Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | JOSÉ LUIS VILLALPANDO-AGUILAR | -- | 4714 | 2023-02-07 20:25:12 | | | |
| 2 | Rita Xu | Meta information modification | 4714 | 2023-02-08 03:34:55 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Villalpando-Aguilar, J.L.; Matos-Pech, G.; López-Rosas, I.; Castelán-Sánchez, H.G.; Alatorre-Cobos, F. Phage Therapy for Crops. Encyclopedia. Available online: https://encyclopedia.pub/entry/40944 (accessed on 25 June 2026).
Villalpando-Aguilar JL, Matos-Pech G, López-Rosas I, Castelán-Sánchez HG, Alatorre-Cobos F. Phage Therapy for Crops. Encyclopedia. Available at: https://encyclopedia.pub/entry/40944. Accessed June 25, 2026.
Villalpando-Aguilar, José Luis, Gilberto Matos-Pech, Itzel López-Rosas, Hugo Gildardo Castelán-Sánchez, Fulgencio Alatorre-Cobos. "Phage Therapy for Crops" Encyclopedia, https://encyclopedia.pub/entry/40944 (accessed June 25, 2026).
Villalpando-Aguilar, J.L., Matos-Pech, G., López-Rosas, I., Castelán-Sánchez, H.G., & Alatorre-Cobos, F. (2023, February 07). Phage Therapy for Crops. In Encyclopedia. https://encyclopedia.pub/entry/40944
Villalpando-Aguilar, José Luis, et al. "Phage Therapy for Crops." Encyclopedia. Web. 07 February, 2023.
Copy Citation
Phage therapy consists of applying bacteriophages, whose natural function is to kill specific bacteria. Bacteriophages are safe, evolve together with their host, and are environmentally friendly. The indiscriminate use of antibiotics and salt minerals (Zn2+ or Cu2+) has caused the emergence of resistant strains that infect crops, causing difficulties and loss of food production. Phage therapy is an alternative that has shown positive results and can improve the treatments available for agriculture.
bacteriophages
genomics
phage therapy
crop
1. Introduction
Earth is home to a great diversity of bacteriophages, which destroy between 4% and 50% of bacteria, thus contributing to natural bacterial control [1].
In 1917, Félix Hubert d’Herelle used the term “bacteriophages” for the first time, focusing on their application for bacterial biocontrol [2]. In the case of phage therapy in plants, in 1924, Mallman and Hermstreet discovered that a filtrate of decomposed cabbage inhibited the growth of Xanthomonas campestris pv. campestris, responsible for cabbage rot disease [3].
In 1926, Moore described experiments that showed successful biocontrol of Erwinia carotovora subsp. atroseptica (currently Pectobacterium carotova subsp. atroseptica), the causative agent of potato tuber rot, and promoted phage therapy as an alternative for disease biocontrol in crops [4]. However, phage therapy was overshadowed by other bacterial disease control alternatives. During 1928–1940, the British scientist Alexander Fleming discovered penicillin, giving rise to the antibiotics era, with a concomitant decreasing interest in applying phages for bacterial control [5]. In 1975, Frederick Williams Twort studied a “bacterial lytic factor” identified as a compound capable of generating bacterial lysis [6].
Since then, antibiotics and mineral salts (Zn2+ or Cu2+) have been indiscriminately employed as bacterial treatments for crops, and have generated resistant strains, thus promoting research on biocontrol alternatives for super-resistant bacteria. One option is phage therapy, which uses bacteriophages as agents that kill bacteria. The advantages are that bacteriophages evolve together with their host; due to their great diversity, it is increasingly possible to find phages for specific diseases; and they are safe and compatible with the environment [7][8].
Therefore, the success of phage therapy is limited by the selection process, in which a high reproduction rate (PFU/mL) and lytic capacity are sought. It is more likely to isolate a lysogenic phage than a lytic phage. Therefore, it is essential to involve modern approaches, such as studying the genetic material of the host (bacteria) and the control agent (phage), which, combined, will increase the possibility of selecting lytic phages for use as bacterial biocontrol agents and reduce the time it takes to do so [9].
It should be noted that it is possible to predict structural characteristics, infection mechanisms, and lytic capacity from phage genomes. On the other hand, bacterial genomes allow the selection of stable hosts for phage propagation, infection susceptibility, and receptor prediction as means of interaction for successful infection. Therefore, complementing experimental strategies with in silico analysis of bacteria and phages can enhance phage therapy for agricultural cultures [10][11].
Bacteriophages are applied as an alternative treatment for bacterial agricultural diseases, and they have shown the ability to reduce Pseudomonas spp., Xanthomonas spp., Pectobacterium spp., Ralstonia spp., Clavibacter michiganensis, and Agrobacterium tumefaciens [12]. In the USA, products approved by the FDA for use in agriculture, such as AgriPhage, have shown positive results in the biocontrol of pepper spot, speck, and tomato canker [13].
2. Bacteriophages Used for Biocontrol of Crop Diseases
The class Caudoviricetes is the most studied among the phage groups, as it is the order in which phages for biocontrol of agricultural crop diseases are found. Its members have an icosahedral capsid and a tail containing trimeric protein fibers in the lower part that serves as a receptor for interaction with the host cell, with stability in L morphology [14]. The class Caudoviricetes include phages according to the tail shape: the myovirus includes phages with long and contractile tails that stand out, the podovirus have short non-contractile tails, and the siphovirus have long and flexible tails (Figure 1) [14][15]. One of the most studied phages is a tailed phage with a dsDNA genome which has three main parts in its structure: a capsid, a tail, and an absorption apparatus. In the assembly process, the capsid of bacteriophages has a structure called a procapsid. Scaffolding proteins have the function of supervising other subunits of the main capsid to facilitate the formation of the icosahedral procapsid, where the dsDNA will be stored. The mechanism of change from procapsid to capsid within the genome is called the ripening process.
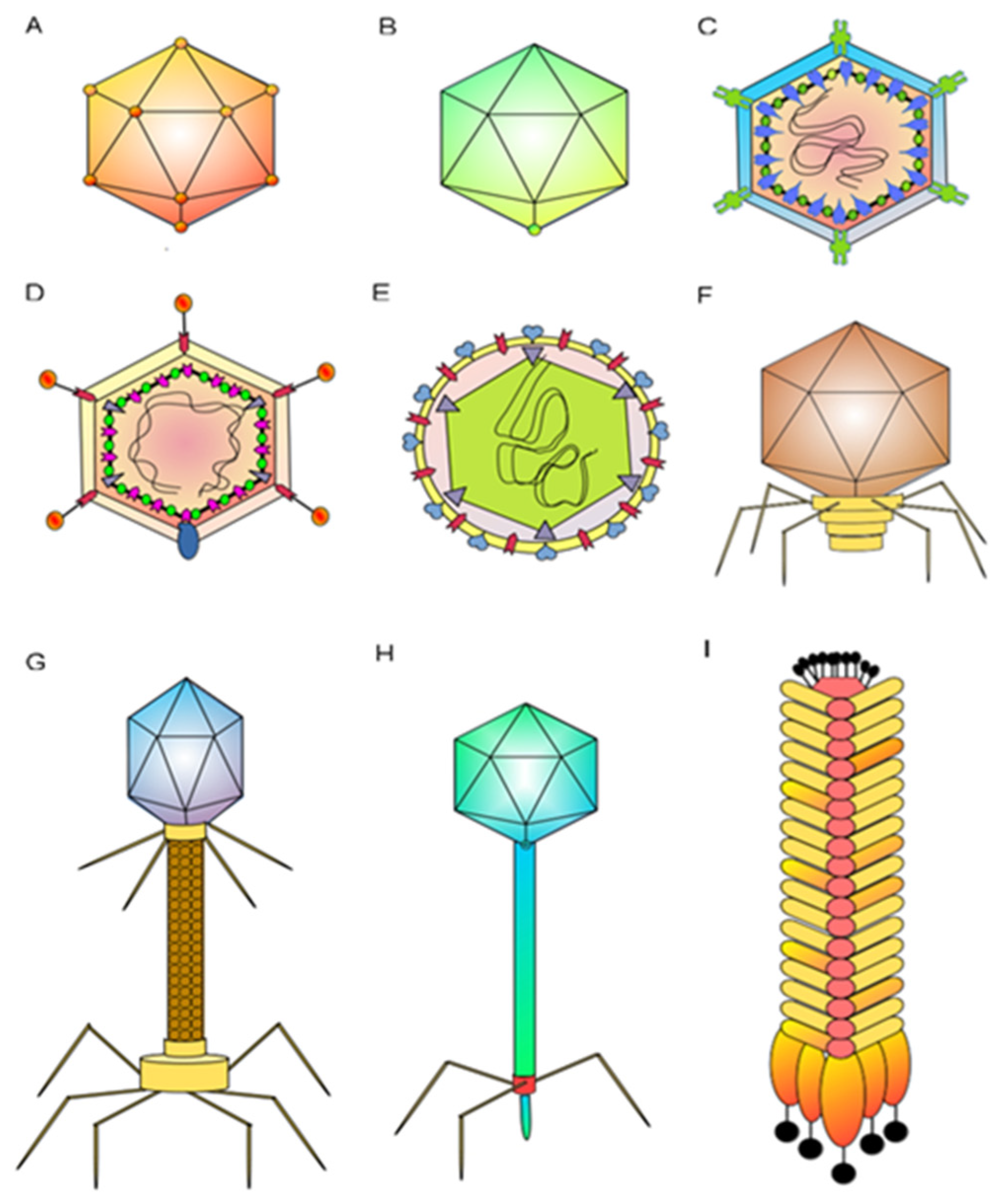
Figure 1. Schematic representation of the most commonly studied phages [16]: (A) levivirus MS2 has a capsid with icosahedral symmetry and a size of about 26 nm; (B) microvirus ϕX174 is a non-evolved icosahedral capsid about 30 nm in size; (C) podovirus T7 is a non-evolved icosahedral capsid about 60 nm in size; (D) tectivirus PRD1 is a non-enveloped icosahedral capsid with a size of about 66 nm; (E) cystovirus phi6 is an enveloped spherical virion 85 nm in diameter; outer and inner capsids have icosahedral symmetry; (F) corticovirusPM2 is an icosahedral capsid 56 nm in diameter; (G) myovirus T4 is non-enveloped, with a morphological head–tail structure about 110 nm in length; (H) siphovirus T5 is non-enveloped, with a head–tail structure; head is about 60 nm in diameter; and (I) inovirus M13 is non-enveloped, with rods of filaments 7 nm in diameter and 700 to 2000 nm in length.
The organization of phage tails is associated with the type of bacteriophage; for instance, siphovirus have long and flexible tails, podovirus have short tails with adhesive properties, and myovirus have long, rigid, contractile tails that shape the rigid internal tube and external contractile sheath. On the other hand, many phages that can infect gram-negative bacteria have an absorption apparatus, which is an oligomeric ring formed by proteins from the distal tail attached to the tailed tube’s last ring. It has the function of recognizing and connecting with receptor-binding proteins [17].
The tectivirus genus structure consists of a rigid protein capsid containing a thick lipoprotein and a flexible vesicle and dsDNA, which gives these phages the ability to infect both gram-positive bacteria, such as the betatectivirus genus, and gram-negative bacteria with the alphatectivirus and PRD1 genus [18], which is important, as gram-negative bacteria cause the most problems in economically important crops [19]. The corticovirus also have lipid layers in the protein capsid; they infect gram-negative Pseudoalteromonas spp. and are associated with a PM2 capsid architecture with fold trimeric proteins containing two β-barrels forming hexagonal capsomeres [20].
In the case of archaeal viruses, they can be divided into two groups: In the first group, the relationships between the morphologic and genetic aspects of these viruses are unique. The second group has clear genetic and structural similarities to bacteriophages and eukaryotic viruses. The majority of archaea viruses have linear or circular dsDNA genomes, while only two families present single DNA genomes. No RNA viruses have been isolated, but they have been detected in metagenomic studies [21].
Currently, studies are focused on the structure and assembly of archaeal viruses, which requires characterization of unexplored host phyla to increase the knowledge of archaeal virus diversity [22].
Proteobacteria are mainly gram-negative and cause many diseases in agricultural crops. From a structural point of view, some examples show how the structure of bacteriophage is related to bacterial species that cause damage to agricultural crops. For instance, Xanthomonas spp. are infected by phage Xaj2 and phage Xf2. Phage RSM3 infects Ralstonia solanacearum. Pseudomonas syringae is infected by like phage phobos or phage MR1-MR18, classified in podovirus or myovirus, and cystovirus as the phage phi6 or phi8. The most studied are myovirus phage T4 and levivirus as phage MS2, which infect E. coli (Figure 1) [23][24][25][26].
3. Bacteriophage Classification
The International Committee on Taxonomy of Viruses (ICTV) has established the official taxonomy. Historically, in the past, viruses were classified based on several criteria, such as propagation characteristics in cell culture, virion morphology, serology, nucleic acid sequence, host range, pathogenicity, and epidemiology or epizootiology [27]. Recently, new proposals have been made to improve virus classification because there is no standardized and automatic universally accepted virus classification. Technological advances in sequencing, and especially metagenomics projects, have increased the available phage genomes and have since been used for phage genomes as a criterion for classification [28]. Essentially, Turner et al. described the abolition of the order Caudovirales and the families Myoviridae, Podoviridae, and Siphoviridae [29]. In bacteriophages, the members of the largest order (Caudovirales) were assigned to the class Caudoviricetes. The old families (Myoviridae, Podoviridae, and Siphoviridae) were replaced by the families Strabovoridae, Drexlerviridae, and Autographiviridae, which have a high similarity to the old families [30][31].
In addition, the order Tubulavirales, which includes the phages of the family Inoviridae, was divided into two families, Inoviridae and Plectroviridae [32][33]. In the family Microviridae, additional subfamilies beyond the existing Gokushovirinae and Bullavirinae have been proposed, namely the subfamilies Alpavirinae, Stokavirinae, Aravirinae, and Pichovirinae based on virome data. Finally, the family Leviviridae, which described a comprehensive identification of ssRNA based on computational approaches, used the phage genome [33][34][35][36].
4. Bacteriophage Infection Mechanisms
Bacteriophages are capable of reproducing by two biological strategies to perpetuate their genetic material. One strategy is the lytic cycle, which is the most aggressive, since it kills the host cell. The first step consists of fixation: the bacteriophage binds to the host cell through a ligand–receptor interaction [37]. This interaction is so specific that it allows differentiation between gram-negative and gram-positive hosts [38].
The first interactions are made by the virus to specific receptors, such as lipopolysaccharides (LPSs) or outer membrane proteins. The binding of two, and even up to six fibers to the receptor sends a signal to the base of the base plate that promotes a conformational change, leading to contraction of the fibers, which readies the bacteriophage into an injection position. Mainly, the stem pierces the membrane, generating a channel that allows the injection of genetic material. This perforation is supported by endopeptidases (N-acetylmuramyl-L-alanine amidase, lysozymes, transglycosylases), which commonly degrade peptidoglycan; these enzymes are part of the fiber structure of bacteriophages [37]. With the genetic material introduced into the bacterium, the host’s replication, transcription, and translation machinery are hijacked to generate the genetic material and to synthesize and obtain viral proteins. First, protein messengers are synthesized to stabilize and protect DNA or RNA molecules from degradation. In DNA viruses, the genetic material is used directly as a template for transcribing genes that code for structural proteins. In RNA viruses, a reverse transcription step is required to achieve the genetic material to be replicated. The genes that encode structural proteins are transcribed, and as soon as the structural proteins are generated, the copies of genetic material begin to assemble into the virion, stabilizing the DNA or RNA molecules with proteins. Then the capsid, the stem, and the fibers are assembled with the base plate. Additionally, lytic enzymes known as lysines are synthesized, which are encoded in the genetic material injected by the bacteriophage, and are the enzymes responsible for breaking the plasma membrane and allowing external liquid to enter the cytosol of the bacterium, which takes it to a point where the membrane breaks and releases the internal content, along with the virions, to start the lytic cycle again [37][38].
An important protein during the lytic cycle is the holin (hole-forming) protein, which exist in double-stranded DNA bacteriophages and control the length of the infectious cycle. Holins are small proteins that accumulate on the membrane and cause permeabilization. These proteins can be classified into three types according to their topology: class I with 95 residues that form three TMDs, class II with 65 to 95 residues that form two TMDs, and class III, with one TMD in the central region of the molecule [39][40].
The second method of reproduction and conservation of viral genetic material bacteriophages that have been developed and are used widely is the lysogenic cycle, which consists of the steps of fixation, injection of genetic material, and the lytic cycle. However, in this case, viral DNA is integrated into the bacterial chromosome and remains there in an inactive form as a prophage. This allows conservation of the bacteriophage sequence, which is replicated and transferred to the bacterial daughter cells through the bacterial chromosome DNA, where the virus is also duplicated. The genetic material is integrated through binding sites located on the bacterial chromosome by factors of the bacteriophage, and not by recombination or the integrated systems of the bacteria [37].
This strategy is more elegant and does not affect the viability of the host. In the literature, it is described as a strategy that seeks to preserve the host. Since bacteriophages are specific, it is believed that they sometimes use the lysogenic cycle to keep host cells alive, because if they carried out the lytic cycle all the time, they could exterminate their hosts and therefore would also be condemned to die [37].
5. Strategies for Bacteriophage Isolation from Plants
In general, the bacteriophage isolation process is as follows: First, the inoculum (phage) must be isolated, and the host bacteria must be identified; it is enough to inoculate a bacterial culture with a phage inoculum and incubate it to obtain a higher bacteriophage titer, which can be clarified by centrifugation or filtration [9].
A challenge is encountered with lytic phages, which is one of the limitations of phage therapy. The characteristics that a phage must have to be a candidate for phage therapy are lytic capacity, high progeny, and host specificity, which means it infects a single species of bacteria while leaving the rest of the microbiome intact [41]. However, in practice, it has been reported that phages can have more than one host and can attack groups of bacterial strains, which would enable solving diseases caused by a variety of bacterial strains by using a single phage with a broad spectrum of hosts. However, this is complicated when different species of bacteria cause disease; in this case, specific phages against specific bacterial strains can no longer solve the problem. A strategy that, in some cases, has made it possible to counteract bacterial growth is the use of a “phage cocktail”, which has shown promising results [19][42].
Therefore, a determining step in the success of phage therapy is isolation and characterization, and taking into consideration the type of sample and the host.
The primary isolation method, which was developed by Félix d’Herelle, consists of an enrichment process [43]. First, a sample of bacteria (host) is mixed with an environmental sample, and this must be close to the area of infection to be treated. In order to obtain phages against a specific disease, it is recommended to take a sample close to the infected site, either from the leaf (5 cm2), stem (2 cm), soil (150 g), or irrigation water (10 mL) of the infected plant. This is then mixed with the host bacterial culture [12][44]. Generally, soil samples have the highest concentration of phages. After the mixture of bacteria (host) with the environmental sample (phages) is prepared, there is an incubation period to 28–37 °C for approximately 16 to 18 h in a shaker (180 to 200 rpm/min). The application and selection of enrichment media will depend on the chosen host bacteria and the cell biomass that must be produced [45].
In general, the most common bacteria that cause plant diseases are Pectobacterium, Pantoea [46], Agrobacterium [47], Pseudomonas [48], Ralstonia [49], Burkholderia [50], Acidovorax, Xanthomonas [51], Clavibacter, Streptomyces [37], Xylella [38], Spiroplasma, and Phytoplasma [40]. The media used for the management of these species are Luria broth (LB), PEB medium, Lennox broth supplemented with calcium, nutrient broth (NB), semi-solid yeast extract agar (NYA), periwinkle wilt (PW) medium, and ATCC medium: 988 Spiroplasma SP-4 medium. Therefore, in the process of obtaining phages, it is necessary to characterize the host bacterial strain to obtain high titers and phage production [38].
Accordingly, bacteria or cell debris are removed from the culture by a physical method such as centrifugation or filtration to analyze the presence of phages. The identified phages are characterized to determine the desired properties for therapy based on their virulence capacity [9]. During the isolation process, a phase of characterizing phage properties should be included in the protocol, focused on determining the lytic capacity and spectrum of host bacteria. In some protocols, the use of chloroform is recommended for the extraction of phages. Currently, it is known that this inactivates enveloped phages, so it is not recommended; in the opposite case, the use of this organic compound would be helpful because the structure of enveloped phages has components of a lipid nature. With the isolation protocol, researchers consider obtaining the concentration that would allow obtaining a high titer of phage that has lytic capacity against a specific bacterial species [39].
However, there are exceptions, such as when phages are found in high titers in environmental samples; researchers can mix the sample with the host bacterial cell culture to achieve sufficient titers that generate inhibition halos during the characterization and phage lytic capacity. In this case, it is recommended to use a limited volume of environmental sample by plaque assay [45].
The phage concentration is generally low in most environmental samples, so adding an enrichment step to the isolation protocol is recommended. In the case of plants, the strategy for obtaining and isolating phages must be focused on taking samples from parts of the infected plant, such as leaves, roots, or stem or from irrigation water or soil [45].
5.1. Obtaining Bacteriophages from Environment Samples
The aim of the process is to find a phage with a high titer on media suitable for reproduction; for instance, tomato diseases have been treated with titers of 106 to 108 plaque-forming units (PFU)/mL [52]. The virus titer, including the phage titer, in seawater can be low, requiring preconcentration of the sample by filtration, precipitation, or both. Multiple studies recommend the use of mineral salts, such as zinc, calcium, or ferric chloride to concentrate phages from samples of seawater, wastewater, or any liquid in which the salt concentration can precipitate the phages, thus avoiding their being salvaged by water molecules, leading to phage precipitation [53][54][55].
On the other hand, bacteriophages in diluted samples can also be concentrated by flocculation, in the form of small insoluble aggregates (flocs) even at low phage concentrations. For wastewater samples, a low-speed centrifugation step is recommended to eliminate large insoluble parts and cells from the sample [56]. When using the filtration method, it is less common to concentrate phages from aqueous samples because the filters clog quickly. Nevertheless, it is an essential step in the phage collection protocol; most protocols end with a filtration step at 0.45 or 0.22 μm to eliminate bacteria, with the choice of filter pore size depending on the need to eliminate all bacteria. To retain very large bacteriophages, it is recommended to use a 0.45 μm membrane [57].
In summary, after sample collection, the phage titer can be evaluated directly, or concentrating it may be recommended, using a step of the protocol to achieve the necessary titer for the infection stage; this is achieved by directly applying the sample to the medium enriched with the host bacteria, considering that the sample must have a high enough titer to achieve successful infection. Another method that is commonly recommended is to add the phage enrichment step prior to infection by precipitation (ZnCl2 or CaCl2, or FeCl3) or floc and, in some cases, to use a filter to concentrate the phages and achieve a successful infection stage [9][39].
5.2. Experimental Detection of Bacteriophages
In this stage, it is recommended to use detection methods for new phage isolates, such as spot test, plaque test, or lysis in cultivation [58][59].
The spot test method consists of inoculating phages with the host bacteria, forming a lawn, and then placing droplets of phage on the plate. This incubation shows a lysis or halo effect related to phage activity. The advantage of this method is that it is simple and allows testing of multiple phages that are filtered on the same plate. A limitation of this method is that it requires growing the host in plate media. It is also prone to false positive results due to the lysis of bacteria by binding media components or by phages that do not lead to productive disease [42].
The plate test method consists of placing high phage dilutions obtained from filtrate together with the bacteria on the surface of the plate by extension or coating of soft agar. The plate is previously incubated with plaques to analyze afterwards. This shows evidence of phage growth, and the plaque indicates the lytic or lysogenic cycle. The size of the lytic plaque may indicate the size of the phage due to diffusion; a disadvantage is that the host must grow to converge on the plate. Most phages cannot be plated, even with highly productive hosts, due to limited agar diffusion [60]. In the culture lysis method, the phage filtrate is added to the bacterial culture broth and incubated for monitoring of cell lysis signals by the turbidity of the culture. Metabolic stains can also be used to measure the level of turbidity associated with metabolic activity. This method is used for bacteria that do not show confluence on the medium plate. Bacteria that grow in broth could be adapted for automation using spectrophotometry based on turbidity [58]. However, a limitation is the occurrence of false positives due to the absence of lysis. Cellular debris can inactivate phages by charge, affecting the infectivity. Hosts that evolve rapidly to become resistant to phages will cause false negatives [61].
In the routine dilution (RTD) method, phages are diluted to a titer that produces minor confluent cell lysis on a plate. This is used for phages that do not show morphological differences on plaques. Unfortunately, this method is inclined to produce false positives due to the fact that the media or components are not diluted [62]. The main challenges of phage detection are the inoculation quantity and specificity of the bacterial host and the viability and disposition of the phage for increasing the probability of successful infection that is possible to detect.
5.3. Detection of Bacteriophages from the Genome for Bacterial Biocontrol
Presently, there are repositories of sequenced genomes of organisms, and information on bacteriophages is available in NCBI’s PhagesDb and GenBank [59][63].
The genome is important in order to determine the constitution of a phage, and the host’s genotype and phenotype may be related. The phage genome is the genetic information that allows reproduction. Tailed dsDNA phages are the most studied, including tailed members of class Caudoviricetes, and families Straboviridae, and Drexlerviridae [31]. Other types of RNA and ssDNA phages make up a small group; there may be more that have not yet been discovered [10].
Another important characteristic is the size of the genetic material described, which can range from ~3300 nucleotides for ssRNA E. coliphages up to 500 kbp for Bacillus megaterium phage G. The size of dsDNA stem phage genomes ranges from ~11.5 kbp (Mycoplasma phage P1) to ~30 kbp (Pasteurella phage F108) for members of the families Drexlerviridae, Autographiviridae, and Straboviridae [31].
The phage genome encodes all the necessary components to generate new virions with structural proteins for the capsid and stem, and ligand proteins that cover the capsid, allowing it to interact with the host cell receptors and introduce genetic material. There may be other proteins that help in the development of infection, such as proteases, which help to evade the host’s immune response [10][41]. These genomes show the complexity of the microorganisms (bacteria and bacteriophages). In addition, researchers present an approach for comparative genome sequencing (Figure 2). Complete genomes of bacteriophages show high variability, which is a characteristic of bacteriophages. In these analyses, the sequence of phage Salvo was used as a template, and only phage Sano had a high conservation sequence >70% (yellow), because it belongs to its homologue; the other sequences added were phage phiXc10 (blue), Ralstonia phage RSM3 (pink), and Agrobacterium phage Atu_phe07 (green), which showed low conservation sequences between bacteriophage genomes (<30%) (Figure 2).
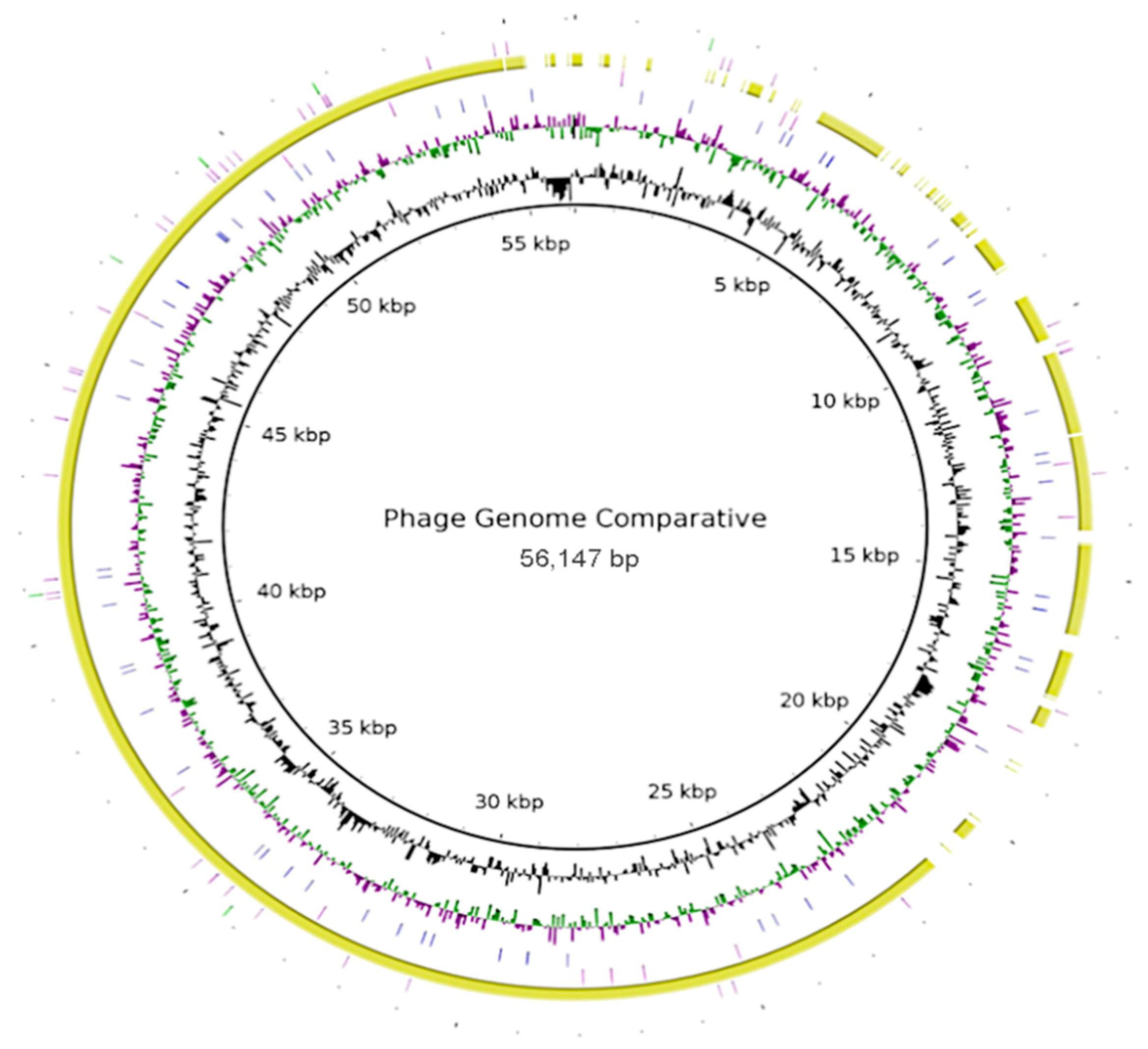
Figure 2. Comparison of bacteriophage genomes used for biocontrol of proteobacteria diseases present in agricultural crops. Complete genomes of bacteriophages were obtained from NCBI. Template sequence was phage Sano; % GC content (black) is shown. Xanthomonas phage phiXc10 (blue) and Ralstonia phage RSM3 (pink) show nucleotide sequence identity conservation <30%. Phage Salvo (yellow) shows identity about 70%, and Agrobacterium phage Atu_ph07 (green). Genomes were compared in Brig version 0.95 software, Brisbane, Australia (http://sourceforge.net/projects/brig/, accessed on 8 September 2022).
The sequences of genomes in the NCBI database showed that there are few sequenced genomes available. For the majority of bacteriophages, the genome is not available. Therefore, it is necessary to increase the number of genomes that can be obtained from other studies (assembly genomes), such as metagenomics studies, and to characterize bacteria and phage genome partners to understand host–phage relationships [64].
Currently, computational studies seek to complement the experimental laboratory studies carried out for the characterization of bacteriophages. Multiple bioinformatics applications have been developed for faster and better determination of candidates for phage therapy. Currently, the availability of viral genomic information has allowed the development of computational tools such as machine learning languages, for an approach called the phage classification tool set (PHACTS), which can identify the type of life cycle of a bacteriophage based on its protein sequences, because it is always required for the selection of lytic phages [65].
Another molecular implementation based on bacterial defense mechanisms is the use of mutations in receptors to prevent phages from interacting, making them unable to penetrate and inject their genetic material. Bacteria can detect regions in their genome that are susceptible to enzymatic cleavage of foreign DNA at specific sites. This is used as a molecular tool known as the Clustered Regularly Interspaced Short Palindromic Repeats/ Caspase 9 system (CRISPR/Cas system), which considers the immune system of bacteria that manage to confer resistance to phages [66].
Currently, genomic information is important for the development of analytical tools at the bioinformatics level. Leite et al. (2018) described an automated phage identification system from a genomic phage library that is capable of detecting effective phages from genomic information through a combination of machine learning and bioinformatics, in order to understand the phage–bacteria relationship by analyzing their genomes [66].
In this approach, a database is created in which the phage sequences are contained, and one for bacterial sequences is also created. Then, a model is created that is aimed at learning the specific characteristics of phages that interact with the bacteria, called positive interactions, and bacteria and phages are used for this probability model. Thus, there is non-experimental evidence that they have interacted, and negative interactions are obtained. Then, after selecting phage candidates based on their characteristics and positive interactions with the bacteria, they are analyzed from the deduced protein sequence at the genomic sequence level, with prediction of functional domains and protein interaction (phage–bacteria), where the focus is on membrane receptors, which is the information that feeds the machine learning model to select phage candidates from theoretical genomic and proteomic information [67].
Recently, Amgarten et al. (2018) developed a computational tool that predicts bacteriophage sequences from metagenomic data called Metagenomic Analysis and Retrieval of Viral Elements (MARVEL), which is based on machine learning. This program feeds on groups of 1247 phage sequences and 1029 bacterial genomes, determined from fragments of sequence counting that identifies sequences corresponding to semi-techniques of phages and bacteria with significant hits for viral proteins. Interestingly, in order to validate the operation and accuracy of MARVEL identification, the authors compared the results of the analysis of the same contigs with VIRsorter and VirFinder against MARVEL functional bases of software recommended to carry out the unknown virus identification. VIRsorter is based on alignments and searches for similarities in databases of known viruses, whereas VirFinder is based on a machine learning algorithm through K-mer frequency profiles that are obtained from the contigs and taken for training of the model [11][68][69]. The results of this study show that in the comparison of the three types of viral sequence analysis software, there were similarities except for two kbp fragments. It should be noted that MARVEL showed significant values (<0.001) for all cases analyzed with positive ranges. Therefore, this new computational tool will be able to support the design and study, as well as the identification, of phages from their genetic information [11].
A new approach, known as VIBRANT, focuses on the annotation of viruses. It is a new hybrid method that uses machine learning and protein similarity to determine genome quality and completeness, and characterizes viral communities from metagenomic assemblies. VIBRANT uses neural networks of protein signatures and newly developed v-score metrics to determine lytic viral genomes or prophages that are integrated. As a platform for evaluating viral community function, it was trained and validated with reference virus datasets, microbiomes, and virome data [70].
References
- Cobián Güemes, A.G.; Youle, M.; Cantú, V.A.; Felts, B.; Nulton, J.; Rohwer, F. Viruses as Winners in the Game of Life. Annu. Rev. Virol. 2016, 3, 197–214.
- Karam, J.D. Bacteriophages: The Viruses for All Seasons of Molecular Biology. Virol. J. 2005, 2, 19.
- Mallman, W.L.; Hemstreet, C. Isolation of an Inhibitory Substance from Plants. J. Agric. Res. 1924, 23, 599–602.
- Moore, E.S. D’Herelle’s Bacteriophage in Relation to Plant Parasites. S. Afr. J. Sci. 1926, 23, 306.
- Dublanchet, A.; Bourne, S. The Epic of Phage Therapy. Can. J. Infect. Dis. Med. Microbiol. 2007, 18, 15–18.
- Lobanovska, M.; Pilla, G. Penicillin’s Discovery and Antibiotic Resistance: Lessons for the Future? Yale J. Biol. Med. 2017, 90, 135–145.
- Aminov, R.I. A Brief History of the Antibiotic Era: Lessons Learned and Challenges for the Future. Front. Microbiol. 2010, 1, 134.
- Luong, T.; Salabarria, A.-C.; Roach, D.R. Phage Therapy in the Resistance Era: Where Do We Stand and Where Are We Going? Clin. Ther. 2020, 42, 1659–1680.
- Sithole-Niang, I.; Muchayi, S.; Mashonganyika, A. Rapid Isolation of Bacteriophages: Steps Towards Phage Therapy. Act Sci. Pharm Sci. 2019, 3, 32–323.
- Hatfull, G.F.; Hendrix, R.W. Bacteriophages and Their Genomes. Curr. Opin. Virol. 2011, 1, 298–303.
- Amgarten, D.; Braga, L.P.P.; da Silva, A.M.; Setubal, J.C. MARVEL, a Tool for Prediction of Bacteriophage Sequences in Metagenomic Bins. Front. Genet. 2018, 9, 304.
- Buttimer, C.; McAuliffe, O.; Ross, R.P.; Hill, C.; O’Mahony, J.; Coffey, A. Bacteriophages and Bacterial Plant Diseases. Front. Microbiol. 2017, 8, 34.
- Obradovic, A.; Jones, J.B.; Momol, M.T.; Olson, S.M.; Jackson, L.E.; Balogh, B.; Guven, K.; Iriarte, F.B. Integration of Biological Control Agents and Systemic Acquired Resistance Inducers Against Bacterial Spot on Tomato. Plant Dis. 2005, 89, 712–716.
- Subirats, J.; Sànchez-Melsió, A.; Borrego, C.M.; Balcázar, J.L.; Simonet, P. Metagenomic Analysis Reveals That Bacteriophages Are Reservoirs of Antibiotic Resistance Genes. Int. J. Antimicrob. Agents 2016, 48, 163–167.
- Ackermann, H.-W. Phage Classification and Characterization. Methods Mol. Biol. 2009, 501, 127–140.
- Rakhuba, D.V.; Kolomiets, E.I.; Dey, E.S.; Novik, G.I. Bacteriophage Receptors, Mechanisms of Phage Adsorption and Penetration into Host Cell. Pol. J. Microbiol. 2010, 59, 145–155.
- Bacteriophages: Their Structural Organisation and Function|IntechOpen. Available online: https://www.intechopen.com/chapters/66740 (accessed on 19 October 2022).
- Sanz-Gaitero, M.; Seoane-Blanco, M.; van Raaij, M.J.; Harper, D.R.; Abedon, S.T.; Burrowes, B.H.; McConville, M.L. Structure and Function of Bacteriophages. In Bacteriophages: Biology, Technology, Therapy; Springer International Publishing: Cham, Switzerland, 2019; pp. 1–73.
- Cieplak, T.; Soffer, N.; Sulakvelidze, A.; Nielsen, D.S. A Bacteriophage Cocktail Targeting Escherichia Coli Reduces E. coli in Simulated Gut Conditions, While Preserving a Non-Targeted Representative Commensal Normal Microbiota. Gut Microbes 2018, 9, 391–399.
- Oksanen, H.M.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Corticoviridae. J. Gen. Virol. 2017, 98, 888–889.
- Wirth, J.; Young, M. The Intriguing World of Archaeal Viruses. PLoS Pathog. 2020, 16, e1008574.
- Pietilä, M.K.; Demina, T.A.; Atanasova, N.S.; Oksanen, H.M.; Bamford, D.H. Archaeal Viruses and Bacteriophages: Comparisons and Contrasts. Trends Microbiol. 2014, 22, 334–344.
- Mäntynen, S.; Sundberg, L.-R.; Poranen, M.M. Recognition of Six Additional Cystoviruses: Pseudomonas Virus Phi6 Is No Longer the Sole Species of the Family Cystoviridae. Arch. Virol. 2018, 163, 1117–1124.
- Amarillas, L.; Estrada-Acosta, M.; León-Chan, R.G.; López-Orona, C.; Lightbourn, L. Complete Genome Sequence of Phobos: A Novel Bacteriophage with Unusual Genomic Features That Infects Pseudomonas Syringae. Arch. Virol. 2020, 165, 1485–1488.
- Nakayinga, R.; Makumi, A.; Tumuhaise, V.; Tinzaara, W. Xanthomonas Bacteriophages: A Review of Their Biology and Biocontrol Applications in Agriculture. BMC Microbiol. 2021, 21, 291.
- Addy, H.S.; Askora, A.; Kawasaki, T.; Fujie, M.; Yamada, T. Utilization of Filamentous Phage ΦRSM3 to Control Bacterial Wilt Caused by Ralstonia Solanacearum. Plant Dis. 2012, 96, 1204–1209.
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus Statement: Virus Taxonomy in the Age of Metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168.
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage Diversity, Genomics and Phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138.
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506.
- Baker, D.N.; Langmead, B. Dashing: Fast and Accurate Genomic Distances with HyperLogLog. Genome Biol. 2019, 20, 265.
- Zhu, Y.; Shang, J.; Peng, C.; Sun, Y. Phage Family Classification under Caudoviricetes: A Review of Current Tools Using the Latest ICTV Classification Framework. arXiv 2022, arXiv:2209.01942.
- Roux, S.; Krupovic, M.; Daly, R.A.; Borges, A.L.; Nayfach, S.; Schulz, F.; Sharrar, A.; Matheus Carnevali, P.B.; Cheng, J.-F.; Ivanova, N.N.; et al. Cryptic Inoviruses Revealed as Pervasive in Bacteria and Archaea across Earth’s Biomes. Nat. Microbiol. 2019, 4, 1895–1906.
- Adriaenssens, E.M.; Sullivan, M.B.; Knezevic, P.; van Zyl, L.J.; Sarkar, B.L.; Dutilh, B.E.; Alfenas-Zerbini, P.; Łobocka, M.; Tong, Y.; Brister, J.R.; et al. Taxonomy of Prokaryotic Viruses: 2018-2019 Update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2020, 165, 1253–1260.
- Krupovic, M.; Dutilh, B.E.; Adriaenssens, E.M.; Wittmann, J.; Vogensen, F.K.; Sullivan, M.B.; Rumnieks, J.; Prangishvili, D.; Lavigne, R.; Kropinski, A.M.; et al. Taxonomy of Prokaryotic Viruses: Update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2016, 161, 1095–1099.
- Quaiser, A.; Dufresne, A.; Ballaud, F.; Roux, S.; Zivanovic, Y.; Colombet, J.; Sime-Ngando, T.; Francez, A.-J. Diversity and Comparative Genomics of Microviridae in Sphagnum-Dominated Peatlands. Front. Microbiol. 2015, 6, 375.
- Krishnamurthy, S.R.; Janowski, A.B.; Zhao, G.; Barouch, D.; Wang, D. Hyperexpansion of RNA Bacteriophage Diversity. PLOS Biol. 2016, 14, e1002409.
- Dowding, J.E. Characterization of a Bacteriophage Virulent for Streptomyces Coelicolor A3(2). J. Gen. Microbiol. 1973, 76, 163–176.
- Chen, J.; Civerolo, E.L. Morphological Evidence for Phages in Xylella Fastidiosa. Virol. J. 2008, 5, 75.
- Ellis, S.D.; Boehm, M.J.; Coplin, D. Bacterial Diseases of Plants. Agric. Nat. Resour. 2008, 4, 401–404.
- Bertaccini, A.; Duduk, B. Phytoplasma and Phytoplasma Diseases: A Review of Recent Research. Phytopathol. Mediterr. 2009, 48, 355–378.
- Bertozzi Silva, J.; Storms, Z.; Sauvageau, D. Host Receptors for Bacteriophage Adsorption. FEMS Microbiol. Lett. 2016, 363, fnw002.
- Hyman, P. Phages for Phage Therapy: Isolation, Characterization, and Host Range Breadth. Pharmaceuticals 2019, 12, E35.
- Van Twest, R.; Kropinski, A.M. Bacteriophage Enrichment from Water and Soil. Methods Mol. Biol. 2009, 501, 15–21.
- Stone, E.; Lhomet, A.; Neve, H.; Grant, I.R.; Campbell, K.; McAuliffe, O. Isolation and Characterization of Listeria Monocytogenes Phage VB_LmoH_P61, a Phage With Biocontrol Potential on Different Food Matrices. Front. Sustain. Food Syst. 2020, 4, 521645.
- Sundin, G.W.; Castiblanco, L.F.; Yuan, X.; Zeng, Q.; Yang, C.-H. Bacterial Disease Management: Challenges, Experience, Innovation and Future Prospects: Challenges in Bacterial Molecular Plant Pathology. Mol. Plant Pathol. 2016, 17, 1506–1518.
- Šimoliūnas, E.; Šimoliūnienė, M.; Kaliniene, L.; Zajančkauskaitė, A.; Skapas, M.; Meškys, R.; Kaupinis, A.; Valius, M.; Truncaitė, L. Pantoea Bacteriophage VB_PagS_Vid5: A Low-Temperature Siphovirus That Harbors a Cluster of Genes Involved in the Biosynthesis of Archaeosine. Viruses 2018, 10, 583.
- Attai, H.; Boon, M.; Phillips, K.; Noben, J.-P.; Lavigne, R.; Brown, P.J.B. Larger Than Life: Isolation and Genomic Characterization of a Jumbo Phage That Infects the Bacterial Plant Pathogen, Agrobacterium Tumefaciens. Front. Microbiol. 2018, 9, 1861.
- Holtappels, D.; Kerremans, A.; Busschots, Y.; Van Vaerenbergh, J.; Maes, M.; Lavigne, R.; Wagemans, J. Preparing for the KIL: Receptor Analysis of Pseudomonas Syringae Pv. Porri Phages and Their Impact on Bacterial Virulence. Int. J. Mol. Sci. 2020, 21, 2930.
- Wang, X.; Wei, Z.; Yang, K.; Wang, J.; Jousset, A.; Xu, Y.; Shen, Q.; Friman, V.-P. Phage Combination Therapies for Bacterial Wilt Disease in Tomato. Nat. Biotechnol. 2019, 37, 1513–1520.
- Guang-Han, O.; Leang-Chung, C.; Vellasamy, K.M.; Mariappan, V.; Li-Yen, C.; Vadivelu, J. Experimental Phage Therapy for Burkholderia Pseudomallei Infection. PLoS ONE 2016, 11, e0158213.
- Gašić, K.; Ivanović, M.M.; Ignjatov, M.; Calić, A.; Obradović, A. Isolation and Characterization of Xanthomonas Euvesicatoria Bacteriophages. J. Plant Pathol. 2011, 93, 415–423.
- Jones, J.B.; Vallad, G.E.; Iriarte, F.B.; Obradović, A.; Wernsing, M.H.; Jackson, L.E.; Balogh, B.; Hong, J.C.; Momol, M.T. Considerations for Using Bacteriophages for Plant Disease Control. Bacteriophage 2012, 2, 208–214.
- Bonilla, N.; Rojas, M.I.; Netto Flores Cruz, G.; Hung, S.-H.; Rohwer, F.; Barr, J.J. Phage on Tap-a Quick and Efficient Protocol for the Preparation of Bacteriophage Laboratory Stocks. PeerJ 2016, 4, e2261.
- Czajkowski, R.; Ozymko, Z.; Lojkowska, E. Application of Zinc Chloride Precipitation Method for Rapid Isolation and Concentration of Infectious Pectobacterium spp. and Dickeya spp. Lytic Bacteriophages from Surface Water and Plant and Soil Extracts. Folia Microbiol. 2016, 61, 29–33.
- John, S.G.; Mendez, C.B.; Deng, L.; Poulos, B.; Kauffman, A.K.M.; Kern, S.; Brum, J.; Polz, M.F.; Boyle, E.A.; Sullivan, M.B. A Simple and Efficient Method for Concentration of Ocean Viruses by Chemical Flocculation. Environ. Microbiol. Rep. 2011, 3, 195–202.
- Beaudoin, R.N.; DeCesaro, D.R.; Durkee, D.L.; Barbaro, S.E. Isolation of a bacteriophage from sewage sludge and characterization of its bacterial host cell. River Acad. J. 2007, 3, 1–8.
- Echeverría-Vega, A.; Morales-Vicencio, P.; Saez-Saavedra, C.; Gordillo-Fuenzalida, F.; Araya, R. A Rapid and Simple Protocol for the Isolation of Bacteriophages from Coastal Organisms. MethodsX 2019, 6, 2614–2619.
- Renu, M.B.; Singh, U.B.; Sahu, U.; Nagrale, D.T.; Sahu, P.K. Characterization of Lytic Bacteriophage XCC9SH3. J. Plant Pathol. 2017, 99, 233–238.
- Brister, J.R.; Ako-Adjei, D.; Bao, Y.; Blinkova, O. NCBI Viral Genomes Resource. Nucleic Acids Res. 2015, 43, D571–D577.
- Abedon, S.T.; Yin, J. Bacteriophage Plaques: Theory and Analysis. Methods Mol. Biol. 2009, 501, 161–174.
- Konopacki, M.; Grygorcewicz, B.; Dołęgowska, B.; Kordas, M.; Rakoczy, R. PhageScore: A Simple Method for Comparative Evaluation of Bacteriophages Lytic Activity. Biochem. Eng. J. 2020, 161, 107652.
- Aucken, H.M.; Westwell, K. Reaction Difference Rule for Phage Typing of Staphylococcus Aureus at 100 Times the Routine Test Dilution. J. Clin. Microbiol. 2002, 40, 292–293.
- Russell, D.A.; Hatfull, G.F. PhagesDB: The Actinobacteriophage Database. Bioinformatics 2017, 33, 784–786.
- Breitwieser, F.P.; Lu, J.; Salzberg, S.L. A Review of Methods and Databases for Metagenomic Classification and Assembly. Brief Bioinform. 2019, 20, 1125–1136.
- McNair, K.; Bailey, B.A.; Edwards, R.A. PHACTS, a Computational Approach to Classifying the Lifestyle of Phages. Bioinformatics 2012, 28, 614–618.
- Leite, D.M.C.; Brochet, X.; Resch, G.; Que, Y.-A.; Neves, A.; Peña-Reyes, C. Computational Prediction of Inter-Species Relationships through Omics Data Analysis and Machine Learning. BMC Bioinform. 2018, 19, 420.
- Klumpp, J.; Fouts, D.E.; Sozhamannan, S. Next Generation Sequencing Technologies and the Changing Landscape of Phage Genomics. Bacteriophage 2012, 2, 190–199.
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining Viral Signal from Microbial Genomic Data. PeerJ 2015, 3, e985.
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A Novel k-Mer Based Tool for Identifying Viral Sequences from Assembled Metagenomic Data. Microbiome 2017, 5, 69.
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated Recovery, Annotation and Curation of Microbial Viruses, and Evaluation of Viral Community Function from Genomic Sequences. Microbiome 2020, 8, 90.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Revisions:
2 times
(View History)
Update Date:
08 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No