, W.M. A Founder Mutation (R254X) of SLC22A5 (OCTN2) in Chinese Primary Carnitine Deficiency Patients. Hum. Mutat. 2002, 20, 232.
- Tang, N.; Hui, J. 20 Years After Discovery of the Causative Gene of Primary Carnitine Deficiency, How Much More Have We Known About the Disease? HK J Paediatr New Ser. 2020, 25, 23–29.
- Liammongkolkul, S.; Sanomcham, K.; Vatanavicharn, N.; Sathienkijkanchai, A.; Ranieri, E.; Wasant, P. AB133. Expanded Newborn Screening Program in Thailand. Ann. Transl. Med. 2017, 5, AB133.
- Padilla, C.D.; Therrell, B.L.; Alcausin, M.M.L.B.; Chiong, M.A.D.; Abacan, M.A.R.; Reyes, M.E.L.; Jomento, C.M.; Dizon-Escoreal, M.T.T.; Canlas, M.A.E.; Abadingo, M.E.; et al. Successful Implementation of Expanded Newborn Screening in the Philippines Using Tandem Mass Spectrometry. Int. J. Neonatal Screen. 2022, 8, 8.
- Mookken, T. Universal Implementation of Newborn Screening in India. Int. J. Neonatal Screen. 2020, 6, 24.
- Wang, L.-Y.; Chen, N.-I.; Chen, P.-W.; Chiang, S.-C.; Hwu, W.-L.; Lee, N.-C.; Chien, Y.-H. Newborn Screening for Citrin Deficiency and Carnitine Uptake Defect Using Second-Tier Molecular Tests. BMC Med. Genet. 2013, 14, 24.
- Huang, X.; Wu, D.; Zhu, L.; Wang, W.; Yang, R.; Yang, J.; He, Q.; Zhu, B.; You, Y.; Xiao, R.; et al. Application of a Next-Generation Sequencing (NGS) Panel in Newborn Screening Efficiently Identifies Inborn Disorders of Neonates. Orphanet J. Rare Dis. 2022, 17, 66.
- Lin, Y.; Zhang, W.; Huang, C.; Lin, C.; Lin, W.; Peng, W.; Fu, Q.; Chen, D. Increased Detection of Primary Carnitine Deficiency through Second-Tier Newborn Genetic Screening. Orphanet J. Rare Dis. 2021, 16, 149.
- Volgina, S.Y.; Sokolov, A.A. An Analysis of Medical Care Services for Children With Rare Diseases in the Russian Federation. Front. Pharmacol. 2021, 12, 754073.
- El-Hattab, A.W.; Li, F.-Y.; Shen, J.; Powell, B.R.; Bawle, E.V.; Adams, D.J.; Wahl, E.; Kobori, J.A.; Graham, B.; Scaglia, F.; et al. Maternal Systemic Primary Carnitine Deficiency Uncovered by Newborn Screening: Clinical, Biochemical, and Molecular Aspects. Genet. Med. 2010, 12, 19–24.
- Lin, H.J.; Neidich, J.A.; Salazar, D.; Thomas-Johnson, E.; Ferreira, B.F.; Kwong, A.M.; Lin, A.M.; Jonas, A.J.; Levine, S.; Lorey, F.; et al. Asymptomatic Maternal Combined Homocystinuria and Methylmalonic Aciduria (CblC) Detected through Low Carnitine Levels on Newborn Screening. J. Pediatr. 2009, 155, 924–927.
- Holme, E.; Jodal, U.; Linstedt, S.; Nordin, I. Effects of Pivalic Acid-Containing Prodrugs on Carnitine Homeostasis and on Response to Fasting in Children. Scand. J. Clin. Lab. Investig. 1992, 52, 361–372.
- Yamada, K.; Kobayashi, H.; Bo, R.; Takahashi, T.; Hasegawa, Y.; Nakamura, M.; Ishige, N.; Yamaguchi, S. Elevation of Pivaloylcarnitine by Sivelestat Sodium in Two Children. Mol. Genet. Metab. 2015, 116, 192–194.
- Lin, Y.; Xu, H.; Zhou, D.; Hu, Z.; Zhang, C.; Hu, L.; Zhang, Y.; Zhu, L.; Lu, B.; Zhang, T.; et al. Screening 3.4 Million Newborns for Primary Carnitine Deficiency in Zhejiang Province, China. Clin. Chim. Acta Int. J. Clin. Chem. 2020, 507, 199–204.
- Peng, G.; Tang, Y.; Cowan, T.M.; Zhao, H.; Scharfe, C. Timing of Newborn Blood Collection Alters Metabolic Disease Screening Performance. Front. Pediatr. 2020, 8, 623184.
- He, F.; Yang, R.; Huang, X.; Tian, Y.; Pei, X.; Bohn, M.K.; Zou, L.; Wang, Y.; Li, H.; Wang, T.; et al. Reference Standards for Newborn Screening of Metabolic Disorders by Tandem Mass Spectrometry: A Nationwide Study on Millions of Chinese Neonatal Populations. Front. Mol. Biosci. 2021, 8, 719866.
- Peng, G.; Tang, Y.; Gandotra, N.; Enns, G.M.; Cowan, T.M.; Zhao, H.; Scharfe, C. Ethnic Variability in Newborn Metabolic Screening Markers Associated with False-positive Outcomes. J. Inherit. Metab. Dis. 2020, 43, 934–943.
More
Information
Subjects:
Pediatrics
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
15 Feb 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No