 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Panagiotis TSIRIGOTIS | -- | 1754 | 2023-02-08 18:49:20 | | | |
| 2 | Catherine Yang | -555 word(s) | 1199 | 2023-02-10 07:22:53 | | |
Video Upload Options
Hemato-poietic stem cell transplantation is associated with significant endothelial dysfunction, which may result in severe complications. Endothelial dysfunction induces the expression of multiple substances including, cell adhesion molecules, pro-inflammatory cytokines, and coagulation factors resulting in activation of the complement system, and promoting a procoagulant and proinflammatory state. The pathophysiological process that mediates the endothelial damage is not clearly understood and the aim of this study is a comprehensive review of the existing knowledge. Moreover, the levels of soluble molecules released in the systemic circulation as a result of endothelial damage may serve as biomarkers for the early diagnosis and the pre-emptive treatment of post-transplant syndromes. The review of the existing literature presented in detail shows that the ideal biomarker with sufficient sensitivity and specificity for the early diagnosis of post-transplant complications does not currently exist. Future studies should focus on the identification of novel biomarkers with sufficient specificity and sensitivity.
1. Endothelial Dysfunction-Related Syndromes Post-Allogeneic Stem Cell Transplantation
The endothelium is a very active organ formed by a thin layer of heterogeneous cells that delignate the barrier between the circulating blood and other tissues. Normal function of the endothelium is of paramount clinical significance, as it plays a key role in maintaining vascular homeostasis and a balanced coagulation but also in host defense, inflammation, and angiogenesis [1][2][3].
1.1. Transplant-Associated Thrombotic Microangiopathy (TA-TMA)
TA-TMA constitutes one of the most severe complications of allogenic hematopoietic stem cell transplantation (allo-HSCT) and is associated with significant morbidity and mortality [3][4]. It is a heterogenous disease, which is characterized by aberrant complement activation, endothelial dysregulation, and microvascular hemolytic anemia [5]. Recently, a three-hit theory was proposed regarding the pathophysiology of the disease [1], which refers to: (1) endothelial vulnerability to damage and complement activation (hit 1), (2) a toxic event (such as the conditioning therapy) injuring the endothelium and initiating the complement cascade (hit 2), and (3) additional insults (such as infection, graft-versus-host disease (GvHD), etc.) exacerbating the complement activation and leading to (widespread) microthrombi formation (hit 3).
1.2. Sinusoidal Obstructive Syndrome (SOS)/Veno-Occlusive Disease (VOD)
Sinusoidal obstructive syndrome (SOS), also known as veno-occlusive disease (VOD), is a life-threatening complication occurring after high-dose chemotherapy and HSCT [6]. It has also been described after high-dose radiotherapy, liver transplantation, and administration of toxic agents [7]. In HSCT, the conditioning regimen causes an initial toxic injury to the sinusoidal endothelium of the liver, disrupting the endothelial cohesions and creating gaps in the sinusoidal barrier. This allows red blood cells, leukocytes, and other debris to pass through and accumulate into the Disse space, leading to dissection of the endothelial lining and downstream embolization and sinusoid flow obstruction. This results in reduced hepatic outflow and post-sinusoidal hypertension with subsequent hepato-renal syndrome and MOD [8].
1.3. Lung Injury Syndromes
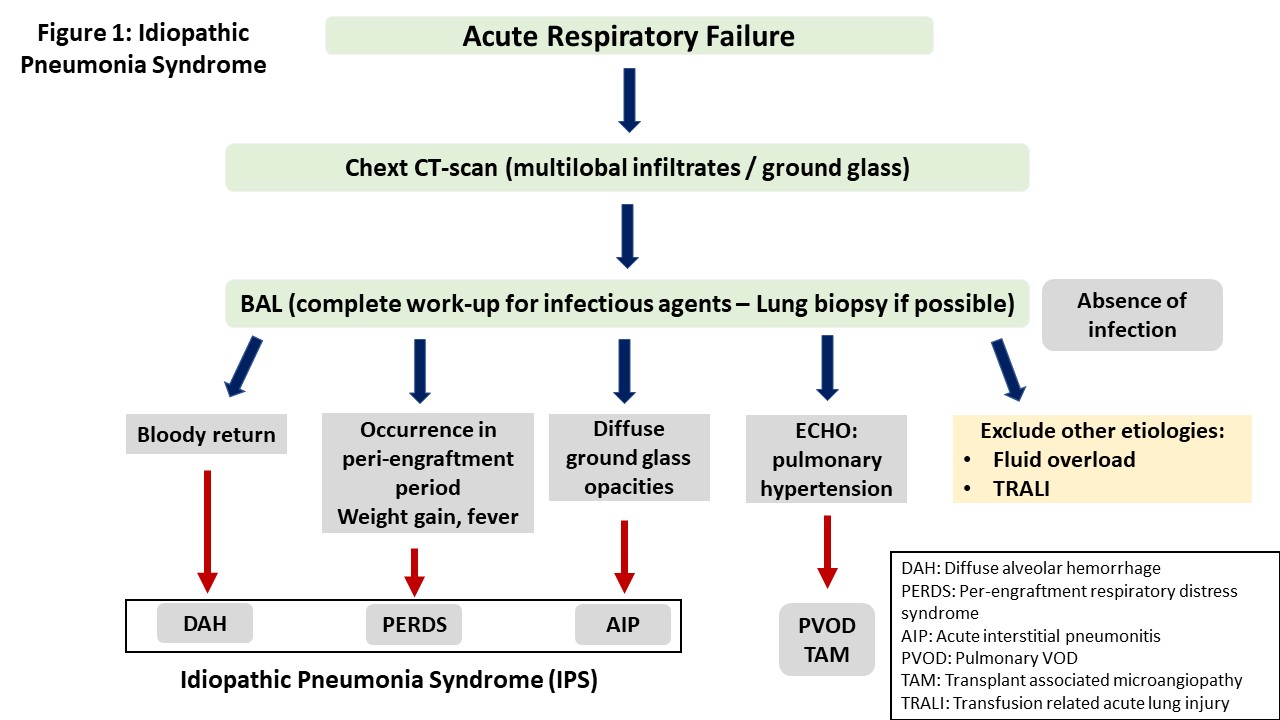
Idiopathic pneumonia syndrome (IPS) was defined in 1993 in a workshop organized by the National Institute of Health as the result of widespread alveoli injury with multi-lobar pulmonary infiltrates and symptoms related to respiratory failure.
IPS may present with a variety of clinical symptoms depending on the site of lung injury. However, the typical presentation is that of acute interstitial pneumonitis. Other manifestations include diffuse alveolar hemorrhage (DAH) and peri-engraftment respiratory distress syndrome (PERDS) [9][10][11]. Table 1 shows the clinical features of lung injury syndromes, while the criteria used for the differential diagnosis between these syndromes are presented in Figure 1.
|
Characteristic |
Idiopathic Pneumonia Syndrome |
Diffuse Alveolar Hemorrhage |
Peri-Engraftment Respiratory Distress Syndrome |
|
Epidemiology |
More common after allo-SCT |
Equal incidence after Auto and allo-SCT |
More common after auto-SCT |
|
Median time of onset |
30–40 days after allo-SCT |
20–25 days after SCT |
From 3 days before to 7 days after engraftment |
|
Relation to engraftment |
No relation |
No relation |
Occurs during the peri-engraftment phase |
|
Clinical features |
Rapid progression to respiratory failure |
Progressively bloodier aliquots of bronchoalveolar lavage |
Systemic manifestations such as fever, rash |
|
Pathology |
Diffuse alveolar damage |
Diffuse alveolar damage |
Diffuse alveolar damage |
|
Pathogenetic drivers* |
TNFα |
Various cytokines |
GM-CSF, G-CSF |
|
Response to corticosteroids |
Poor, some response after anti-TNF agents |
Moderate response to high dose steroids |
Excellent response |
|
Prognosis |
Favorable |
Moderate to Poor |
Very poor |
|
*TNFα, tumor necrosis factor α; GM-CSF, granulocyte monocyte colony stimulating factor; G-CSF, granulocyte colony stimulating factor. |
|||
2. Vascular Endothelium: A Target of Graft-Versus-Host Disease
Traditionally, GvHD has been considered as an epithelial cell disease, with the three most commonly involved organs being the skin, intestinal tract, and liver. However, experimental and clinical data allow us to reconsider the epithelium as being the single GVH target.
Indeed, host vascular ECs is the first cell type that the alloreactive donor T lymphocytes attack after allo-HSCT, potentially inducing endothelial GvHD. In accordance with the previous notion, many clinical syndromes observed after allo-HSCT are pathogenetically considered as syndromes of endothelial cell dysfunction.
The term “endothelial cell dysfunction” is poorly defined and is used for the description of certain changes in endothelial cells (EC). There are several stimuli that have the potential to induce EC dysfunction especially in the setting of allo-SCT, such as infections, toxins, chemotherapeutic agents, donor cell alloreactivity, etc. [12]. Intensive and persistence activating stimuli can produce either organ-localized or systemic EC dysfunction. EC dysfunction in the setting of allo-HSCT can present with the form of specific endothelial syndromes such as transplant associated microangiopathy, engraftment syndrome, diffuse alveolar hemorrhage, etc. EC dysfunction results in distortion of normal homeostasis, increase in capillary permeability, and extravasation of fluid, plasma proteins, and immune cells from the vascular space to extracellular tissue, thus creating an inflammatory milieu.
The pathophysiology is not clearly delineated and may involve interactions of EC with T cells, monocytes, complement components, and proinflammatory cytokines [13].
Experimental data have shown that EC are immune targets of alloreactive donor T cells, and EC dysfunction syndromes observed after allo-HSCT can be viewed as “graft-versus-EC” phenomena [14].
3. Atheromatosis: A Late GVHD Manifestation
Long-term survivors after allo-HSCT have an increased incidence of cardiovascular events when compared with healthy controls. An obvious explanation is the presence of metabolic syndrome occurring frequently in patients treated with HCT. Allogeneic HSCT survivors were more likely to report hypertension, diabetes, and dyslipidemia than patients after autologous HSCT [15]. The increased occurrence of dyslipidemia, hypertension, and diabetes after allo-HSCT can be attributed to prolonged administration of calcineurin inhibitors and corticosteroids or can be the result of subclinical organ damage such as hypothyroidism or decreased growth factor hormone [16].
Although supported by observational studies and multiple case reports, the direct association between GVHD and premature atheromatosis has not been definitely established. A schematic overview of the endothelial dysfunction-associated syndromes after allo-HSCT is given in Figure 2.
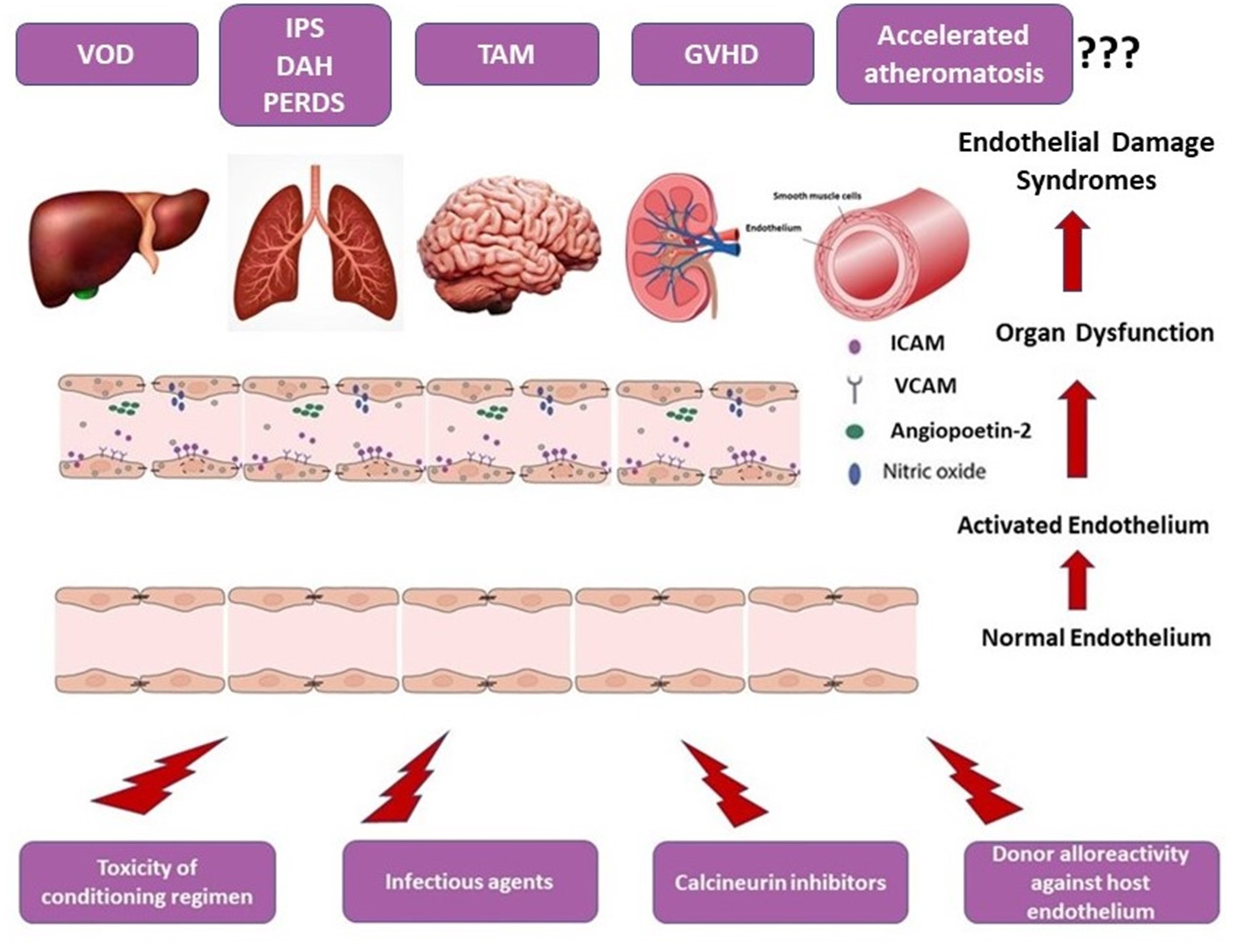
4. Soluble Biomarkers of Endothelial Dysfunction
Endothelial damage syndromes occurring after allo-HSCT usually overlap with other complications, and therefore, biomarkers are needed for early identification of endothelial-cell injury. Endothelial dysfunction results in loss of vascular integrity and increase in capillary permeability. EC damage induces changes in various protein expression, shedding of these proteins from damaged endothelial cells, and release in the systemic circulation. Monitoring of released proteins in the peripheral blood may help in the early identification of endothelial cell damage.
The ideal biomarker for EC dysfunction should be characterized by high sensitivity, specificity, and predictive value. Although an ideal biomarker does not currently exist, there is an enormous activity in the field, with various soluble molecules, circulating endothelial cells, coagulation factors, cytokines, etc., being actively tested.
References
- Luft, T.; Dreger, P.; Radujkovic, A. Endothelial cell dysfunction: A key determinant for the outcome of allogeneic stem cell transplantation. Bone Marrow Transplant. 2021, 56, 2326–2335. https://doi.org/10.1038/s41409-021-01390-y.
- Varma, A.; Rondon, G.; Srour, S.A.; Chen, J.; Ledesma, C.; Champlin, R.E.; Ciurea, S.O.; Saliba, R.M. Endothelial Activation and Stress Index (EASIX) at Admission Predicts Fluid Overload in Recipients of Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2020, 26, 1013–1020. https://doi.org/10.1016/j.bbmt.2020.01.028.
- Sakellari, I.; Gavriilaki, E.; Boussiou, Z.; Batsis, I.; Mallouri, D.; Constantinou, V.; Kaloyannidis, K.; Yannaki, E.; Bamihas, G.; Anagnostopoulos, A. Transplant-associated thrombotic microangiopathy: An unresolved complication of unrelated allogeneic transplant for hematologic diseases. Hematol. Oncol. 2016, 35, 932–934. https://doi.org/10.1002/hon.2346.
- Postalcioglu, M.; Kim, H.T.; Obut, F.; Yilmam, O.A.; Yang, J.; Byun, B.C.; Kupiec-Weglinski, S.; Soiffer, R.; Ritz, J.; Antin, J.H.; et al. Impact of Thrombotic Microangiopathy on Renal Outcomes and Survival after Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2018, 24, 2344–2353. https://doi.org/10.1016/j.bbmt.2018.05.010.
- Pagliuca, S.; Michonneau, D.; de Fontbrune, F.S.; del Galy, A.S.; Xhaard, A.; Robin, M.; de Latour, R.P.; Socie, G. Allogeneic reactivity–mediated endothelial cell complications after HSCT: A plea for consensual definitions. Blood Adv. 2019, 3, 2424–2435. https://doi.org/10.1182/bloodadvances.2019000143.
- Kumar, S.; Deleve, L.D.; Kamath, P.S.; Tefferi, A. Hepatic Veno-occlusive Disease (Sinusoidal Obstruction Syndrome) after Hematopoietic Stem Cell Transplantation. Mayo Clin. Proc. 2003, 78, 589–598. https://doi.org/10.4065/78.5.589.
- Valla, D.; Cazals-Hatem, D. Sinusoidal obstruction syndrome. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 378–385. https://doi.org/10.1016/j.clinre.2016.01.006.
- Fan, C.Q.; Crawford, J.M. Sinusoidal Obstruction Syndrome (Hepatic Veno-Occlusive Disease). J. Clin. Exp. Hepatol. 2014, 4, 332–346. https://doi.org/10.1016/j.jceh.2014.10.002.
- Panoskaltsis-Mortari, A.; Griese, M.; Madtes, D.K.; Belperio, J.A.; Haddad, I.Y.; Folz, R.J.; Cooke, K.R. An Official American Thoracic Society Research Statement: Noninfectious Lung Injury after Hematopoietic Stem Cell Transplantation: Idiopathic Pneumonia Syndrome. Am. J. Respir. Crit. Care Med. 2011, 183, 1262–1279. https://doi.org/10.1164/rccm.2007-413st.
- Afessa, B.; Peters, S.G. Noninfectious pneumonitis after blood and marrow transplant. Curr. Opin. Oncol. 2008, 20, 227–233. https://doi.org/10.1097/cco.0b013e3282f50ff5.
- Watkins, T.R.; Chien, J.W.; Crawford, S.W. Graft versus Host-Associated Pulmonary Disease and other Idiopathic Pulmonary Complications after Hematopoietic Stem Cell Transplant. Semin. Respir. Crit. Care Med. 2005, 26, 482–489. https://doi.org/10.1055/s-2005-922031.
- Biedermann, B.C. Vascular endothelium and graft-versus-host disease. Best Pr. Res. Clin. Haematol. 2008, 21, 129–138. https://doi.org/10.1016/j.beha.2008.02.003.
- Tichelli, A.; Gratwohl, A. Vascular endothelium as ‘novel’ target of graft-versus-host disease. Best Pr. Res. Clin. Haematol. 2008, 21, 139–148. https://doi.org/10.1016/j.beha.2008.02.002.
- Nomura, S.; Ishii, K.; Fujita, S.; Nakaya, A.; Satake, A.; Ito, T. Associations between acute GVHD-related biomarkers and endothelial cell activation after allogeneic hematopoietic stem cell transplantation. Transpl. Immunol. 2017, 43-44, 27–32. https://doi.org/10.1016/j.trim.2017.06.004.
- Baker, K.S.; Ness, K.K.; Steinberger, J.; Carter, A.; Francisco, L.; Burns, L.J.; Sklar, C.; Forman, S.; Weisdorf, D.; Gurney, J.G.; et al. Diabetes, hypertension, and cardiovascular events in survivors of hematopoietic cell transplantation: A report from the bone marrow transplantation survivor study. Blood 2006, 109, 1765–1772. https://doi.org/10.1182/blood-2006-05-022335.
- Couriel, D.R.; Saliba, R.; Escalon, M.P.; Hsu, Y.; Ghosh, S.; Ippoliti, C.; Hicks, K.; Donato, M.; Giralt, S.; Khouri, I.F.; et al. Sirolimus in combination with tacrolimus and corticosteroids for the treatment of resistant chronic graft-versus-host disease. Br. J. Haematol. 2005, 130, 409–417. https://doi.org/10.1111/j.1365-2141.2005.05616.x.


