Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anatoly Vereshchagin | -- | 2477 | 2023-02-08 15:14:14 | | | |
| 2 | Jessie Wu | Meta information modification | 2477 | 2023-02-09 03:22:51 | | | | |
| 3 | Jessie Wu | Meta information modification | 2477 | 2023-02-09 03:25:33 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Frolov, N.A.; Vereshchagin, A.N. Pharmacological Applications of Piperidine Derivatives. Encyclopedia. Available online: https://encyclopedia.pub/entry/40989 (accessed on 08 August 2026).
Frolov NA, Vereshchagin AN. Pharmacological Applications of Piperidine Derivatives. Encyclopedia. Available at: https://encyclopedia.pub/entry/40989. Accessed August 08, 2026.
Frolov, Nikita A., Anatoly N. Vereshchagin. "Pharmacological Applications of Piperidine Derivatives" Encyclopedia, https://encyclopedia.pub/entry/40989 (accessed August 08, 2026).
Frolov, N.A., & Vereshchagin, A.N. (2023, February 08). Pharmacological Applications of Piperidine Derivatives. In Encyclopedia. https://encyclopedia.pub/entry/40989
Frolov, Nikita A. and Anatoly N. Vereshchagin. "Pharmacological Applications of Piperidine Derivatives." Encyclopedia. Web. 08 February, 2023.
Copy Citation
Piperidine is a six-membered heterocycle including one nitrogen atom and five carbon atoms in the sp3-hybridized state. Piperidine-containing compounds represent one of the most important synthetic medicinal blocks for drugs construction, and their synthesis has long been widespread. It can be unequivocally stated that heterocyclic compounds play a significant part in the pharmaceutical industry, and one of the most common in their structure is the piperidine cycle.
piperidines
derivatives
biological
1. Introduction
The piperidine cycle is utterly common in pharmaceuticals. Its derivatives are used in over twenty drug classes [1], including anticancer agents [2][3][4][5][6][7], drugs for Alzheimer’s disease therapy [8], antibiotics [9], analgesics [10][11], antipsychotics [12][13][14], antioxidants [15][16], etc. (Figure 1).
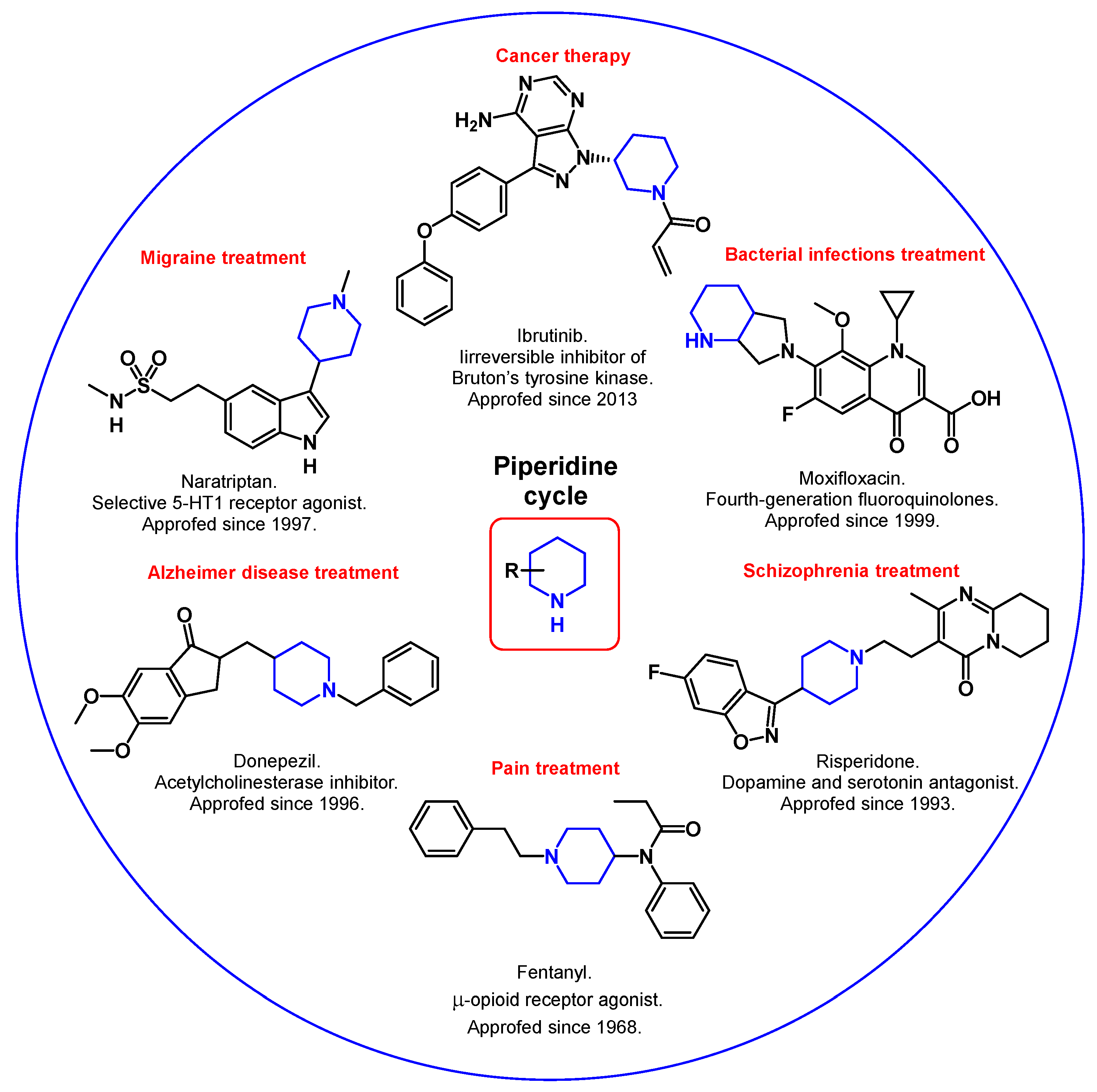
Figure 1. Synthetic piperidine derivatives in medicine.
Moreover, piperidines are also a part of many alkaloids showing biological activity (Figure 2). For example, the well-known atropine (used clinically for the treatment of vomiting, nausea, and bradycardia [17]; an effective agent for slowing the development of myopia [18]) and morphine (analgesic for severe pain relief [19]; used as a third-line therapy in the treatment of neuropathic pain [20]) contain a fused piperidine ring.
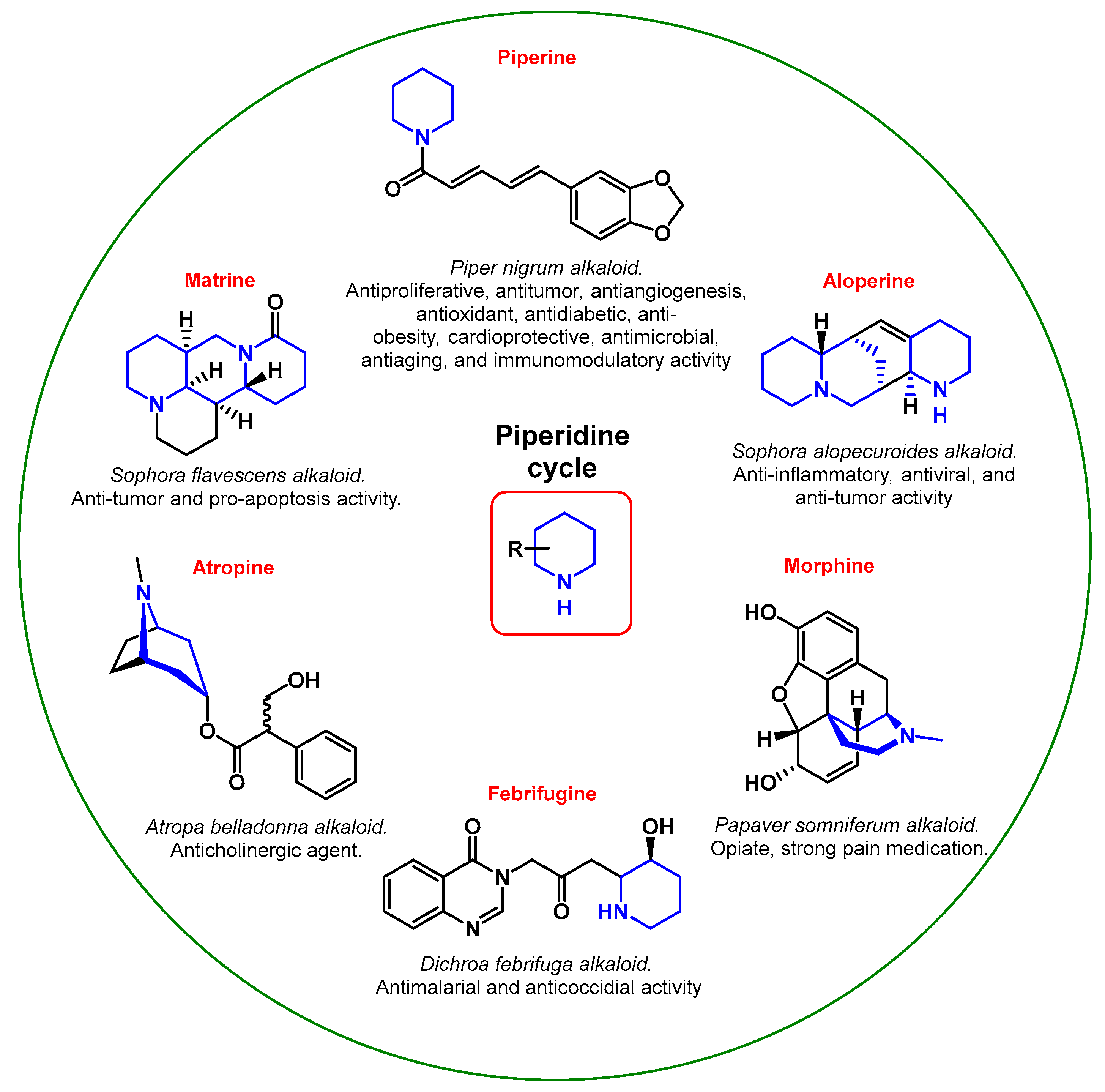
Figure 2. Pharmacological properties of natural piperidine derivatives.
Piperine, a derivative of piperidine and the main active chemical component of black pepper, is attracting more and more attention from researchers, despite the fact that it was discovered more than 200 years ago. It is believed that piperine has a broad scope of beneficial biological properties, from antibacterial to anticancer [21][22][23][24][25]. Aloperine and Matrine—an alkaloid of the Sophora containing two fused piperidine rings at once—and their derivatives showed antiviral, anti-inflammatory, and antitumor properties [26][27]. Febrifugine and its synthetic analog halofuginone are efficiently used as antiparasitic drugs [28].
Along with already known drugs, the scientific community constantly proposes new biologically active piperidine scaffolds. Further, researchers will discuss recent discoveries in the biological evaluation of synthetic potential drugs containing the piperidine moiety. Particular attention was paid to four pharmaceutical groups: cancer (pro-tumorigenic receptor inhibitors, apoptosis initiators), infectious and parasitic diseases (biocides), Alzheimer’s disease (anticholinergics), and neuropathic pain (analgesics). The choice of drug groups was based on current trends and relevance in the medical community.
2. Cancer Therapy
Cancer is one of the biggest health problems worldwide, with nearly 10 million deaths reported in 2020 according to WHO. A lot of resources are spent on the development of new drugs for fighting cancer, but despite all efforts, innate and acquired resistance mechanisms are often observed [29]. Therefore, screening for new developments and breakthroughs in this area is very important and relevant.
Piperidine moieties are often used in anticancer drug construction [3][7]. Herein, the recent proposals and developments of scientists on this subject will be briefly discussed.
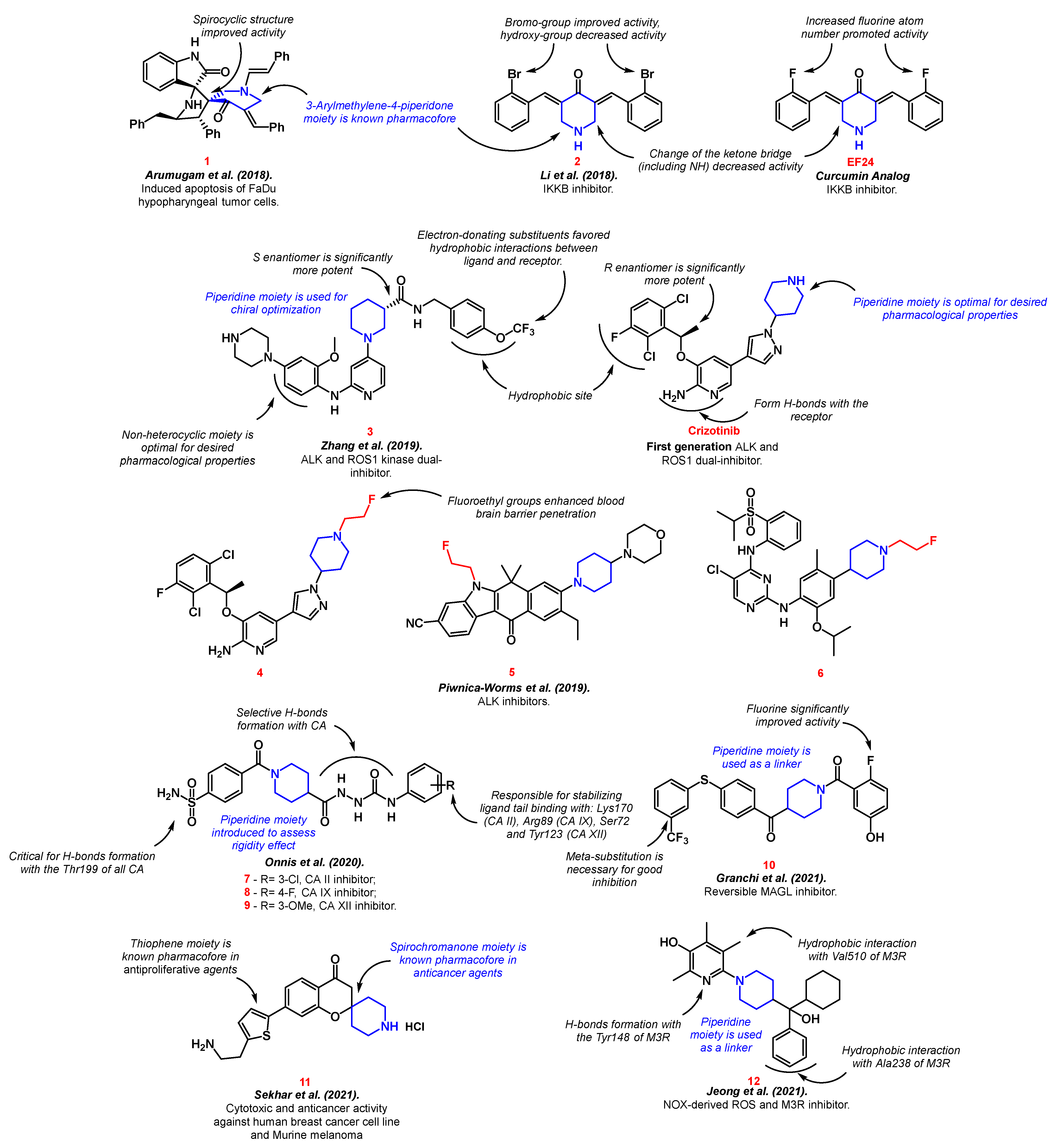
Arumugam et al. synthesized spirooxindolopyrrolidine-embedded piperidinone 1 with potential anticancer activity through three-component 1,3-dipolar cycloaddition and subsequent enamine reaction [30]. The resulting product showed slightly better cytotoxicity and apoptosis induction in the FaDu hypopharyngeal tumor cells model than the reference drug bleomycin. The authors followed the “escape from flatland” approach, which was popularized throughout recent years [31][32][33] and was successfully used in the development of anti-cancer agents [34][35][36]. This approach suggests that more saturated and three-dimensional structures will interact better with binding sites of proteins. Therefore, the authors reasoned that the spirocyclic structure played a key role in the biological activity of compound 1.
Li et al. developed IκB kinase (IKKb) inhibitor 2 as an EF24 analog [37]. EF24 is a piperidinone derivative with potential activity against lung, breast, ovarian, and cervical cancer [38][39]. The activation of IKKb is one of the major factors of NF-κB transcription, which induces chronic inflammation in carcinomas, leading to desmoplasia and neoplastic progression [40]. The new analog 2 possessed better IKKb inhibitory properties than the reference drug. The active component with the piperidine moiety developed a stable hydrophobic interaction with the IKKb catalytic pocket. The structure–activity relationship shows that the presence of a nitrogen atom in the cycle is optimal, and any substitution in the ketone bridge is not favorable for IKKb inhibition.
A series of 2-amino-4-(1-piperidine) pyridine derivatives 3, as the clinically Crizotinib-resistant anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 kinase (ROS1) dual inhibitor, was designed by Zhang et al. [41]. ALK was originally discovered in anaplastic large cell lymphoma as a transmembrane receptor tyrosine kinase [42]. It was found that ALK is involved in the development of non-small cell lung cancer, neuroblastoma, diffuse large B-cell lymphoma, anaplastic thyroid cancer, rhabdomyosarcoma, ovarian cancer, esophageal squamous cell, colorectal, and breast carcinomas, etc. [43][44]. ROS1 was discovered more recently as a similar enzyme to ALK. ROS1 rearrangements were identified in glioblastoma, cholangiocarcinoma, gastric cancer, ovarian cancer, soft-tissue sarcomas, breast cancer etc. [45]. Crizotinib—the first approved ALK/ROS1 dual inhibitor—also includes the piperidine moiety [46]. Despite the fact that the piperidine fragment did not form any bonds with the active site of the receptor, its introduction was the most optimal for the desired pharmacological properties compared to other substituents. In the case of substance 3, piperidine derivatives (namely, (S or R)-ethyl piperidine-3-carboxylate) were used as the starting components for chiral optimization [41]. The piperidine ring was essential for chiral optimization. Piwnica-Worms et al. obtained radiolabeled fluoro-analogs of the commercial ALK inhibitors crizotinib 4, alectinib 5, and ceritinib 6 [47]. The products have a potential for brain metastases treatment due to their enhanced CNS pharmacokinetic properties. It is worth noting that the introduction of fluoroethyl groups did not affect the inhibitory properties of parent drugs, while it enhanced their ability to pass through blood–brain barrier.
Onnis et al. conducted a synthesis of benzenesulfonamide with a piperidinyl-hydrazidoureido linker as potent carbonic anhydrase (CA) II (7), IX (8), and XII (9) inhibitors [48]. CAs are metalloenzymes localized in the cytosol, mitochondria, membranes, and secreted substances of living organisms. CAs are involved in the catalysis of chemical processes (the hydration of carbon dioxide to bicarbonate, the conversion of cyanate to carbamic acid, etc.) and esterase activity [49]. Two out of the sixteen known types of CA (CA IX and CA XII) are found in vertebrate tumor cells. Their inhibition is an effective way to control the growth, progression, and metastasis of cancerous tumors [50]. Currently, the leading compound among CA IX and XII inhibitors is SLC-0111, which is in phase I/II of clinical trials for the management of hypoxic tumors [51][52]. The authors used SLC-0111 as the parent drug, incorporating a piperidinyl-hydrazidoureido linker in its structure to improve binding selectivity with CA. The piperidine residue was also introduced to assess rigidity [48].
Benzoylpiperidine scaffold 10 with antitumor activity via monoacylglycerol lipase (MAGL) inhibition was constructed by Granchi et al. [53][54]. Fluorine atoms and the meta-substitution of the benzene ring enhanced the inhibition properties. MAGL is responsible for the inactivation of the brain’s endocannabinoid 2-arachidonoylglycerol. Moreover, MAGL indirectly controls the levels of free fatty acids, as well as other lipids with pro-inflammatory or pro-oncogenic effects, therefore causing pain and cancer progression [55]. Sekhar et al. developed spirochromanone analog 11 with significant activity against the breast cancer cell line and Murine melanoma, as well as the ability to induce apoptosis [56]. The authors combined known pharmacophore structures to achieve the best anti-proliferative and anti-cancer effects.
Jeong et al. synthesized piperidine-embedded anticancer agents with particularly good activity on androgen-refractory cancer cell lines (ARPC) [57]. The authors showed that compound 12 was a ligand to the M3 muscarinic acetylcholine receptor (M3R), which is presented in ARPC (Figure 3). M3R activation stimulates cell proliferation, resistance to apoptosis, and metastasis and is responsible for the early progression and invasion of colorectal cancer tumors [58][59][60].
3. Alzheimer Disease Therapy
Alzheimer disease is one of the most lethal and burdening illnesses of the last century. It has no definite treatment other than symptomatic treatment and results in death 6 years after diagnosis, on average [61]. The oldest theory of Alzheimer’s disease is the cholinergic hypothesis, which suggests that the illness is caused by the loss of cholinergic innervation [62].
The neurotransmitter acetylcholine is one of many vital components for normal brain function. Deficiency of the cholinergic system has been observed in the brains of Alzheimer’s disease patients, leading to the pathophysiology of learning and memory impairment [63]. The main goal of modern therapy is to maintain the level of acetylcholine through the inhibition of cholinesterases: acetylcholinesterase (ACHe) and butyrylcholinesterase (BuCHe) [64]. Currently, the leading drug among acetylcholinesterase inhibitors is Donepezil, a piperidine derivative.
Martinez et al. proposed indolylpiperidine analog 13 of Donepezil [65]. The active agent was capable of inhibiting both acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) enzymes. Moreover, the authors discovered unusual conformational changes in the molecule depending on the binding site. Thus, compound 13 was extended in AChE interaction and flopped in BuChE interaction. Liu et al. expanded this field with 4-N-phenylaminoquinoline derivative 14 [66] via piperidine moiety introduction to a previously reported lead compound [67]. Piperidine incorporation improved the brain exposure of the resulting dual inhibitor. In addition, the compound showed antioxidant and metal chelating properties.
In 2018, Gobec et al. designed selective BuChe inhibitor 15 [68]. Two cationic nitrogen atoms were essential for selectivity and good inhibition properties. Further, the authors conducted a detailed structure–activity relationship study of N-alkylpiperidine carbamates [69]. Structures with an N-benzyl moiety were superior in cholinesterase inhibition, and a terminal alkyne group was essential for efficient monoamine oxidase B inhibition. Thus, compounds 16–18 were selected as the best in the series. Moreover, a study by Malawska and Gobec outlined a multi-targeted approach to Alzheimer’s disease treatment. Novel 1-Benzylpyrrolidine-3-amine derivatives with piperidine groups 19 and 20 expressed both antiaggregatory and antioxidant effects [70]. Along with dual cholinesterase inhibition, compounds 19–20 also targeted the beta secretase enzyme.
Beta secretase is also known as beta-site amyloid precursor protein cleaving enzyme-1 (BACE-1). It has been established that the inhibition of BACE-1 prevents the accumulation of amyloid beta [71][72]. According to the current concept of Alzheimer’s disease based on the amyloid hypothesis, deposits of amyloid beta and tau proteins cause neurodegeneration and cognitive impairment [73].
The benzyl-piperidine group (Donepezil-like) is often a necessary part for the successful inhibition of cholinesterase receptors. The AChE enzyme includes two active anionic binding sites: catalytic and peripheral. The benzyl-piperidine group provides good binding to the catalytic site, interacting with Trp84, Trp279, Phe330, and Phe331 [74]. Therefore, the selection of various substituents on top of the benzyl-piperidine residue is a well-established approach in the synthesis of new active agents for combatting Alzheimer’s disease. Thus, pyrrolizine 21 [75], fluorine 22 [76], thiazole 23 [77], indoline 24 [78], benzofuran 25 [79], thiophene 26 [80], and chromene 27 [81] groups have been effectively incorporated and biologically evaluated by various authors. The structure–activity relationship is shown in Figure 4.
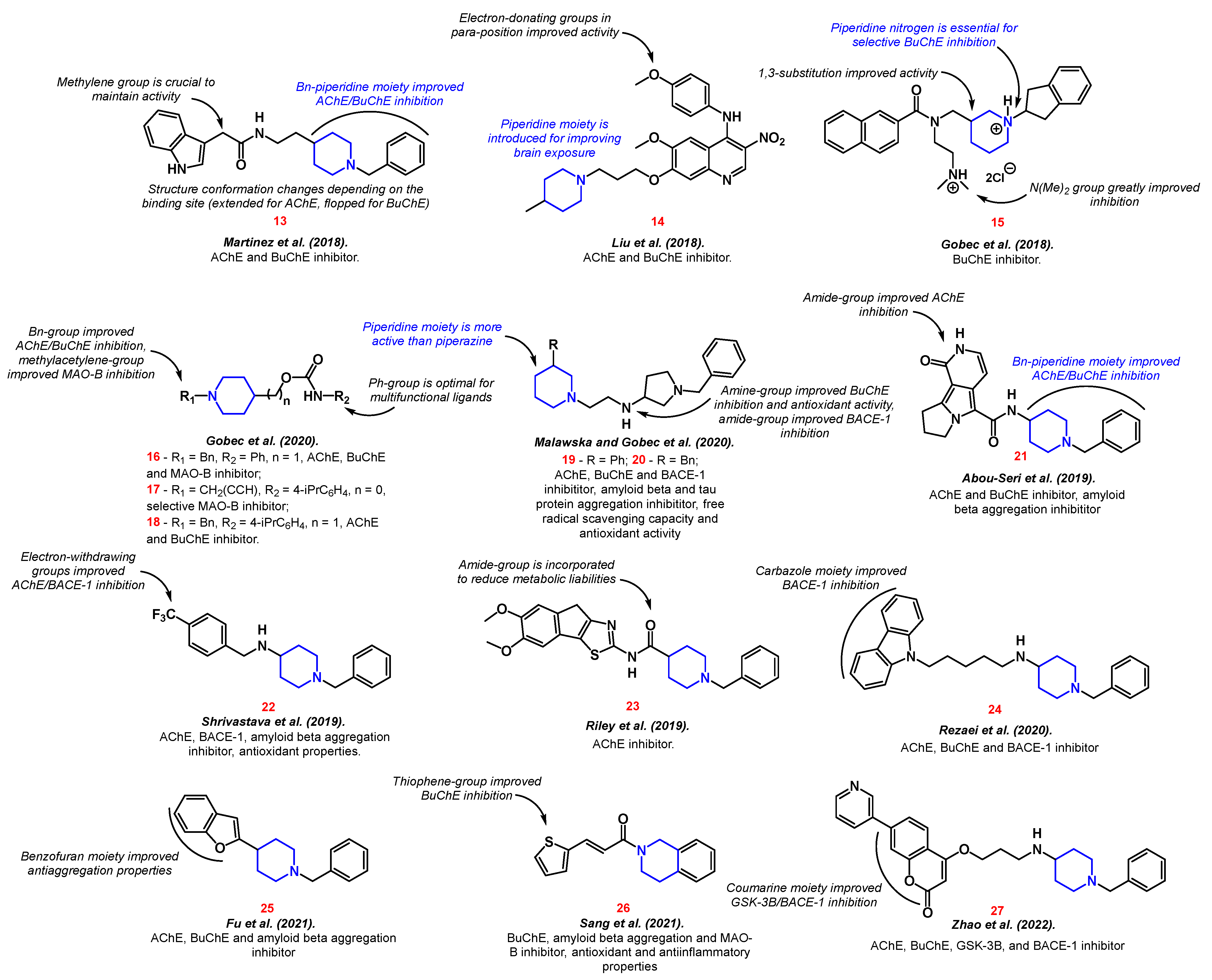
It is worth noting that research on multifunctional active agents is prevalent compared to compounds that affect only one target. Therefore, along with inhibitors of cholinesterase receptors, attention is also paid to inhibitors of monoamine oxidase 16–17, 26 [69][80], amyloid beta and tau protein aggregation 19–20, 22, 25–26 [70][76][79][80], BACE-1 19–20, 22, 24, 27 [70][76][78][81], as well as the presence of anti-inflammatory 26 [80], anti-radical 19–20 [70], and antioxidant properties 19–20, 22, 26 [70][76][80].
Drawing conclusions from the analyzed data, it can be said that the piperidine group affects the inhibition of cholinesterase receptors and serves as a constructing (linker) part.
4. Biocides
Biocides are chemical compounds designed to neutralize, suppress, or prevent the action of harmful organisms, namely, pathogenic bacteria, fungi, viruses, parasites, etc. [82]. As noted earlier, piperidine derivatives find use in this class of pharmaceuticals.
In recent years, a number of works on the topic can be noted. However, due to the wide variety of human pathogens, it is not possible to point out one template structure for all types of activities.
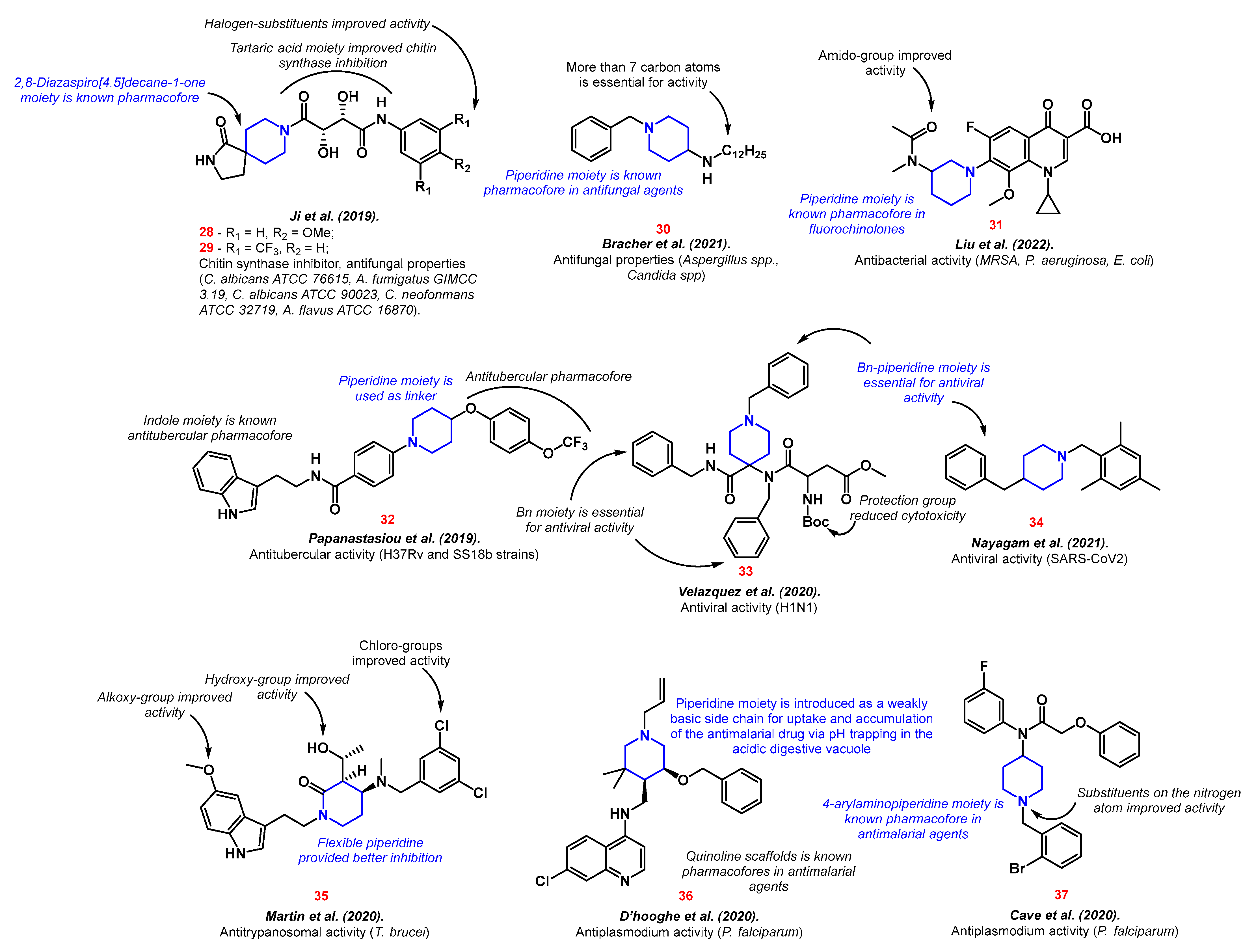
Thus, piperidine moieties were represented in structures with antifungal properties 28–30. Compounds containing tartaric acid fragment 28–29 inhibited chitin synthase, therefore suppressing a growth of five fungi strains (C. albicans ATCC 76615, A. fumigatus GIMCC 3.19, C. albicans ATCC 90023, C. neofonmans ATCC 32719, A. flavus ATCC 16870) [83]. The resulting compounds combined two pharmacophores: 2,8-Diazaspiro [4.5]decane-1-one and a tartaric acid residue with a substituted aminobenzene. When designing the structure, the authors were guided by the “escape from flatness” theory (which was mentioned earlier) and the enzyme inhibition potential of the chosen moieties. Long-tailed 4-aminopiperidines 30 have proven to be effective against fungi of the genus Aspergillus and Candida via fungal ergosterol biosynthesis inhibition [84]. Ergosterol is one of the most abundant fungal cell membrane sterols. It is responsible for membrane permeability and fluidity [85].
Piperidine-containing fluoroquinolones analogs, namely, bafloxacins 31, were proposed by Liu et al. [86]. Along with the good inhibition values on MRSA, P. aeruginosa, and E. coli, the new compounds showed good biocompatibility and potential two-targeted action via cell walls destruction and interaction with IV-DNA and DNA gyrase. Piperidinyl “tails” structures 32 possessed inhibition properties against streptomycin-starved Mycobacterium tuberculosis 18b (SS18b) and H37Rv strains [87]. Compound 32 consists of various known tubercular pharmacofores with piperidine as a linker.
The benzyl-piperidines activity against different viruses was shown. Thus, 4,4-disubstituted N-benzyl piperidines 33 inhibited the H1N1 influenza virus through specific hemagglutinin fusion peptide interaction [88]. Nayagam et al. discovered the potential inhibitor of SARS-CoV2 with piperidine core 34 [89]. Compound 34 possessed a better binding affinity with the SARS-CoV2 main protease than Remdesivir, with five binding pockets interaction compared to two.
Compounds with a piperidine backbone structure have shown antiparasitic properties on T. brucei (the main cause of African trypanosomiasis) 35 [90] and P. falciparum (the cause of the deadliest type of malaria) 36–37 [91][92] (Figure 5).
5. Neuropathic Pain Therapy
Neuropathic pain occurs as a result of the pathological excitation of neurons in the peripheral or central nervous system, which is caused by neurological diseases with damage to peripheral fibers and central neurons [93]. The modern approach to the treatment of neuropathic pain includes three lines of pharmacotherapy. Most of the piperidine derivatives are part of opioids, which are the second and, in some cases, the third line of treatment [20].
Opioid receptors are divided into four similar types: μ-opioid (MOR), δ-opioid (DOR), κ-opioid (KOR), and nociceptin/orphanin FQ peptide receptor (NOP) [94][95]. MOR and DOR are the main targets of opioid agonists. MOR agonists cause euphoria and help with coping with stress; however, their use causes serious side effects and physical dependence, leading to overdose [96]. One of the main synthetic piperidine-containing opioids is fentanyl.
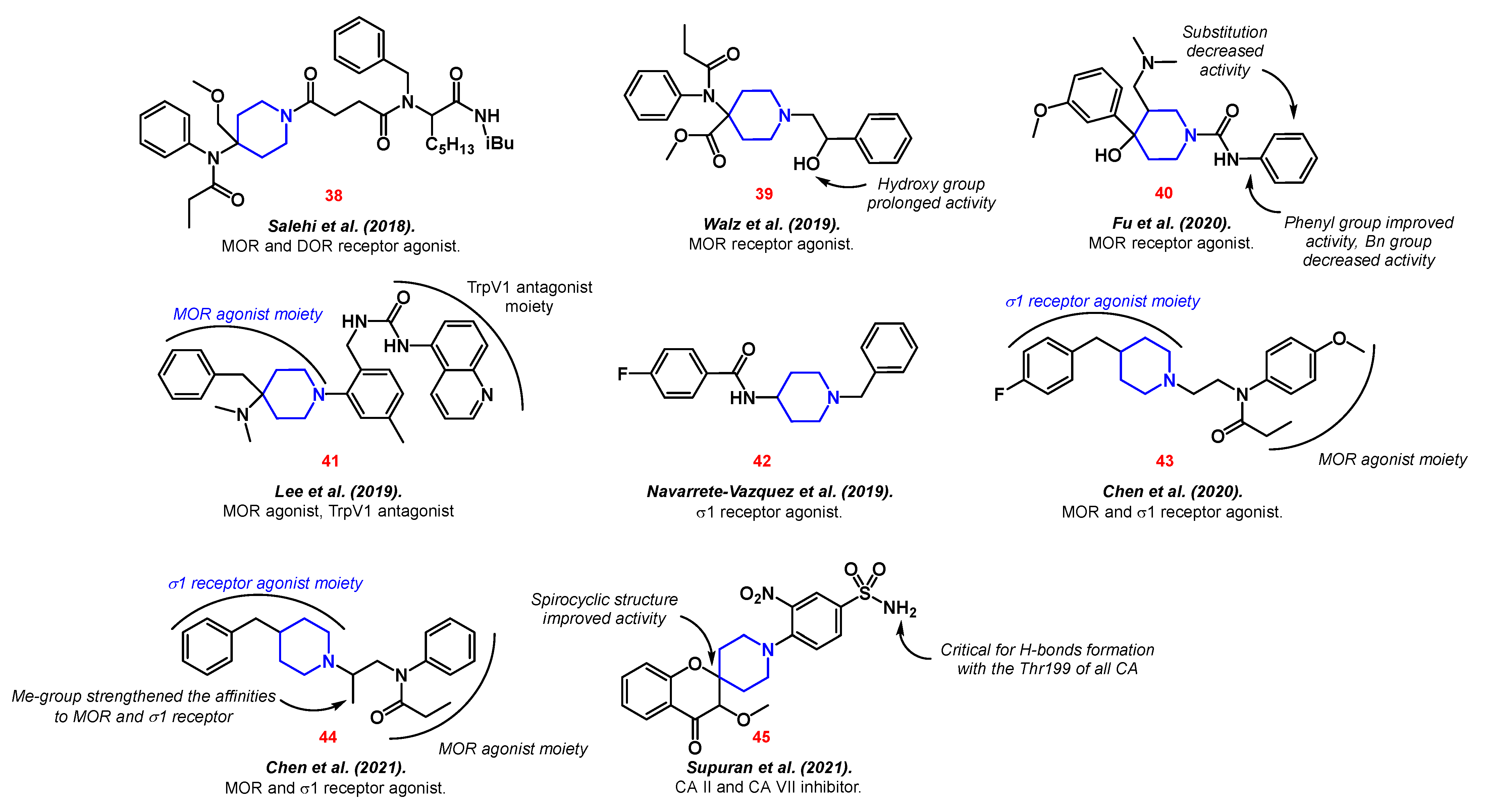
Most often, opioid derivatives serve as the starting point for the discovery of new types of analgesics. Thus, derivatives of norsufentanil with amino acids 38 were developed [97], the synthesis of the main metabolites of carfentanil 39 was reproduced [98], and new analogs of tramadol 40 were proposed [99]. All compounds showed a strong affinity for MOR.
In order to achieve a dual effect ligand, Lee et al. created a hybrid based on Pethidine, also known as meperidine, and a transient receptor potential cation channel subfamily V member 1 (TrpV1) antagonist 41 [100]. TrpV1 functions are widely linked to the generation of pain [101]. This combination potentially increases the anti-inflammatory effect and treatment efficiency.
Navarrete-Vázquez et al. developed a haloperidol analog 42 as a σ1 receptor antagonist [102]. The σ1 receptor plays a role in various regulatory processes, including pain reduction [103]. Therefore, Chen et al. proposed novel piperidine propionamide derivatives 43–44 as dual agonists of μ-opioid and σ1 receptors [104][105].
Lastly, CA inhibition is another prominent therapeutic target in neuropathic pain treatment. Thus, Supuran et al. synthetized piperidine-embedded 4-oxo-spirochromanes 45 with high activity against CA II and CA VII (Figure 6) [106].
References
- Vardanyan, R. Chapter 10–Classes of Piperidine-Based Drugs. In Piperidine-Based Drug Discovery; Vardanyan, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 299–332.
- McLornan, D.P.; Pope, J.E.; Gotlib, J.; Harrison, C.N. Current and future status of JAK inhibitors. Lancet 2021, 398, 803–816.
- Goel, P.; Alam, O.; Naim, M.J.; Nawaz, F.; Iqbal, M.; Alam, M.I. Recent advancement of piperidine moiety in treatment of cancer- A review. Eur. J. Med. Chem. 2018, 157, 480–502.
- Charalambous, A.; Schwarzbich, M.-A.; Witzens-Harig, M. Ibrutinib. In Small Molecules in Hematology; Martens, U.M., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 133–168.
- Milling, R.V.; Grimm, D.; Krüger, M.; Grosse, J.; Kopp, S.; Bauer, J.; Infanger, M.; Wehland, M. Pazopanib, Cabozantinib, and Vandetanib in the Treatment of Progressive Medullary Thyroid Cancer with a Special Focus on the Adverse Effects on Hypertension. Int. J. Mol. Sci. 2018, 19, 3258.
- Coricello, A.; Mesiti, F.; Lupia, A.; Maruca, A.; Alcaro, S. Inside Perspective of the Synthetic and Computational Toolbox of JAK Inhibitors: Recent Updates. Molecules 2020, 25, 3321.
- Shaw, V.; Srivastava, S.; Srivastava, S.K. Repurposing antipsychotics of the diphenylbutylpiperidine class for cancer therapy. Semin. Cancer Biol. 2021, 68, 75–83.
- Li, Q.; He, S.; Chen, Y.; Feng, F.; Qu, W.; Sun, H. Donepezil-based multi-functional cholinesterase inhibitors for treatment of Alzheimer′s disease. Eur. J. Med. Chem. 2018, 158, 463–477.
- Ezelarab, H.A.A.; Abbas, S.H.; Hassan, H.A.; Abuo-Rahma, G.E.-D.A. Recent updates of fluoroquinolones as antibacterial agents. Arch. Der Pharm. 2018, 351, 1800141.
- Martinelli, D.; Bitetto, V.; Tassorelli, C. Lasmiditan: An additional therapeutic option for the acute treatment of migraine. Expert Rev. Neurother. 2021, 21, 491–502.
- Ye, N.; Qin, W.; Tian, S.; Xu, Q.; Wold, E.A.; Zhou, J.; Zhen, X.-C. Small Molecules Selectively Targeting Sigma-1 Receptor for the Treatment of Neurological Diseases. J. Med. Chem. 2020, 63, 15187–15217.
- Rathore, A.; Asati, V.; Kashaw, K.S.; Agarwal, S.; Parwani, D.; Bhattacharya, S.; Mallick, C. The Recent Development of Piperazine and Piperidine Derivatives as Antipsychotic Agents. Mini-Rev. Med. Chem. 2021, 21, 362–379.
- Friedman, J.H. Pharmacological interventions for psychosis in Parkinson’s disease patients. Expert Opin. Pharmacother. 2018, 19, 499–505.
- Kantrowitz, J.T. Targeting Serotonin 5-HT2A Receptors to Better Treat Schizophrenia: Rationale and Current Approaches. CNS Drugs 2020, 34, 947–959.
- Mezeiova, E.; Spilovska, K.; Nepovimova, E.; Gorecki, L.; Soukup, O.; Dolezal, R.; Malinak, D.; Janockova, J.; Jun, D.; Kuca, K.; et al. Profiling donepezil template into multipotent hybrids with antioxidant properties. J. Enzym. Inhib. Med. Chem. 2018, 33, 583–606.
- Rk, M.; Begum, S.; Begum, A.; Koganti, B. Antioxidant potential of piperidine containing compounds–A short review. Asian J. Pharm. Clin. Res. 2018, 11, 66.
- Lakstygal, A.M.; Kolesnikova, T.O.; Khatsko, S.L.; Zabegalov, K.N.; Volgin, A.D.; Demin, K.A.; Shevyrin, V.A.; Wappler-Guzzetta, E.A.; Kalueff, A.V. DARK Classics in Chemical Neuroscience: Atropine, Scopolamine, and Other Anticholinergic Deliriant Hallucinogens. ACS Chem. Neurosci. 2019, 10, 2144–2159.
- Vagge, A.; Ferro Desideri, L.; Nucci, P.; Serafino, M.; Giannaccare, G.; Traverso, C.E. Prevention of Progression in Myopia: A Systematic Review. Diseases 2018, 6, 92.
- Devereaux, A.L.; Mercer, S.L.; Cunningham, C.W. DARK Classics in Chemical Neuroscience: Morphine. ACS Chem. Neurosci. 2018, 9, 2395–2407.
- Cavalli, E.; Mammana, S.; Nicoletti, F.; Bramanti, P.; Mazzon, E. The neuropathic pain: An overview of the current treatment and future therapeutic approaches. Int. J. Immunopathol. Pharmacol. 2019, 33, 2058738419838383.
- Shityakov, S.; Bigdelian, E.; Hussein, A.A.; Hussain, M.B.; Tripathi, Y.C.; Khan, M.U.; Shariati, M.A. Phytochemical and pharmacological attributes of piperine: A bioactive ingredient of black pepper. Eur. J. Med. Chem. 2019, 176, 149–161.
- Manayi, A.; Nabavi, M.S.; Setzer, N.W.; Jafari, S. Piperine as a Potential Anti-cancer Agent: A Review on Preclinical Studies. Curr. Med. Chem. 2018, 25, 4918–4928.
- Smilkov, K.; Ackova, G.D.; Cvetkovski, A.; Ruskovska, T.; Vidovic, B.; Atalay, M. Piperine: Old Spice and New Nutraceutical? Curr. Pharm. Des. 2019, 25, 1729–1739.
- Afreen; Salahuddin; Mazumder, A.; Joshi, S.; Kumar, R.; Yar, S.M.; Ahsan, J.M. Insight into the Isolation, Synthesis, and Structure-Activity Relationship of Piperine Derivatives for the Development of New Compounds: Recent Updates. Curr. Top. Med. Chem. 2021, 21, 2715–2751.
- Haq, I.-U.; Imran, M.; Nadeem, M.; Tufail, T.; Gondal, T.A.; Mubarak, M.S. Piperine: A review of its biological effects. Phytother. Res. 2021, 35, 680–700.
- Cheng, Y.; Rauf, A.; Pan, X. Research Progress on the Natural Product Aloperine and Its Derivatives. Mini-Rev. Med. Chem. 2022, 22, 729–742.
- Singh, L.; Upadhyay, K.A.; Dixit, P.; Singh, A.; Yadav, D.; Chhavi, A.; Konar, S.; Srivastava, P.R.; Pandey, S.; Devkota, P.H.; et al. A Review of Chemistry and Pharmacology of Piperidine Alkaloids of Pinus and Related Genera. Curr. Pharm. Biotechnol. 2022, 23, 1132–1141.
- Gill, J.; Sharma, A. Prospects of halofuginone as an antiprotozoal drug scaffold. Drug Discov. Today 2022, 27, 2586–2592.
- Ward, R.A.; Fawell, S.; Floc′h, N.; Flemington, V.; McKerrecher, D.; Smith, P.D. Challenges and Opportunities in Cancer Drug Resistance. Chem. Rev. 2021, 121, 3297–3351.
- Arumugam, N.; Almansour, A.I.; Kumar, R.S.; Al-thamili, D.M.; Periyasami, G.; Periasamy, V.S.; Athinarayanan, J.; Alshatwi, A.A.; Mahalingam, S.M.; Menéndez, J.C. Regio and stereoselective synthesis of anticancer spirooxindolopyrrolidine embedded piperidone heterocyclic hybrids derived from one-pot cascade protocol. Chem. Cent. J. 2018, 12, 95.
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756.
- Lovering, F. Escape from Flatland 2: Complexity and promiscuity. MedChemComm 2013, 4, 515–519.
- Harmata, A.S.; Roldan, B.J.; Stephenson, C.R.J. Formal Cycloadditions Driven by the Homolytic Opening of Strained, Saturated Ring Systems. Angew. Chem. Int. Ed. 2023, 62, e202213003.
- Dayal, N.; Wang, M.; Sintim, H.O. HSD1787, a Tetrahydro-3H-PyrazoloQuinoline Compound Synthesized via Povarov Reaction, Potently Inhibits Proliferation of Cancer Cell Lines at Nanomolar Concentrations. ACS Omega 2020, 5, 23799–23807.
- Irie, T.; Asami, T.; Sawa, A.; Uno, Y.; Taniyama, C.; Funakoshi, Y.; Masai, H.; Sawa, M. Discovery of AS-0141, a Potent and Selective Inhibitor of CDC7 Kinase for the Treatment of Solid Cancers. J. Med. Chem. 2021, 64, 14153–14164.
- Kuznetcova, I.; Ostojić, M.; Gligorijević, N.; Aranđelović, S.; Arion, V.B. Enriching Chemical Space of Bioactive Scaffolds by New Ring Systems: Benzazocines and Their Metal Complexes as Potential Anticancer Drugs. Inorg. Chem. 2022, 61, 20445–20460.
- Yin, D.-L.; Liang, Y.-J.; Zheng, T.-S.; Song, R.-P.; Wang, J.-B.; Sun, B.-S.; Pan, S.-H.; Qu, L.-D.; Liu, J.-R.; Jiang, H.-C.; et al. EF24 inhibits tumor growth and metastasis via suppressing NF-kappaB dependent pathways in human cholangiocarcinoma. Sci. Rep. 2016, 6, 32167.
- He, Y.; Li, W.; Hu, G.; Sun, H.; Kong, Q. Bioactivities of EF24, a Novel Curcumin Analog: A Review. Front. Oncol. 2018, 8, 614.
- Bazzaro, M.; Linder, S. Dienone Compounds: Targets and Pharmacological Responses. J. Med. Chem. 2020, 63, 15075–15093.
- Khurana, N.; Dodhiawala, P.B.; Bulle, A.; Lim, K.-H. Deciphering the Role of Innate Immune NF-ĸB Pathway in Pancreatic Cancer. Cancers 2020, 12, 2675.
- Liu, S.; Jiang, Y.; Yan, R.; Li, Z.; Wan, S.; Zhang, T.; Wu, X.; Hou, J.; Zhu, Z.; Tian, Y.; et al. Design, synthesis and biological evaluations of 2-amino-4-(1-piperidine) pyridine derivatives as novel anti crizotinib-resistant ALK/ROS1 dual inhibitors. Eur. J. Med. Chem. 2019, 179, 358–375.
- Golding, B.; Luu, A.; Jones, R.; Viloria-Petit, A.M. The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer (NSCLC). Mol. Cancer 2018, 17, 52.
- Kong, X.; Pan, P.; Sun, H.; Xia, H.; Wang, X.; Li, Y.; Hou, T. Drug Discovery Targeting Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2019, 62, 10927–10954.
- Du, X.; Shao, Y.; Qin, H.-F.; Tai, Y.-H.; Gao, H.-J. ALK-rearrangement in non-small-cell lung cancer (NSCLC). Thorac. Cancer 2018, 9, 423–430.
- Guaitoli, G.; Bertolini, F.; Bettelli, S.; Manfredini, S.; Maur, M.; Trudu, L.; Aramini, B.; Masciale, V.; Grisendi, G.; Dominici, M.; et al. Deepening the Knowledge of ROS1 Rearrangements in Non-Small Cell Lung Cancer: Diagnosis, Treatment, Resistance and Concomitant Alterations. Int. J. Mol. Sci. 2021, 22, 12867.
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.-P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure Based Drug Design of Crizotinib (PF-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal–Epithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363.
- Radaram, B.; Pisaneschi, F.; Rao, Y.; Yang, P.; Piwnica-Worms, D.; Alauddin, M.M. Novel derivatives of anaplastic lymphoma kinase inhibitors: Synthesis, radiolabeling, and preliminary biological studies of fluoroethyl analogues of crizotinib, alectinib, and ceritinib. Eur. J. Med. Chem. 2019, 182, 111571.
- Moi, D.; Nocentini, A.; Deplano, A.; Osman, S.M.; AlOthman, Z.A.; Piras, V.; Balboni, G.; Supuran, C.T.; Onnis, V. Appliance of the piperidinyl-hydrazidoureido linker to benzenesulfonamide compounds: Synthesis, in vitro and in silico evaluation of potent carbonic anhydrase II, IX and XII inhibitors. Bioorganic Chem. 2020, 98, 103728.
- Mishra, C.B.; Tiwari, M.; Supuran, C.T. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: Where are we today? Med. Res. Rev. 2020, 40, 2485–2565.
- Krasavin, M.; Kalinin, S.; Sharonova, T.; Supuran, C.T. Inhibitory activity against carbonic anhydrase IX and XII as a candidate selection criterion in the development of new anticancer agents. J. Enzym. Inhib. Med. Chem. 2020, 35, 1555–1561.
- McDonald, P.C.; Chia, S.; Bedard, P.L.; Chu, Q.; Lyle, M.; Tang, L.; Singh, M.; Zhang, Z.; Supuran, C.T.; Renouf, D.J.; et al. A Phase 1 Study of SLC-0111, a Novel Inhibitor of Carbonic Anhydrase IX, in Patients With Advanced Solid Tumors. Am. J. Clin. Oncol. 2020, 43, 484–490.
- Ciccone, V.; Filippelli, A.; Angeli, A.; Supuran, C.T.; Morbidelli, L. Pharmacological Inhibition of CA-IX Impairs Tumor Cell Proliferation, Migration and Invasiveness. Int. J. Mol. Sci. 2020, 21, 2983.
- Bononi, G.; Tonarini, G.; Poli, G.; Barravecchia, I.; Caligiuri, I.; Macchia, M.; Rizzolio, F.; Demontis, G.C.; Minutolo, F.; Granchi, C.; et al. Monoacylglycerol lipase (MAGL) inhibitors based on a diphenylsulfide-benzoylpiperidine scaffold. Eur. J. Med. Chem. 2021, 223, 113679.
- Granchi, C.; Bononi, G.; Ferrisi, R.; Gori, E.; Mantini, G.; Glasmacher, S.; Poli, G.; Palazzolo, S.; Caligiuri, I.; Rizzolio, F.; et al. Design, synthesis and biological evaluation of second-generation benzoylpiperidine derivatives as reversible monoacylglycerol lipase (MAGL) inhibitors. Eur. J. Med. Chem. 2021, 209, 112857.
- Gil-Ordóñez, A.; Martín-Fontecha, M.; Ortega-Gutiérrez, S.; López-Rodríguez, M.L. Monoacylglycerol lipase (MAGL) as a promising therapeutic target. Biochem. Pharmacol. 2018, 157, 18–32.
- Chitti, S.; Pulya, S.; Nandikolla, A.; Patel, T.K.; Karan Kumar, B.; Murugesan, S.; Ghosh, B.; Sekhar, K.V.G.C. Design, synthesis and biological evaluation of 7–(5–((substituted–amino)-methyl)-thiophen–2–yl)-spiro-–4–one hydrochloride analogues as anticancer agents. Bioorganic Chem. 2021, 112, 104865.
- Karmacharya, U.; Chaudhary, P.; Lim, D.; Dahal, S.; Awasthi, B.P.; Park, H.D.; Kim, J.-A.; Jeong, B.-S. Synthesis and anticancer evaluation of 6-azacyclonol-2,4,6-trimethylpyridin-3-ol derivatives: M3 muscarinic acetylcholine receptor-mediated anticancer activity of a cyclohexyl derivative in androgen-refractory prostate cancer. Bioorganic Chem. 2021, 110, 104805.
- Felton, J.; Hu, S.; Raufman, J.-P. Targeting M3 Muscarinic Receptors for Colon Cancer Therapy. Curr. Mol. Pharmacol. 2018, 11, 184–190.
- Tolaymat, M.; Larabee, S.M.; Hu, S.; Xie, G.; Raufman, J.-P. The Role of M3 Muscarinic Receptor Ligand-Induced Kinase Signaling in Colon Cancer Progression. Cancers 2019, 11, 308.
- Ali, O.; Tolaymat, M.; Hu, S.; Xie, G.; Raufman, J.-P. Overcoming Obstacles to Targeting Muscarinic Receptor Signaling in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 716.
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer′s disease. Lancet 2021, 397, 1577–1590.
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Khachaturian, A.S.; Vergallo, A.; Farlow, M.R.; Snyder, P.J.; Giacobini, E.; Khachaturian, Z.S.; Cholinergic System Working, G.; et al. Revisiting the Cholinergic Hypothesis in Alzheimer’s Disease: Emerging Evidence from Translational and Clinical Research. J. Prev. Alzheimer’s Dis. 2019, 6, 2–15.
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer′s disease. Neuropharmacology 2021, 190, 108352.
- Stanciu, G.D.; Luca, A.; Rusu, R.N.; Bild, V.; Beschea Chiriac, S.I.; Solcan, C.; Bild, W.; Ababei, D.C. Alzheimer’s Disease Pharmacotherapy in Relation to Cholinergic System Involvement. Biomolecules 2020, 10, 40.
- Chierrito, T.P.C.; Pedersoli-Mantoani, S.; Roca, C.; Sebastian-Pérez, V.; Martínez-Gonzalez, L.; Pérez, D.I.; Perez, C.; Canales, A.; Cañada, F.J.; Campillo, N.E.; et al. Chameleon-like behavior of indolylpiperidines in complex with cholinesterases targets: Potent butyrylcholinesterase inhibitors. Eur. J. Med. Chem. 2018, 145, 431–444.
- Cai, R.; Wang, L.-N.; Fan, J.-J.; Geng, S.-Q.; Liu, Y.-M. New 4-N-phenylaminoquinoline derivatives as antioxidant, metal chelating and cholinesterase inhibitors for Alzheimer’s disease. Bioorganic Chem. 2019, 93, 103328.
- Zhu, J.; Wang, L.-N.; Cai, R.; Geng, S.-Q.; Dong, Y.-F.; Liu, Y.-M. Design, synthesis, evaluation and molecular modeling study of 4-N-phenylaminoquinolines for Alzheimer disease treatment. Bioorganic Med. Chem. Lett. 2019, 29, 1325–1329.
- Košak, U.; Brus, B.; Knez, D.; Žakelj, S.; Trontelj, J.; Pišlar, A.; Šink, R.; Jukič, M.; Živin, M.; Podkowa, A.; et al. The Magic of Crystal Structure-Based Inhibitor Optimization: Development of a Butyrylcholinesterase Inhibitor with Picomolar Affinity and in Vivo Activity. J. Med. Chem. 2018, 61, 119–139.
- Košak, U.; Strašek, N.; Knez, D.; Jukič, M.; Žakelj, S.; Zahirović, A.; Pišlar, A.; Brazzolotto, X.; Nachon, F.; Kos, J.; et al. N-alkylpiperidine carbamates as potential anti-Alzheimer’s agents. Eur. J. Med. Chem. 2020, 197, 112282.
- Wichur, T.; Więckowska, A.; Więckowski, K.; Godyń, J.; Jończyk, J.; Valdivieso, Á.d.R.; Panek, D.; Pasieka, A.; Sabaté, R.; Knez, D.; et al. 1-Benzylpyrrolidine-3-amine-based BuChE inhibitors with anti-aggregating, antioxidant and metal-chelating properties as multifunctional agents against Alzheimer’s disease. Eur. J. Med. Chem. 2020, 187, 111916.
- Prati, F.; Bottegoni, G.; Bolognesi, M.L.; Cavalli, A. BACE-1 Inhibitors: From Recent Single-Target Molecules to Multitarget Compounds for Alzheimer’s Disease. J. Med. Chem. 2018, 61, 619–637.
- Gehlot, P.; Kumar, S.; Kumar Vyas, V.; Singh Choudhary, B.; Sharma, M.; Malik, R. Guanidine-based β amyloid precursor protein cleavage enzyme 1 (BACE-1) inhibitors for the Alzheimer′s disease (AD): A review. Bioorganic Med. Chem. 2022, 74, 117047.
- Frisoni, G.B.; Altomare, D.; Thal, D.R.; Ribaldi, F.; van der Kant, R.; Ossenkoppele, R.; Blennow, K.; Cummings, J.; van Duijn, C.; Nilsson, P.M.; et al. The probabilistic model of Alzheimer disease: The amyloid hypothesis revised. Nat. Rev. Neurosci. 2022, 23, 53–66.
- Sugimoto, H.; Yamanish, Y.; Iimura, Y.; Kawakami, Y. Donepezil Hydrochloride (E2020) and Other Acetylcholinesterase Inhibitors. Curr. Med. Chem. 2000, 7, 303–339.
- El-Sayed, N.A.-E.; Farag, A.E.-S.; Ezzat, M.A.F.; Akincioglu, H.; Gülçin, İ.; Abou-Seri, S.M. Design, synthesis, in vitro and in vivo evaluation of novel pyrrolizine-based compounds with potential activity as cholinesterase inhibitors and anti-Alzheimer′s agents. Bioorganic Chem. 2019, 93, 103312.
- Sharma, P.; Tripathi, A.; Tripathi, P.N.; Prajapati, S.K.; Seth, A.; Tripathi, M.K.; Srivastava, P.; Tiwari, V.; Krishnamurthy, S.; Shrivastava, S.K. Design and development of multitarget-directed N-Benzylpiperidine analogs as potential candidates for the treatment of Alzheimer′s disease. Eur. J. Med. Chem. 2019, 167, 510–524.
- van Greunen, D.G.; Johan van der Westhuizen, C.; Cordier, W.; Nell, M.; Stander, A.; Steenkamp, V.; Panayides, J.-L.; Riley, D.L. Novel N-benzylpiperidine carboxamide derivatives as potential cholinesterase inhibitors for the treatment of Alzheimer′s disease. Eur. J. Med. Chem. 2019, 179, 680–693.
- Sadeghian, B.; Sakhteman, A.; Faghih, Z.; Nadri, H.; Edraki, N.; Iraji, A.; Sadeghian, I.; Rezaei, Z. Design, synthesis and biological activity evaluation of novel carbazole-benzylpiperidine hybrids as potential anti Alzheimer agents. J. Mol. Struct. 2020, 1221, 128793.
- Chowdhury, S.R.; Gu, J.; Hu, Y.; Wang, J.; Lei, S.; Tavallaie, M.S.; Lam, C.; Lu, D.; Jiang, F.; Fu, L. Synthesis, biological evaluation and molecular modeling of benzofuran piperidine derivatives as Aβ antiaggregant. Eur. J. Med. Chem. 2021, 222, 113541.
- Wang, K.; Shi, J.; Zhou, Y.; He, Y.; Mi, J.; Yang, J.; Liu, S.; Tang, X.; Liu, W.; Tan, Z.; et al. Design, synthesis and evaluation of cinnamic acid hybrids as multi-target-directed agents for the treatment of Alzheimer′s disease. Bioorganic Chem. 2021, 112, 104879.
- Liu, W.; Wu, L.; Liu, W.; Tian, L.; Chen, H.; Wu, Z.; Wang, N.; Liu, X.; Qiu, J.; Feng, X.; et al. Design, synthesis and biological evaluation of novel coumarin derivatives as multifunctional ligands for the treatment of Alzheimer′s disease. Eur. J. Med. Chem. 2022, 242, 114689.
- Vereshchagin, A.N.; Frolov, N.A.; Egorova, K.S.; Seitkalieva, M.M.; Ananikov, V.P. Quaternary Ammonium Compounds (QACs) and Ionic Liquids (ILs) as Biocides: From Simple Antiseptics to Tunable Antimicrobials. Int. J. Mol. Sci. 2021, 22, 6793.
- Li, B.; Wang, K.; Zhang, R.; Li, B.; Shen, Y.; Ji, Q. Design, synthesis and biological evaluation of novel diazaspirodecan-1-one derivatives as potential chitin synthase inhibitors and antifungal agents. Eur. J. Med. Chem. 2019, 182, 111669.
- Krauß, J.; Müller, C.; Klimt, M.; Valero, L.J.; Martínez, J.F.; Müller, M.; Bartel, K.; Binder, U.; Bracher, F. Synthesis, Biological Evaluation, and Structure & ndash; Activity Relationships of 4-Aminopiperidines as Novel Antifungal Agents Targeting Ergosterol Biosynthesis. Molecules 2021, 26, 7208.
- Rodrigues Marcio, L. The Multifunctional Fungal Ergosterol. Mbio 2018, 9, e01755-18.
- Hong, G.; Li, W.; Mao, L.; Wang, J.; Liu, T. Synthesis and antibacterial activity evaluation of N (7) position-modified balofloxacins. Front. Chem. 2022, 10, 963442.
- Karoutzou, O.; Benaki, D.; Papanastasiou, I.; Vocat, A.; Cole, S.T. Synthesis of New Indole and Adamantane Amido Derivatives with Pharmacological Interest. ChemistrySelect 2019, 4, 8727–8730.
- de Castro, S.; Ginex, T.; Vanderlinden, E.; Laporte, M.; Stevaert, A.; Cumella, J.; Gago, F.; Camarasa, M.J.; Luque, F.J.; Naesens, L.; et al. N-benzyl 4,4-disubstituted piperidines as a potent class of influenza H1N1 virus inhibitors showing a novel mechanism of hemagglutinin fusion peptide interaction. Eur. J. Med. Chem. 2020, 194, 112223.
- Nandini Asha, R.; Ravindran Durai Nayagam, B.; Bhuvanesh, N. Synthesis, molecular docking, and in silico ADMET studies of 4-benzyl-1-(2,4,6-trimethyl-benzyl)-piperidine: Potential Inhibitor of SARS-CoV2. Bioorganic Chem. 2021, 112, 104967.
- Lepovitz, L.T.; Meis, A.R.; Thomas, S.M.; Wiedeman, J.; Pham, A.; Mensa-Wilmot, K.; Martin, S.F. Design, synthesis, and evaluation of novel anti-trypanosomal compounds. Tetrahedron 2020, 76, 131086.
- Van de Walle, T.; Boone, M.; Van Puyvelde, J.; Combrinck, J.; Smith, P.J.; Chibale, K.; Mangelinckx, S.; D′hooghe, M. Synthesis and biological evaluation of novel quinoline-piperidine scaffolds as antiplasmodium agents. Eur. J. Med. Chem. 2020, 198, 112330.
- Seck, R.; Gassama, A.; Cojean, S.; Cavé, C. Synthesis and Antimalarial Activity of 1,4-Disubstituted Piperidine Derivatives. Molecules 2020, 25, 299.
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002.
- Valentino, R.J.; Volkow, N.D. Untangling the complexity of opioid receptor function. Neuropsychopharmacology 2018, 43, 2514–2520.
- Günther, T.; Dasgupta, P.; Mann, A.; Miess, E.; Kliewer, A.; Fritzwanker, S.; Steinborn, R.; Schulz, S. Targeting multiple opioid receptors–improved analgesics with reduced side effects? Br. J. Pharmacol. 2018, 175, 2857–2868.
- Armenian, P.; Vo, K.T.; Barr-Walker, J.; Lynch, K.L. Fentanyl, fentanyl analogs and novel synthetic opioids: A comprehensive review. Neuropharmacology 2018, 134, 121–132.
- Nami, M.; Salehi, P.; Dabiri, M.; Bararjanian, M.; Gharaghani, S.; Khoramjouy, M.; Al-Harrasi, A.; Faizi, M. Synthesis of novel norsufentanil analogs via a four-component Ugi reaction and in vivo, docking, and QSAR studies of their analgesic activity. Chem. Biol. Drug Des. 2018, 91, 902–914.
- Hsu, F.-L.; Walz, A.J.; Myslinski, J.M.; Kong, L.; Feasel, M.G.; Goralski, T.D.P.; Rose, T.; Cooper, N.J.; Roughley, N.; Timperley, C.M. Synthesis and μ-Opioid Activity of the Primary Metabolites of Carfentanil. ACS Med. Chem. Lett. 2019, 10, 1568–1572.
- Huang, H.; Wang, W.; Xu, X.; Zhu, C.; Wang, Y.; Liu, J.; Li, W.; Fu, W. Discovery of 3-((dimethylamino)methyl)-4-hydroxy-4-(3-methoxyphenyl)-N-phenylpiperidine-1-carboxamide as novel potent analgesic. Eur. J. Med. Chem. 2020, 189, 112070.
- Lee, H.; Ahn, S.; Ann, J.; Ha, H.; Yoo, Y.D.; Kim, Y.H.; Hwang, J.-Y.; Hur, K.-H.; Jang, C.-G.; Pearce, L.V.; et al. Discovery of dual-acting opioid ligand and TRPV1 antagonists as novel therapeutic agents for pain. Eur. J. Med. Chem. 2019, 182, 111634.
- Benítez-Angeles, M.; Morales-Lázaro, S.L.; Juárez-González, E.; Rosenbaum, T. TRPV1: Structure, Endogenous Agonists, and Mechanisms. Int. J. Mol. Sci. 2020, 21, 3421.
- Déciga-Campos, M.; Melo-Hernández, L.A.; Torres-Gómez, H.; Wünsch, B.; Schepmann, D.; González-Trujano, M.E.; Espinosa-Juárez, J.; López-Muñoz, F.J.; Navarrete-Vázquez, G. Design and synthesis of N-(benzylpiperidinyl)-4-fluorobenzamide: A haloperidol analog that reduces neuropathic nociception via σ1 receptor antagonism. Life Sci. 2020, 245, 117348.
- Johnson, A.C.; Greenwood-Van Meerveld, B. Chapter Ten–The Pharmacology of Visceral Pain. In Advances in Pharmacology; Barrett, J.E., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 75, pp. 273–301.
- Xiong, J.; Jin, J.; Gao, L.; Hao, C.; Liu, X.; Liu, B.-F.; Chen, Y.; Zhang, G. Piperidine propionamide as a scaffold for potent sigma-1 receptor antagonists and mu opioid receptor agonists for treating neuropathic pain. Eur. J. Med. Chem. 2020, 191, 112144.
- Xiong, J.; Zhuang, T.; Ma, Y.; Xu, J.; Ye, J.; Ma, R.; Zhang, S.; Liu, X.; Liu, B.-F.; Hao, C.; et al. Optimization of bifunctional piperidinamide derivatives as σ1R Antagonists/MOR agonists for treating neuropathic pain. Eur. J. Med. Chem. 2021, 226, 113879.
- Kalisha Vali, Y.; Gundla, R.; Singh, O.V.; Tamboli, Y.; Di Cesare Manelli, L.; Ghelardini, C.; Al-Tamimi, A.-M.S.; Carta, F.; Angeli, A.; Supuran, C.T. Spirocyclic sulfonamides with carbonic anhydrase inhibitory and anti-neuropathic pain activity. Bioorganic Chem. 2019, 92, 103210.
More
Information
Subjects:
Chemistry, Medicinal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
17.2K
Revisions:
3 times
(View History)
Update Date:
09 Feb 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No