 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yong Wu | -- | 2949 | 2023-02-14 07:07:29 | | | |
| 2 | Conner Chen | Meta information modification | 2949 | 2023-02-15 07:14:22 | | | | |
| 3 | Conner Chen | + 2 word(s) | 2951 | 2023-02-16 02:35:59 | | |
Video Upload Options
Diabetes is not only a risk factor for breast cancer but also worsens its prognosis. Patients with diabetes usually show hyperglycemia and hyperinsulinemia, which are accompanied by different glucose, protein, and lipid metabolism disorders. Metabolic abnormalities observed in diabetes can induce the occurrence and development of breast cancer. The changes in substrate availability and hormone environment not only create a favorable metabolic environment for tumorigenesis but also induce metabolic reprogramming events required for breast cancer cell transformation. Metabolic reprogramming is the basis for the development, swift proliferation, and survival of cancer cells. Metabolism must also be reprogrammed to support the energy requirements of the biosynthetic processes in cancer cells. In addition, metabolic reprogramming is essential to enable cancer cells to overcome apoptosis signals and promote invasion and metastasis.
1. Background
2. MRS Targeting Glucose Metabolism
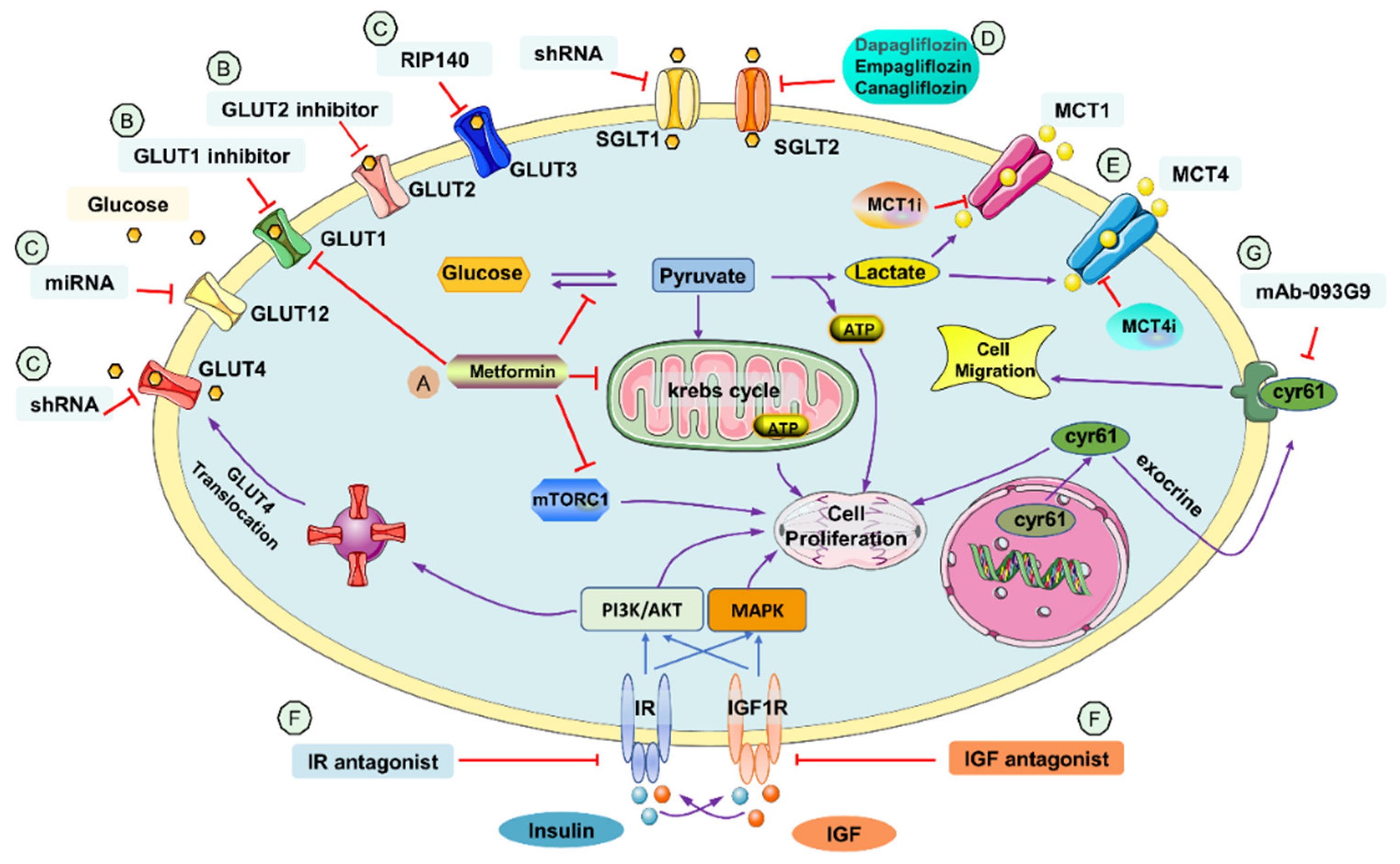
2.1. Metformin
2.2. GLUT Inhibitor
2.3. SGLT Inhibitor
| Title | Status | Study Results |
Conditions | Interventions | Phases | NCT Number | |
|---|---|---|---|---|---|---|---|
| 1 | Alpelisib, Fulvestrant and Dapagliflozin for the Treatment of HR+, HER2-, PIK3CA Mutant Metastatic Breast Cancer | Recruiting | No Results Available | Metastatic BC HER2-negative BC |
Dapagliflozin 10 Mg Tab | Phase 2 | NCT05025735 |
| 2 | A Phase 1b/2 Study of Serabelisib in Combination with Canagliflozin in Patients with Advanced Solid Tumors | Unknown status | No Results Available | BC Endometrial Cancer Lung Cancer Colorectal Cancer Head and Neck Cancer |
Serabelisib Canagliflozin 300 mg |
Phase 1 Phase 2 |
NCT04073680 |
| 3 | Preventing High Blood Sugar in People Being Treated for Metastatic Breast Cancer | Recruiting | No Results Available | BC BC Stage IV Metastatic BC |
Dietary Supplement: Ketogenic Diet Dietary Supplement: Low Carbohydrate Diet Drug: Alpelisib Drug: Fulvestrant Drug: Canagliflozin |
Phase 2 | NCT05090358 |
| 4 | Study of Safety and Efficacy of Dapagliflozin + Metformin XR Versus Metformin XR in Participants With HR+, HER2-, Advanced Breast Cancer While on Treatment with Alpelisib and Fulvestrant | Recruiting | No Results Available | BC | Alpelisib Fulvestrant Metformin XR Dapagliflozin + metformin XR Dapagliflozin |
Phase 2 | NCT04899349 |
2.4. MCT Inhibitor
2.5. Insulin Growth Factor Receptor (IGF1R) Antagonist and Insulin Receptor (IR) Antagonist
References
- Centers for Disease Control and Prevention. National Diabetes Statistics Report website. 2022. Available online: https://www.cdc.gov/diabetes/data/statistics-report/index.html (accessed on 25 November 2022).
- American Cancer Society. Cancer Facts & Figures. 2022, 10. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2022/2022-cancer-facts-and-figures.pdf (accessed on 25 November 2022).
- Hardefeldt, P.J.; Edirimanne, S.; Eslick, G.D. Diabetes increases the risk of breast cancer: A meta-analysis. Endocr. Relat. Cancer 2012, 19, 793.
- Zhao, X.-B.; Ren, G.-S. Diabetes mellitus and prognosis in women with breast cancer: A systematic review and meta-analysis. Medicine 2016, 95, e5602.
- Martin, S.D.; McGee, S.L. Metabolic reprogramming in type 2 diabetes and the development of breast cancer. J. Endocrinol. 2018, 237, R35–R46.
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314.
- Dancey, J. mTOR signaling and drug development in cancer. Nat. Rev. Clin. Oncol. 2010, 7, 209–219.
- Tomas, N.M.; Masur, K.; Piecha, J.C.; Niggemann, B.; Zänker, K.S. Akt and phospholipase Cγ are involved in the regulation of growth and migration of MDA-MB-468 breast cancer and SW480 colon cancer cells when cultured with diabetogenic levels of glucose and insulin. BMC Res. Notes 2012, 5, 1–7.
- Faria, J.; Negalha, G.; Azevedo, A.; Martel, F. Metformin and breast cancer: Molecular targets. J. Mammary Gland Biol. Neoplasia 2019, 24, 111–123.
- Goodwin, P.J.; Chen, B.E.; Gelmon, K.A.; Whelan, T.J.; Ennis, M.; Lemieux, J.; Ligibel, J.A.; Hershman, D.L.; Mayer, I.A.; Hobday, T.J.; et al. Effect of Metformin vs Placebo on Invasive Disease-Free Survival in Patients With Breast Cancer: The MA.32 Randomized Clinical Trial. JAMA 2022, 327, 1963–1973.
- Chae, Y.K.; Arya, A.; Malecek, M.K.; Shin, D.S.; Carneiro, B.; Chandra, S.; Kaplan, J.; Kalyan, A.; Altman, J.K.; Platanias, L.; et al. Repurposing metformin for cancer treatment: Current clinical studies. Oncotarget 2016, 7, 40767–40780.
- Tanaka, Y.; Iwaya, C.; Kawanami, T.; Hamaguchi, Y.; Horikawa, T.; Shigeoka, T.; Yanase, T.; Kawanami, D.; Nomiyama, T. Combined treatment with glucagon-like peptide-1 receptor agonist exendin-4 and metformin attenuates breast cancer growth. Diabetol. Int. 2022, 13, 480–492.
- Iwaya, C.; Nomiyama, T.; Komatsu, S.; Kawanami, T.; Tsutsumi, Y.; Hamaguchi, Y.; Horikawa, T.; Yoshinaga, Y.; Yamashita, S.; Tanaka, T.; et al. Exendin-4, a Glucagonlike Peptide-1 Receptor Agonist, Attenuates Breast Cancer Growth by Inhibiting NF-κB Activation. Endocrinology 2017, 158, 4218–4232.
- Hao, Q.; Huang, Z.; Li, Q.; Liu, D.; Wang, P.; Wang, K.; Li, J.; Cao, W.; Deng, W.; Wu, K.; et al. A Novel Metabolic Reprogramming Strategy for the Treatment of Diabetes-Associated Breast Cancer. Adv. Sci. 2022, 9, e2102303.
- Singh, S.V.; Chaube, B.; Mayengbam, S.S.; Singh, A.; Malvi, P.; Mohammad, N.; Deb, A.; Bhat, M.K. Metformin induced lactic acidosis impaired response of cancer cells towards paclitaxel and doxorubicin: Role of monocarboxylate transporter. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2021, 1867, 166011.
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138.
- Szablewski, L. Expression of glucose transporters in cancers. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2013, 1835, 164–169.
- Silva, C.; Andrade, N.; Guimarães, J.T.; Patrício, E.; Martel, F. The in vitro effect of the diabetes-associated markers insulin, leptin and oxidative stress on cellular characteristics promoting breast cancer progression is GLUT1-dependent. Eur. J. Pharmacol. 2021, 898, 173980.
- Wu, Q.; Ba-Alawi, W.; Deblois, G.; Cruickshank, J.; Duan, S.; Lima-Fernandes, E.; Haight, J.; Tonekaboni, S.A.M.; Fortier, A.M.; Kuasne, H.; et al. GLUT1 inhibition blocks growth of RB1-positive triple negative breast cancer. Nat. Commun. 2020, 11, 4205.
- Barbosa, A.M.; Martel, F. Targeting Glucose Transporters for Breast Cancer Therapy: The Effect of Natural and Synthetic Compounds. Cancers 2020, 12, 154.
- Uldry, M.; Ibberson, M.; Hosokawa, M.; Thorens, B. GLUT2 is a high affinity glucosamine transporter. FEBS Lett. 2002, 524, 199–203.
- Wu, K.-H.; Ho, C.-T.; Chen, Z.-F.; Chen, L.-C.; Whang-Peng, J.; Lin, T.-N.; Ho, Y.-S. The apple polyphenol phloretin inhibits breast cancer cell migration and proliferation via inhibition of signals by type 2 glucose transporter. J. Food Drug Anal. 2018, 26, 221–231.
- Azevedo, C.; Correia-Branco, A.; Araújo, J.R.; Guimaraes, J.T.; Keating, E.; Martel, F. The chemopreventive effect of the dietary compound kaempferol on the MCF-7 human breast cancer cell line is dependent on inhibition of glucose cellular uptake. Nutr. Cancer 2015, 67, 504–513.
- Jacquier, V.; Gitenay, D.; Fritsch, S.; Bonnet, S.; Győrffy, B.; Jalaguier, S.; Linares, L.K.; Cavaillès, V.; Teyssier, C. RIP140 inhibits glycolysis-dependent proliferation of breast cancer cells by regulating GLUT3 expression through transcriptional crosstalk between hypoxia induced factor and p53. Cell. Mol. Life Sci. 2022, 79, 1–17.
- Tsai, T.H.; Yang, C.C.; Kou, T.C.; Yang, C.E.; Dai, J.Z.; Chen, C.L.; Lin, C.W. Overexpression of GLUT3 promotes metastasis of triple-negative breast cancer by modulating the inflammatory tumor microenvironment. J. Cell. Physiol. 2021, 236, 4669–4680.
- Bryant, N.J.; Govers, R.; James, D.E. Regulated transport of the glucose transporter GLUT4. Nat. Rev. Mol. Cell Biol. 2002, 3, 267–277.
- Garrido, P.; Osorio, F.G.; Morán, J.; Cabello, E.; Alonso, A.; Freije, J.M.; González, C. Loss of GLUT4 induces metabolic reprogramming and impairs viability of breast cancer cells. J. Cell. Physiol. 2015, 230, 191–198.
- Stenbit, A.E.; Tsao, T.S.; Li, J.; Burcelin, R.; Geenen, D.L.; Factor, S.M.; Houseknecht, K.; Katz, E.B.; Charron, M.J. GLUT4 heterozygous knockout mice develop muscle insulin resistance and diabetes. Nat. Med. 1997, 3, 1096–1101.
- Alam, F.; Islam, M.A.; Khalil, M.I.; Gan, S.H. Metabolic Control of Type 2 Diabetes by Targeting the GLUT4 Glucose Transporter: Intervention Approaches. Curr. Pharm. Des. 2016, 22, 3034–3049.
- Rogers, S.; Macheda, M.L.; Docherty, S.E.; Carty, M.D.; Henderson, M.A.; Soeller, W.C.; Gibbs, E.M.; James, D.E.; Best, J.D. Identification of a novel glucose transporter-like protein-GLUT-12. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E733–E738.
- Shi, Y.; Zhang, Y.; Ran, F.; Liu, J.; Lin, J.; Hao, X.; Ding, L.; Ye, Q. Let-7a-5p inhibits triple-negative breast tumor growth and metastasis through GLUT12-mediated warburg effect. Cancer Lett. 2020, 495, 53–65.
- Matsui, C.; Takatani-Nakase, T.; Maeda, S.; Nakase, I.; Takahashi, K. Potential Roles of GLUT12 for Glucose Sensing and Cellular Migration in MCF-7 Human Breast Cancer Cells Under High Glucose Conditions. Anticancer Res. 2017, 37, 6715–6722.
- Purcell, S.H.; Aerni-Flessner, L.B.; Willcockson, A.R.; Diggs-Andrews, K.A.; Fisher, S.J.; Moley, K.H. Improved insulin sensitivity by GLUT12 overexpression in mice. Diabetes 2011, 60, 1478–1482.
- Hsia, D.S.; Grove, O.; Cefalu, W.T. An update on sodium-glucose co-transporter-2 inhibitors for the treatment of diabetes mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73–79.
- Poulsen, S.B.; Fenton, R.A.; Rieg, T. Sodium-glucose cotransport. Curr. Opin. Nephrol. Hypertens. 2015, 24, 463–469.
- Padda, I.S.; Mahtani, A.U.; Parmar, M. Sodium-Glucose Transport Protein 2 (SGLT2) Inhibitors; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022.
- Komatsu, S.; Nomiyama, T.; Numata, T.; Kawanami, T.; Hamaguchi, Y.; Iwaya, C.; Horikawa, T.; Fujimura-Tanaka, Y.; Hamanoue, N.; Motonaga, R.; et al. SGLT2 inhibitor ipragliflozin attenuates breast cancer cell proliferation. Endocr. J. 2020, 67, 99–106.
- Zhou, J.; Zhu, J.; Yu, S.J.; Ma, H.L.; Chen, J.; Ding, X.F.; Chen, G.; Liang, Y.; Zhang, Q. Sodium-glucose co-transporter-2 (SGLT-2) inhibition reduces glucose uptake to induce breast cancer cell growth arrest through AMPK/mTOR pathway. Biomed. Pharm. 2020, 132, 110821.
- Nasiri, A.R.; Rodrigues, M.R.; Li, Z.; Leitner, B.P.; Perry, R.J. SGLT2 inhibition slows tumor growth in mice by reversing hyperinsulinemia. Cancer Metab. 2019, 7, 10.
- Akingbesote, N.D.; Norman, A.; Zhu, W.; Halberstam, A.A.; Zhang, X.; Foldi, J.; Lustberg, M.B.; Perry, R.J. A precision medicine approach to metabolic therapy for breast cancer in mice. Commun. Biol. 2022, 5, 478.
- Song, P.; Onishi, A.; Koepsell, H.; Vallon, V. Sodium glucose cotransporter SGLT1 as a therapeutic target in diabetes mellitus. Expert Opin. Ther. Targets 2016, 20, 1109–1125.
- Zambrowicz, B.; Freiman, J.; Brown, P.M.; Frazier, K.S.; Turnage, A.; Bronner, J.; Ruff, D.; Shadoan, M.; Banks, P.; Mseeh, F.; et al. LX4211, a dual SGLT1/SGLT2 inhibitor, improved glycemic control in patients with type 2 diabetes in a randomized, placebo-controlled trial. Clin Pharm. 2012, 92, 158–169.
- Cherney, D.Z.I.; Ferrannini, E.; Umpierrez, G.E.; Peters, A.L.; Rosenstock, J.; Carroll, A.K.; Lapuerta, P.; Banks, P.; Agarwal, R. Efficacy and safety of sotagliflozin in patients with type 2 diabetes and severe renal impairment. Diabetes Obes. Metab. 2021, 23, 2632–2642.
- Niu, X.; Ma, J.; Li, J.; Gu, Y.; Yin, L.; Wang, Y.; Zhou, X.; Wang, J.; Ji, H.; Zhang, Q. Sodium/glucose cotransporter 1-dependent metabolic alterations induce tamoxifen resistance in breast cancer by promoting macrophage M2 polarization. Cell Death Dis. 2021, 12, 509.
- Liu, H.; Ertay, A.; Peng, P.; Li, J.; Liu, D.; Xiong, H.; Zou, Y.; Qiu, H.; Hancock, D.; Yuan, X.; et al. SGLT1 is required for the survival of triple-negative breast cancer cells via potentiation of EGFR activity. Mol. Oncol. 2019, 13, 1874–1886.
- Wang, J.; Ji, H.; Niu, X.; Yin, L.; Wang, Y.; Gu, Y.; Li, D.; Zhang, H.; Lu, M.; Zhang, F.; et al. Sodium-Dependent Glucose Transporter 1 (SGLT1) Stabled by HER2 Promotes Breast Cancer Cell Proliferation by Activation of the PI3K/Akt/mTOR Signaling Pathway in HER2+ Breast Cancer. Dis. Mrk. 2020, 2020, 6103542.
- Halestrap, A.P.; Wilson, M.C. The monocarboxylate transporter family--role and regulation. IUBMB Life 2012, 64, 109–119.
- Baenke, F.; Dubuis, S.; Brault, C.; Weigelt, B.; Dankworth, B.; Griffiths, B.; Jiang, M.; Mackay, A.; Saunders, B.; Spencer-Dene, B.; et al. Functional screening identifies MCT4 as a key regulator of breast cancer cell metabolism and survival. J. Pathol. 2015, 237, 152–165.
- Pinheiro, C.; Albergaria, A.; Paredes, J.; Sousa, B.; Dufloth, R.; Vieira, D.; Schmitt, F.; Baltazar, F. Monocarboxylate transporter 1 is up-regulated in basal-like breast carcinoma. Histopathology 2010, 56, 860–867.
- Benyahia, Z.; Blackman, M.; Hamelin, L.; Zampieri, L.X.; Capeloa, T.; Bedin, M.L.; Vazeille, T.; Schakman, O.; Sonveaux, P. In Vitro and In Vivo Characterization of MCT1 Inhibitor AZD3965 Confirms Preclinical Safety Compatible with Breast Cancer Treatment. Cancers 2021, 13, 569.
- Silva, A.; Antunes, B.; Batista, A.; Pinto-Ribeiro, F.; Baltazar, F.; Afonso, J. In Vivo Anticancer Activity of AZD3965: A Systematic Review. Molecules 2021, 27, 181.
- Padilla, J.; Lee, B.S.; Zhai, K.; Rentz, B.; Bobo, T.; Dowling, N.M.; Lee, J. A Heme-Binding Transcription Factor BACH1 Regulates Lactate Catabolism Suggesting a Combined Therapy for Triple-Negative Breast Cancer. Cells 2022, 11, 1177.
- Duan, X.; Xie, Y.; Yu, J.; Hu, X.; Liu, Z.; Li, N.; Qin, J.; Lan, L.; Yuan, M.; Pan, Z.; et al. MCT4/Lactate Promotes PD-L1 Glycosylation in Triple-Negative Breast Cancer Cells. J. Oncol. 2022, 2022, 3659714.
- Luo, E.; Wang, D.; Yan, G.; Qiao, Y.; Zhu, B.; Liu, B.; Hou, J.; Tang, C. The NF-κB/miR-425-5p/MCT4 axis: A novel insight into diabetes-induced endothelial dysfunction. Mol. Cell Endocrinol. 2020, 500, 110641.
- Nadai, T.; Narumi, K.; Furugen, A.; Saito, Y.; Iseki, K.; Kobayashi, M. Pharmacological Inhibition of MCT4 Reduces 4-Hydroxytamoxifen Sensitivity by Increasing HIF-1α Protein Expression in ER-Positive MCF-7 Breast Cancer Cells. Biol. Pharm. Bull. 2021, 44, 1247–1253.
- Ekyalongo, R.C.; Yee, D. Revisiting the IGF-1R as a breast cancer target. NPJ Precis. Oncol. 2017, 1, 14.
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928.
- Tseng, C.-H. Prolonged use of human insulin increases breast cancer risk in Taiwanese women with type 2 diabetes. BMC Cancer 2015, 15, 846.
- Platts, J. Insulin therapy and cancer risk in diabetes mellitus. Clin. Med. 2010, 10, 509–512.
- Sarkissyan, S.; Sarkissyan, M.; Wu, Y.; Cardenas, J.; Koeffler, H.P.; Vadgama, J.V. IGF-1 Regulates Cyr61 Induced Breast Cancer Cell Proliferation and Invasion. PLoS ONE 2014, 9, e103534.
- Hellinger, J.W.; Hüchel, S.; Goetz, L.; Bauerschmitz, G.; Emons, G.; Gründker, C. Inhibition of CYR61-S100A4 Axis Limits Breast Cancer Invasion. Front. Oncol. 2019, 9, 1074.
- Lin, J.; Huo, R.; Wang, L.; Zhou, Z.; Sun, Y.; Shen, B.; Wang, R.; Li, N. A novel anti-Cyr61 antibody inhibits breast cancer growth and metastasis in vivo. Cancer Immunol. Immunother. 2012, 61, 677–687.
- Cao, J.; Yee, D. Disrupting Insulin and IGF Receptor Function in Cancer. Int. J. Mol. Sci. 2021, 22, 555.
- Wenbin, K.; Xiaoqin, L.; Qiuchan, D.; Xinwen, Z.; Xiaoqin, X.; Fangyuan, S.; Dabao, H.; Shuangjiu, Z. Development of a novel insulin receptor (IR) antagonist that exhibits anti-breast tumor activity. Hum. Cell 2020, 33, 1204–1217.
- Rostoker, R.; Bitton-Worms, K.; Caspi, A.; Shen-Orr, Z.; LeRoith, D. Investigating new therapeutic strategies targeting hyperinsulinemia’s mitogenic effects in a female mouse breast cancer model. Endocrinology 2013, 154, 1701–1710.
- Hamilton, N.; Márquez-Garbán, D.; Rogers, B.; Austin, D.; Foos, K.; Tong, A.; Adams, D.; Vadgama, J.; Brecht, M.-L.; Pietras, R. Dual Therapy with Insulin-Like Growth Factor-I Receptor/Insulin Receptor (IGF1R/IR) and Androgen Receptor (AR) Antagonists Inhibits Triple-Negative Breast Cancer Cell Migration In Vitro. SPG BioMed 2019.
- Hamilton, N.M.; Marquez-Garban, D.C.; Burton, L.P.; Comin-Anduix, B.; Garcia, A.J.; Vadgama, J.V. Abstract LB-391: Combination of insulin-like growth factor-1 receptor/insulin receptor (IGF1R/IR) antagonist with anti-PD-L1 antibody blocks triple-negative breast cancer (TNBC) progression. Cancer Res. 2020, 80, LB-391.
- Aguirre, G.A.; De Ita, J.R.; de la Garza, R.G.; Castilla-Cortazar, I. Insulin-like growth factor-1 deficiency and metabolic syndrome. J. Transl. Med. 2016, 14, 3.


