 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marziale Comito | -- | 2775 | 2023-02-07 18:08:49 | | | |
| 2 | Jason Zhu | Meta information modification | 2775 | 2023-02-08 02:36:55 | | |
Video Upload Options
Continuous-flow chemistry has become a mainstream process and a notable trend among emerging technologies for drug synthesis. It is routinely used in academic and industrial laboratories to generate a wide variety of molecules and building blocks. The advantages it provides, in terms of safety, speed, cost efficiency and small-equipment footprint compared to analog batch processes, have been known for some time. What has become even more important is its compliance with the quality objectives that are required by drug-development protocols that integrate inline analysis and purification tools. There can be no doubt that worldwide government agencies have strongly encouraged the study and implementation of this innovative, sustainable and environmentally friendly technology.
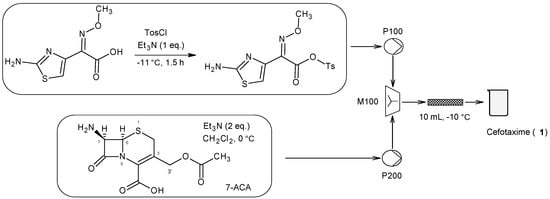
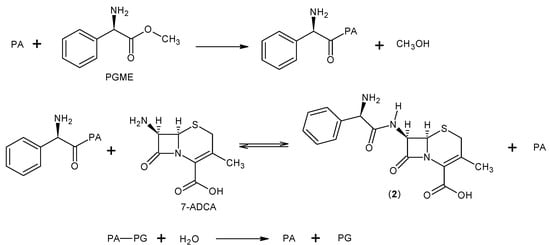
1. Cefotaxime
2. Cephalexin
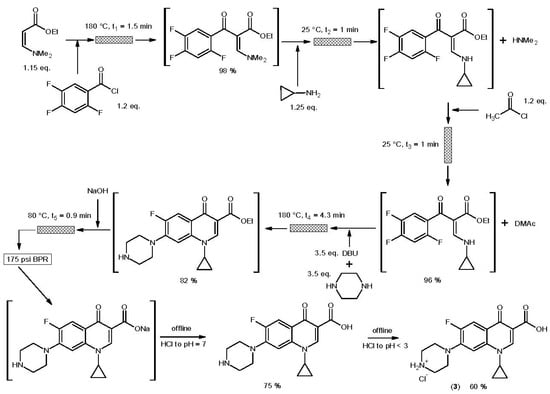
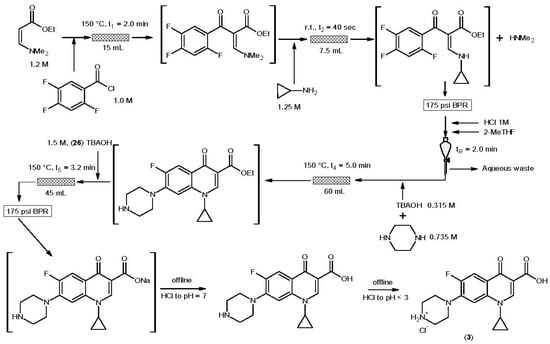
3. Ciprofloxacin Hydrochloride
4. Linezolid
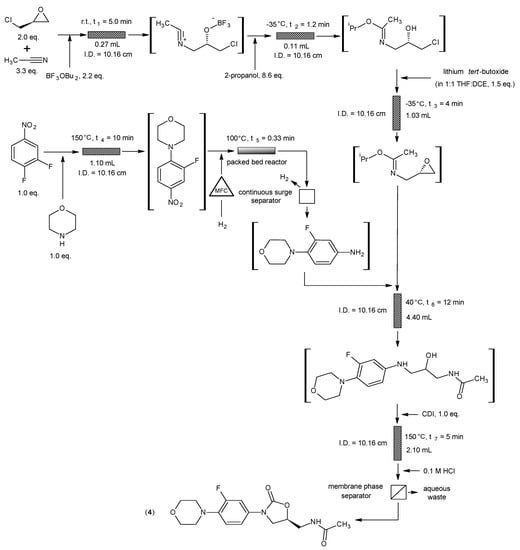
5. Tazobactam
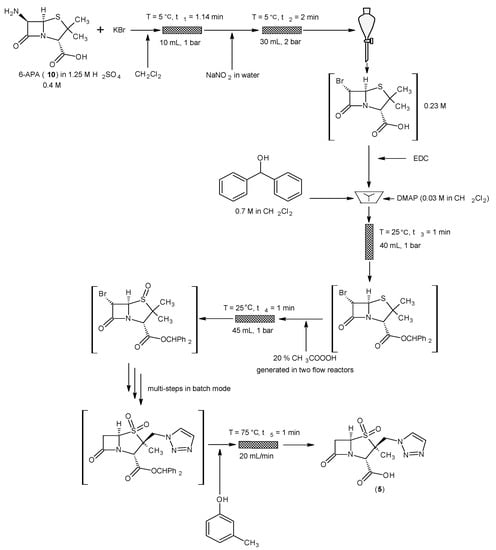
6. Key Vaborbactam Intermediate
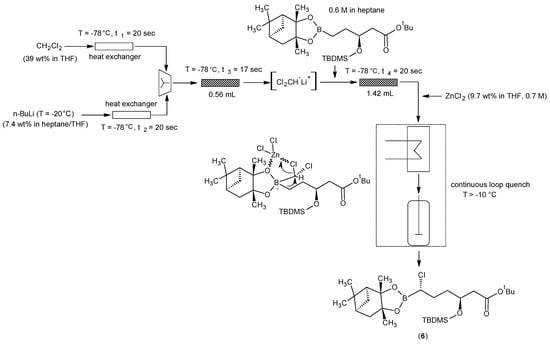
7. Key Cefodizime Intermediate
References
- Todd, P.A.; Brogden, R.N. Cefotaxime. An update of its pharmacology and therapeutic use. Drugs 1990, 40, 608–651.
- PLoSker, G.L.; Foster, R.H.; Benfield, P. Cefotaxime. A pharmacoeconomic review of its use in the treatment of infections. PharmacoEconomics 1998, 13, 91–106.
- Carmine, A.A.; Brogden, R.N.; Heel, R.C.; Speight, T.M.; Avery, G.S. Cefotaxime. A review of its antibacterial activity, pharmacological properties and therapeutic use. Drugs 1983, 25, 223–289.
- LeFrock, J.L.; Prince, R.A.; Leff, R.D. Mechanism of action, antimicrobial activity, pharmacology, adverse effects, and clinical efficacy of cefotaxime. Pharmacotherapy 1982, 2, 174–184.
- Gonik, B.; Cotton, D.B.; Feldman, S.; Cleary, T.G.; Pickering, L.K. Pharmacokinetics of cefotaxime in the postpartum patient. Am. J. Perinatol. 1985, 2, 114–117.
- Sader, H.S.; Jones, R.N. Cefotaxime is extensively used for surgical prophylaxis. Am. J. Surg. 1992, 164, 28S–38S.
- WHO Model List of Essential Medicines. Available online: https://en.wikipedia.org/wiki/WHO_Model_List_of_Essential_Medicines (accessed on 22 November 2022).
- Fanelli, F.; Parisi, G.; Degennaro, L.; Luisi, R. Contribution of microreactor technology and flow chemistry to the development of green and sustainable synthesis. Beilstein J. Org. Chem. 2017, 13, 520–542.
- Hessel, W.; Löwe, H. Organic synthesis with microstructured reactors. Chem. Eng. Technol. 2005, 28, 267–284.
- Pieper, M.; Kumpert, M.; König, B.; Schleich, H.; Bayer, T.; Gröger, H. Process development of synthesizing the cephalosporin antibiotic cefotaxime in batch mode and flow mode. Org. Process. Res. Dev. 2018, 22, 947–954.
- Hessel, V.; Noël, T. Micro process technology, 1. Introduction. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2012.
- The Top 300 of 2020. Available online: https://clincal.com/DrugStats/Top300Drugs.aspx (accessed on 30 November 2022).
- Cephalexin. Drug Usage Statistics. Available online: https://clincal.com/DrugsStats/Drugs/Cephalexin (accessed on 30 November 2022).
- Bailey, A.; Walker, A.; Hadley, A.; James, D.G. Cephalexin. A new oral antibiotic. Postgrad. Med. J. 1970, 46, 157–158.
- Gwee, A.; Autmizguine, J.; Curtis, N.; Duffull, S.B. Twice-and thrice-daily cephalexin dosing for Staphyloccus aureus infections in children. Pediatr. Infect. Dis. J. 2020, 39, 519–522.
- Valent, A.M.; DeArmond, C.; Houston, j.M.; Reddy, S.; Masters, H.R.; Gold, A.; Boldt, M.; DeFranco, M.; Evans, A.T.; Warshak, C.R. Effect of post-cesarean delivery oral cephalexin and metronidazole on surgical site infection among obese women: A randomized clinical trial. JAMA 2017, 318, 1026–1034.
- Gill, C.L. Cephalexin. J. Am. Dent. Assoc. 1980, 100, 172–174.
- Speight, T.M.; Brogden, R.N.; Avery, G.S. Cephalexin: A review of its antibacterial, pharmacological and therapeutic properties. Drugs 1972, 3, 9–78.
- Morin, R.B.; Jackson, B.G. Certain 3-Methyl-Cephalosporin Compounds. U.S. Patent 3,507,861, 21 April 1970.
- Vobeckà, L.; Tichà, L.; Atanasova, A.; Slouka, Z.; Hasal, P.; Přibyl, M. Enzyme synthesis of cephalexin in continuous-flow microfluidic device in ATPS environment. Chem. Eng. J. 2020, 396, 125236.
- Sharma, P.C.; Jain, A.; Jain, S.; Pahwa, R.; Yar, M.S. Ciprofloxacin: Review on developments in synthetic, analytical, and medicinal aspects. J. Enzyme Inhib. Med. Chem. 2010, 25, 577–589.
- Meyerhoff, A.; Albrecht, R.; Meyer, J.M.; Dionne, P.; Higgins, K.; Murphy, D. US Food and Drug Administration approval of ciprofloxacin hydrochloride for management of postexposure inhalational anthrax. Clin. Infect. Dis. 2004, 39, 303–308.
- Campoli-Richards, D.M.; Monk, J.P.; Price, A.; Benfield, P.; Todd, P.A.; Ward, A. Ciprofloxacin. A review of its antibacterial activity, pharmacokinetic properties and therapeutic use. Drugs 1988, 35, 373–447.
- Apangu, T.; Griffith, K.; Abaru, J.; Candini, G.; Apio, H.; Okoth, F.; Okello, R.; Kaggwa, J.; Acayo, S.; Ezama, G.; et al. Successful treatment of human plague with oral ciprofloxacin. Emerg. Infect. Dis. 2017, 23, 553–555.
- Zhang, H.L.; Tan, M.; Qiu, A.M.; Tao, Z.; Wang, C.H. Antibiotics for treatment of acute exacerbation of chronic obstructive pulmonary disease: A network meta-analysis. BMC Pulm. Med. 2017, 17, 196.
- Zhang, G.F.; Liu, X.; Zhang, S.; Pan, B.; Liu, M.L. Ciprofloxacin derivates and their antibacterial activities. Eur. J. Med. Chem. 2018, 146, 599–612.
- Terp, D.K.; Ryback, M.J. Ciprofloxacin. Drug Intell. Clin. Pharm. 1987, 21, 568–574.
- Lin, H.; Dai, C.; Jamison, T.; Jensen, K.F. A rapid total synthesis of ciprofloxacin hydrochloride in continuous flow. Angew. Chem. Int. Ed. 2017, 56, 8870–8873.
- Armstrong, C.; Miyai, Y.; Formosa, A.; Thomas, D.; Chen, E.; Hart, T.; Schultz, V.; Desai, B.K.; Cai, A.Y.; Almasay, A.; et al. On-demand continuous manufacturing of ciprofloxacin in portable plug-and-play factories: Development of a highly efficient synthesis for ciprofloxacin. Org. Process. Res. Dev. 2021, 25, 1524–1533.
- Schwalbe, T.; Kadzimirsz, D.; Jas, G. Synthesis of library of ciprofloxacin analogues by means of sequential organic synthesis in microreactors. QSAR Comb. Sci. 2005, 24, 758–768.
- Adamo, A.; Beingessner, R.L.; Behnam, M.; Chen, J.; Jamison, T.F.; Jensen, K.F.; Monbaliu, J.C.; Myerson, A.S.; Revalor, E.M.; Snead, D.R.; et al. On-demand continuous-flow production of pharmaceuticals in a compact, reconfigurable system. Science 2016, 352, 61–67.
- Rogers, L.; Briggs, N.; Acherman, R.; Adamo, A.; Azad, M.; Brancazio, D.; Capellades, G.; Hammersmith, G.; Hart, T.; Imbrogno, J.; et al. Continuous production of five active pharmaceutical ingredients in flexible plug-and-play modules: A demonstration campaign. Org. Process. Res. Dev. 2020, 24, 2183–2196.
- Zhang, P.; Weeranoppanant, N.; Thomas, D.A.; Tahara, K.; Stelzer, T.; Russell, M.G.; O’Mahony, M.; Myerson, A.S.; Lin, H.; Kelly, L.P.; et al. Advanced continuous flow platform for on-demand pharmaceutical manufacturing. Chem. Eur. J. 2018, 24, 1–10.
- Capellades, G.; Neurohr, C.; Briggs, N.; Rapp, K.; Hammersmith, G.; Brancazio, D.; Derksen, B.; Myerson, A.S. On-demand continuous manufacturing of ciprofloxacin in portable plug-and-play factories: Implementation and in situ control of downstream production. Org. Process. Res. Dev. 2021, 25, 1534–1546.
- Perry, C.M.; Jarvis, B. Linezolid: A review of its use in the management of serious gram-positive infections. Drugs 2001, 61, 525–551.
- Hashemian, S.M.R.; Farhadi, T.; Ganjparvar, M. Linezolid: A review of its property, function, and use in critical care. Drug Des. Dev. Ther. 2018, 12, 1759–1767.
- Wilcox, M.H. Efficacy of linezolid versus comparator therapies in gram-positive infections. J. Antimicrob. Chemother. 2003, 51, 27–35.
- Livermore, D.M. Linezolid in vitro: Mechanism and antibacterial spectrum. J. Antimicrob. Chemother. 2003, 51, 9–16.
- Clemett, D.; Markham, A. Linezolid. Drugs 2000, 59, 815–827.
- Chien, J.W.; Kucia, M.L.; Salata, R.A. Use of linezolid, an oxazolidinone, in the treatment of multidrug-resistant gram-positive bacterial infections. Clin. Infect. Dis. 2000, 30, 146–151.
- Bai, P.Y.; Qin, S.S.; Chu, W.C.; Yang, Y.; Cui, D.Y.; Hua, Y.G.; Yang, Q.Q.; Zhang, E. Synthesis and antibacterial bioactivities of cationic deacetyl linezolid amphiphiles. Eur. J. Med. Chem. 2018, 155, 925–945.
- Russell, M.G.; Jamison, T.F. Seven-step continuous flow synthesis of linezolid without intermediate purification. Angew. Chem. Int. Ed. 2019, 58, 7678–7681.
- Brickner, S.J.; Hutchinson, D.K.; Barbachyn, M.R.; Manninen, P.R.; Ulanowicz, D.A.; Garmon, S.A.; Grega, K.C.; Hendges, S.K.; Toops, D.S.; Ford, C.W.; et al. Synthesis of antibacterial activity of U-100592 and U-100766, two oxazolidinone antibacterial agents for the potential treatment of multidrug-resistant gram-positive bacterial infections. J. Med. Chem. 1996, 39, 673–679.
- Ramgren, S.D.; Silberstein, A.L.; Yang, Y.; Garg, N.K. Nickel-catalyzed amination of aryl sulfamates. Angew. Chem. Int. Ed. Engl. 2011, 50, 2171–2173.
- Perrault, W.R.; Pearlman, B.A.; Godrej, D.B.; Jeganathan, A.; Yamagata, K.; Chen, J.J.; Lu, C.V.; Herrinton, P.M.; Gadwood, R.C.; Chan, L.; et al. The synthesis of N-aryl-5(S)-aminomethyl-2-oxazolidinone antibacterials and derivates in one step from aryl carbamates. Org. Process Res. Dev. 2003, 7, 533–546.
- Sheldon, R.A. The E factor: Fifteen years on. Green Chem. 2007, 9, 1273–1283.
- Kadima, T.A.; Weiner, J.H. Mechanism of suppression of piperacillin resistance in enterobacteria by tazobactam. Antimicrob. Agents Chemother. 1997, 41, 2177–2183.
- Perry, C.M.; Markham, A. Piperacillin/tazobactam: An update review of its use in the treatment of bacterial infections. Drugs 1999, 57, 805–843.
- Yang, Y.; Rasmussen, B.A.; Shlaes, D.M. Class A beta-lactamases-enzyme-inhibitor interactions and resistance. Pharmacol. Ther. 1999, 83, 141–151.
- Lizza, B.D.; Betthauser, K.D.; Ritchie, D.J.; Micek, S.T.; Kollef, M.H. New perspectives on antimicrobial agents: Ceftolozane-tazobactam. Antimicrob. Agents Chemother. 2021, 65, e02318-20.
- Zhou, S.; Xin, Y.; Wang, J.; Wu, C.; Sun, T. Application of continuous flow in tazobactam synthesis. Org. Process Res. Dev. 2021, 25, 1648–1657.
- Hecker, S.J.; Reddy, K.R.; Totrov, M.; Hirst, G.C.; Lomovskaya, O.; Griffith, D.C.; King, P.; Tsivkovski, R.; Sun, D.; Sabet, M.; et al. Discovery of a cyclic boronic acid β-lactamase inhibitor (RPX7009) with utility vs. class a serine carbapenemases. J. Med. Chem. 2015, 58, 3682–3692.
- Novelli, A.; Del Giacomo, P.; Rossolini, G.M.; Tumbarello, M. Meropenem/vaborbactam: A next generation β-lactam β-lactamase inhibitor combination. Expert Rev. Anti-Infect. Ther. 2020, 18, 643–655.
- Bhowmick, T.; Weinstein, M.P. Microbiology of meropenem-vaborbactam: A novel carbapenem beta-lactamase inhibitor combination for carbapenem-resistant enterobacterales infections. Infect. Dis. Ther. 2020, 9, 757–767.
- FDA Approves New Antibacterial Drug. Available online: https://fda.gov/news-events/press-announcements/fda-approves-new-antibacterial-drug (accessed on 7 December 2022).
- Lee, Y.R.; Baker, N.T. Meropenem-vaborbactam: A carbapenem and beta-lactamase inhibitor with activity against carbapenem-resistant Enterobacteriaceae. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1411–1419.
- Cho, J.C.; Zmarlicka, M.T.; Shaeer, K.M.; Pardo, J. Meropenem/vaborbactam, the first carbapenem/β-lactamase inhibitor combination. Ann. Pharmacother. 2018, 52, 769–779.
- Burgos, R.M.; Biagi, M.J.; Rodvold, K.A.; Danziger, L.H. Pharmacokinetic evaluation of meropenem and vaborbactam for the treatment of urinary tract infection. Expert Opin. Drug Metab. Toxicol. 2018, 14, 1007–1021.
- Stueckler, C.; Hermsen, P.; Ritzen, B.; Vasiloiu, M.; Poechlauer, P.; Steinhofer, S.; Pelz, A.; Zinganell, C.; Felfer, U.; Boyer, S.; et al. Development of a continuous flow process for a matteson reaction: From lab scale to full-scale production of a pharmaceutical intermediate. Org. Process Res. Dev. 2019, 23, 1069–1077.
- Felfer, U.; Stueckler, C.; Steinhofer, S.; Pelz, A.; Hanacek, M.; Pabst, T.H.; Winkler, G.; Poechlauer, P.; Ritzen, B.; Gold-Bach, M. Apparatus and Continuous Flow Process for Production of Boronic Acid Derivates. WO 2016/100043, 23 June 2016.
- Barradell, L.B.; Brogden, R.N. Cefodizime. A review of its antibacterial activity, pharmacokinetic properties and therapeutic use. Drugs 1992, 44, 800–834.
- Brockmeier, D.; Dagrosa, E.E. Pharmacokinetic profile of cefodizime. Infection 1992, 20, S14–S17.
- Song, H.; Li, G.; Ye, J.; Wan, F.; Qian, Y. Immunomodulating effects of cefodizime on Klebsiella pneumoniae-stimulated neutrophils. Immunobiology 2004, 209, 277–282.
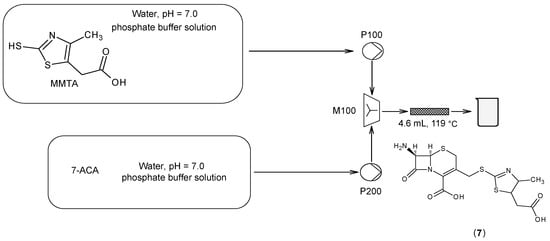
- Wirth, V.; Volkmar, J.; Bayer, T. Synthesis of the antibiotic precursor thiazolyl-7-aminocephalosporanic acid in a microstructured flow system. Chem. Eng. Technol. 2019, 42, 2035–2043.

