Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sanjukta Chakraborty | -- | 3300 | 2023-02-07 13:53:59 | | | |
| 2 | Jessie Wu | Meta information modification | 3300 | 2023-02-08 06:05:22 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Banerjee, P.; Gaddam, N.; Pandita, T.K.; Chakraborty, S. Senescence and Senescence-Associated Secretory Phenotype. Encyclopedia. Available online: https://encyclopedia.pub/entry/40927 (accessed on 26 May 2026).
Banerjee P, Gaddam N, Pandita TK, Chakraborty S. Senescence and Senescence-Associated Secretory Phenotype. Encyclopedia. Available at: https://encyclopedia.pub/entry/40927. Accessed May 26, 2026.
Banerjee, Priyanka, Niyanshi Gaddam, Tej K. Pandita, Sanjukta Chakraborty. "Senescence and Senescence-Associated Secretory Phenotype" Encyclopedia, https://encyclopedia.pub/entry/40927 (accessed May 26, 2026).
Banerjee, P., Gaddam, N., Pandita, T.K., & Chakraborty, S. (2023, February 07). Senescence and Senescence-Associated Secretory Phenotype. In Encyclopedia. https://encyclopedia.pub/entry/40927
Banerjee, Priyanka, et al. "Senescence and Senescence-Associated Secretory Phenotype." Encyclopedia. Web. 07 February, 2023.
Copy Citation
Cellular senescence—the irreversible cell cycle arrest driven by a variety of mechanisms and, more specifically, the senescence-associated secretory phenotype (SASP)—is an important area of research in the context of different age-related diseases, such as cardiovascular disease and cancer. SASP factors play both beneficial and detrimental roles in age-related disease progression depending on the source of the SASPs, the target cells, and the microenvironment.
senescence
cancer
chemotherapy
chemoresistance
signal transduction
senolytics
inflammation
tumor microenvironment
1. Senescence
Cellular senescence is defined as a stress-responsive stable cell cycle arrest [1][2], which has been associated with shortened telomeres [3]. At senescence, cells are metabolically active and viable but no longer divide. This was first characterized by Hayflick and Moorhead [4]. Senescent cells are distinguished by their dynamic pathophysiology resulting from their departure from the cell cycle and their morphological and metabolic reconfiguration that enable them to differentially contribute to various pathological conditions, aging, and tissue remodeling [5]. As telomeres intrinsically shorten and modify conformations through the natural passage of time, replicative senescence (RS) culminates through the DNA damage response (DDR) to exposed chromosomal ends. This occurrence is widely thought to be a driving factor in aging, with the deleterious accumulation of senescent cells in age-related pathologies remaining unchecked by evolutionary processes of selection [6]. In addition to RS, senescence can also be induced by internal and external stimuli such as genotoxic agents, stress, mitochondrial dysfunction, oncogene activation, and chemotherapy, and is referred to as stress-induced premature senescence (SIPS) [7].
Senescent cells secrete a cocktail of proinflammatory cytokines, chemokines, growth factors, proangiogenic factors, reactive oxygen species (ROS), and proteases that represent the senescence-associated secretory phenotype (SASP) [7]. Senescent cells further communicate with neighboring cells and the immune system via the SASP in an autocrine or paracrine manner. The cytokines and chemokines secreted by senescent cells recruit T-cells, macrophages, and natural killer cells that, in turn, aid the removal of the senescent cells [7] to maintain tissue homeostasis. With the increase in age, the weakened immune system, or manifestation of ‘immunosenescence’, fails to clear the senescent cells, which results in their accumulation over time. In addition to the crosstalk with the surrounding immune cells, senescent cells also further induce senescence in neighboring cells by the secretion of SASP factors and extracellular vesicles (EVs).
Cellular Senescence: Double Edged Sword for Cancer
The role of the SASP on the surrounding cells in cancer progression or cancer prevention is very much context dependent. The SASP results in the secretion of numerous proinflammatory cytokines and chemokines promoting the dedifferentiation and proliferation of neighboring metastatic cells. These chemokines and cytokines in turn attract immune cells to the tumor site and help with the immune clearance of the malignant cells [8]. On the other hand, the SASP can serve as an inducer of tumorigenesis. In an in vitro model of ovarian cancer, it has been shown that when non-neoplastic cells were treated with the conditioned media (CM) from senescent fibroblasts, neoplastic transformation was induced in those cells [9]. Interleukin-6 (IL-6) and Interleukin-8 (IL-8) are some of the common SASP factors inducing tumorigenesis in breast, prostate, and lung cancers [10][11][12][13]. Further, senescent cells share some common characteristics of cancer-associated fibroblasts (CAFs) [14][15][16]. Further, senescent cells also promote tumorigenesis via the production of matrix metalloproteinases (MMPs) as SASP factors that enable the restructuring of the extracellular matrix (ECM) and facilitate tumor growth [17][18][19]. Senescent cells, via SASP factors, can induce the epithelial-to-mesenchymal transition (EMT) [8][20] and also promote an immunosuppressive tumorigenic microenvironment. Senescent hepatocytes in hepatocellular carcinoma attract immunosuppressive Cd11b+Gr1 myeloid cells that inhibit T-cell proliferation and contribute to tumor progression [21]. Additionally, the SASP can also induce chemoresistance in cancer cells. When malignant pleural mesothelioma (MPM) cells that show significant chemoresistance were treated with pemetrexed for 96 h in vitro, 60% of the cells became senescent. The conditioned media (CM) from the pemetrexed-treated senescent MPM cells induced EMT with an increased expression of vimentin, fibronectin, slug, and snail in previously nonsenescent cells [20]. In the process of cancer progression and metastasis, senescent cells also increased the angiogenesis by producing angiogenic factors, such as vascular endothelial growth factor (VEGF) and connective tissue growth factor (CTGF) [22][23][24].
Interestingly, senescent cells also acquire a higher potential of invasiveness and lymphangiogenesis. In several metastatic cancers, lymph node metastasis and increased lymphangiogenesis are one of the prognostic factors [25][26][27]. Kim et al. showed that there was an accumulation of senescent cells in the front region of a collective invasion of papillary thyroid carcinoma (PTC), and also within the lymphatic vessels and the metastatic lymph node, which indicate their role in tumor progression and LN metastasis [28]. Further, it also lends credence to the hypothesis that metastatic tumor cells within the lymph nodes acquire vulnerabilities that help to evade traditional therapies and become more aggressive. Importantly, a new role for senescent cells in tumorigenesis has recently emerged, demonstrating that therapy-induced senescent cells can acquire stemness (SAS: senescence-associated stemness) [2][29], and that acquired stemness assists senescent cells in escaping from cell cycle arrest and harnessing an aggressive growth potential (Figure 1).
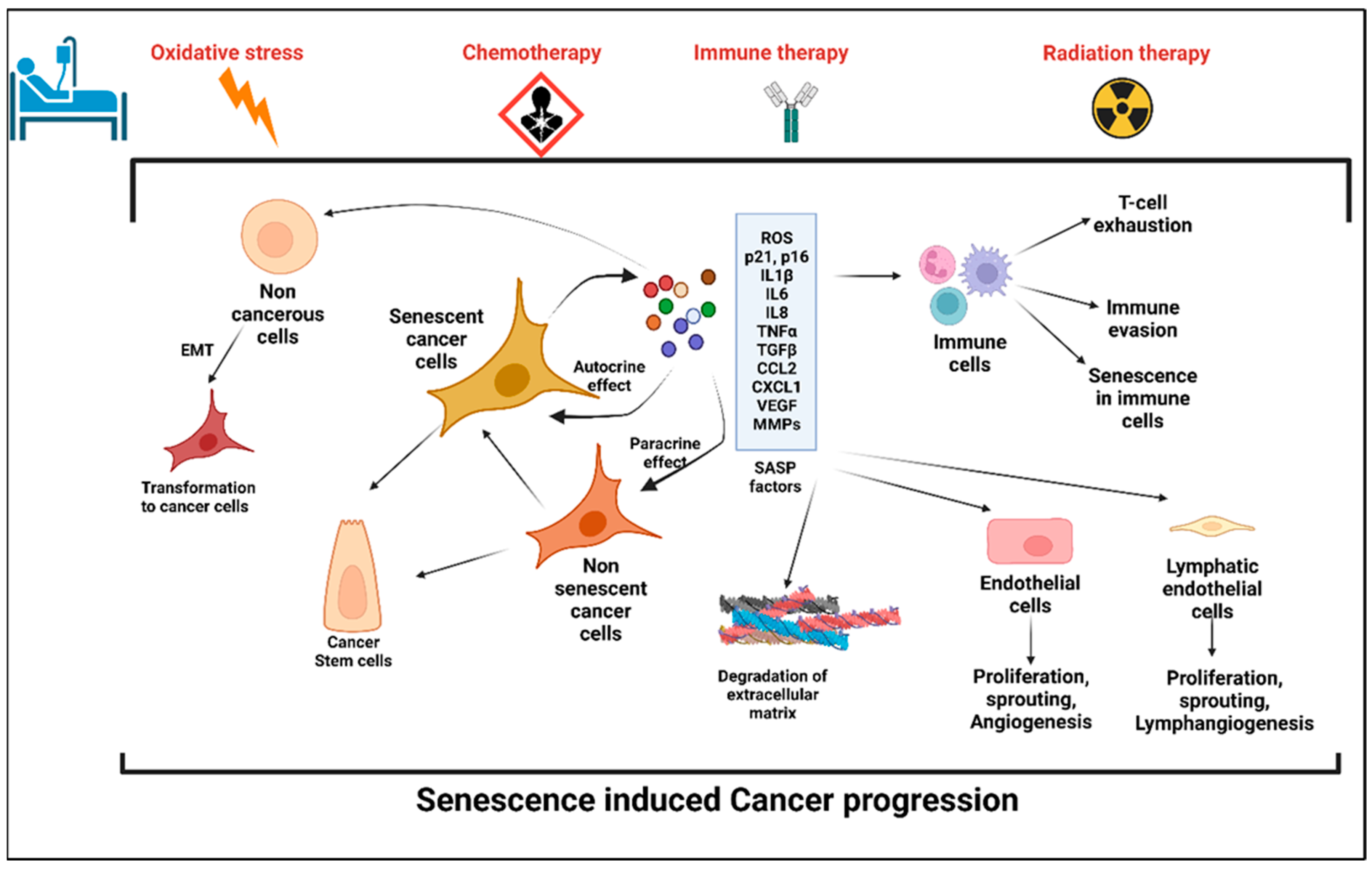
Figure 1. Stress-induced senescence, SASP, and cancer progression. Anticancer therapy including chemotherapy, radiation therapy, and immune therapy induced premature senescence in cancer cells and the cells in the tumor microenvironment (TME). Senescent cancer cells secrete plethora of proinflammatory cytokines, chemokines, matrix metalloproteinases, angiogenic factors, and reactive oxygen species, which are collectively known as senescence-associated secretory phenotype (SASP). SASP factors induce senescence in the cells in the TME, which in turn evade the immune response. Factors secreted by senescent cells promote angiogenesis and lymphangiogenesis by its effects on neighboring endothelial cells and hence enhances cancer cell metastasis. SASP-induced cancer stemness increases the proliferation and self-renewal properties of the cancer cells.
2. Anticancer Treatment: A Potential Trigger to Cellular Senescence
The role of conventional anticancer therapies in the induction of cellular senescence in cancerous or noncancerous tissues is an important area of research. The use of chemotherapies, radiation therapy, and also immunotherapy significantly induce senescence.
- A.
-
Chemotherapy and senescence
The general mode of action of chemotherapy is to impair the mitosis in cancer cells and disrupt the DDR [30]. Doxorubicin, Etoposide, and Camptothecin are common topoisomerase inhibitors that inhibit the progression of replication and are used as chemotherapeutic agents for colon cancers, breast cancer, hepatocellular carcinoma, lung cancers, and acute lymphocytic leukemia [30][31][32][33]. A recent study by Karabicici et al. showed that Doxorubicin treatment induced senescence in both liver cancer stem cells (EpCAM+/CD133+) as well as in a non-stem-cell population (EpCAM-/CD133-nonstem) in the Huh7 cell line with a concomitant increase in the reprogramming genes (SOX2, KLF4, c-MYC), liver stemness-related genes, (EpCAM, CK19), and ANXA3 in those cell populations [34]. The conditioned media from the Doxorubicin-treated cells contained high levels of inflammatory cytokines, IL8, and Interferon Gamma Induced Protein-10 (IP10) [34]. Doxorubicin treatment also caused cardiotoxicity in cancer survivors by inducing cardiomyocyte senescence [35]. Along with the topoisomerase inhibitors, alkylating agents are another group of chemotherapies used for several cancers as they inhibit DNA replication [35]. Two commonly used alkylating agents for cancer treatment are Cisplatin and Temozolomide. Temozolomide is the first-line therapeutic for high-grade glioblastoma. A recent study showed that temozolomide induced cellular senescence in the glioblastoma cells at a four-fold higher level than cellular apoptosis. Interestingly, compared to the primary tumors, the population of senescent cells was significantly higher in the recurrent cancer tissues. The high population of senescent cells upon temozolomide treatment contained elevated levels of proinflammatory cytokines including IL-1α, IL-1β, IL-6, and IL-8, as well as CCL2, CCL8, and CXCL1. The proinflammatory cytokine cocktail present in the tumor microenvironment of temozolomide glioblastoma potentially accelerated tumor growth and relapse [36]. Cisplatin-induced cellular senescence in cancer has been well reported in several articles [37][38][39][40]. Microtubule inhibitors such as Paclitaxel arrest the cells at mitosis by interfering with microtubule dynamics [41]; however, like other chemotherapeutic agents, microtubule inhibitors have been reported to induce cellular senescence in cancer as well as noncancer cells [42]. Table 1 summarizes the role of chemotherapies on the induction of cellular senescence in cancer patients.
Table 1. List of anticancer therapies that induce senescence.
| Class | Name of the Drug | Cancer Types | Reference |
|---|---|---|---|
| Chemotherapy | Doxorubicin | Cervical cancer (HeLa cells), hepatocellular carcinoma (HuH7), colorectal carcinoma, breast cancer | [34][43] |
| Etoposide | Adrenocortical H295R cells, epithelial carcinoma (A549), adrenocortical tumor cells | [44][45] | |
| Bleomycin | Pulmonary fibrosis, alveolar epithelial cells | [46] | |
| Cisplatin | Ovarian cancer, nasopharyngeal carcinoma cells, lung cancer | [40][47] | |
| Mitoxantrone | Dermal fibroblasts, prostate cancer | [48][49] | |
| Temozolomide | Glioma, melanoma | [50][51] | |
| Paclitaxel | Non-small-cell lung cancer cells, breast cancer | [52][53] | |
| Methotrexate | Breast cancer, colon cancer, adenocarcinoma | [53][54] | |
| Camptothecin | Colorectal cancer | [55] | |
| Radiation therapy | Breast cancer, glioblastoma, non-small-cell lung cancer | [56][57] | |
| Immune therapy | Rituximab | B-cell lymphoma | [58][59] |
B. Radiation-induced senescence
Radiation therapy (RT) is used to kill the cancer cells by inducing irreparable DNA damages in a nonspecific manner. Ionizing radiation (IR) is one of the potent RTs for cancer patients with a wide range of cancers, including lymphoma, soft tissue sarcoma, head–neck cancer, breast cancer, and lung cancer [60]. Depending on the dose and fraction of the IR regimen, cancer cells exposed to IR are arrested at different stages of the cell cycle, i.e., in the G1, G2, or S phase [61]. The dose of IR also determines the induction of cellular senescence or apoptosis. Studies have shown that a high dose of IR (>10 Gy) to endothelial cells (ECs) induced apoptosis while a moderate dose (>0.5 Gy) of IR induced senescence [62]. As listed in Table 1, IR can induce senescence with the overexpression of p16, p21, and beta-galactosidase activity in the exposed cells.
- C.
-
Immunotherapy-induced senescence
Immunotherapy is currently a promising anticancer therapy for several cancers. For antitumor immunity, both the Th1 and Th2 CD4+-T-helper cells play a crucial role by inducing cellular and humoral immunity, respectively [63][64]. A study of the carcinogenesis in pancreatic islets showed that T-antigen-specific CD4+ Th cells induced growth arrest of proliferating tumor cells without any significant cytotoxic effects [65]. That study also highlighted the possibility of a noncytotoxic way to induce cellular growth arrest or cellular senescence mediated by Th1, Th2 cytokine immunotherapy [65]. In invasive β-cell cancers, Interferon-gamma (IFN-ϒ)- and Tumor Necrosis Factor (TNF)-producing CD4+ Th1 cells induce senescence in β-cells via the STAT1- and TNFR1-dependent stabilization of the p16INK4a–Rb [66]. In triple negative and HER2+ breast cancer cells, treatment with either CD4+ Th1 cells or Th1 cytokines TNF-α and IFN-γ induced apoptosis and tumor senescence [67]. In B cell lymphoma, CD20-targeted immunotherapy induced senescence in the cancer cells by enhancing the levels of cellular reactive oxygen species (ROS), which is an important SASP factor and also sensitizes the cells to the DDR [58]. The senescent cancer cells can attract the other immune cells in the primary tumor site and help with the immune-mediated clearance of cancer cells via the SASP (Figure 1).
3. The Cellular and Molecular Mechanism of SIPS
3.1. Mitochondrial Dysfunction and SIPS
Mitochondrial dysfunction contributes to premature senescence [68]. One of the potential mechanisms of dysfunctional mitochondria-induced senescence is excessive ROS production. Excessive ROS can lead to DNA damage and induce senescence [69]. A recent study by Kotla et al. showed that cancer treatment with IR or Doxorubicin (i) increased mitochondrial ROS (mtROS) production and (ii) caused mitochondrial stunning (the reversible mt dysfunction), (iii) which then activated the p90RSK/ERK5-S496 complex and decreased the nuclear factor erythroid 2-related factor 2 (NRF2) transcriptional activity, and (iv) the reduced NRF2 transcriptional activity reduced the expression of antioxidant genes (HO1 and Trx1). ROS further cause telomeric DNA damages in the nucleus via poly (ADP-ribose) polymerase (PARP) activation and consequently deplete the NAD+ level and lead to further mtROS production. Altogether, a positive feedback loop is established between the nucleus and mitochondria, which reprograms the neighboring myeloid cells to induce a sustained SASP state [70].
3.2. Molecular Pathways
- A.
-
Cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) (cGAS-STING) pathway
SASP factors can be expressed by various mechanism depending on the cell types [71]. The nuclei of the primary senescent cells released fragmented genomic DNA into the cytoplasm. The cyclic guanosine monophosphate (GMP)–adenosine monophosphate (AMP) synthase (cGAS) senses the cytoplasmic DNA in the form of cyclic dinucleotides. The cyclic GMP-AMP (cGAMP) complex then activates a stimulator of interferon genes (STING) located in the endoplasmic reticulum. The activated STING combines with TANK-binding kinase 1 (TBK1) and phosphorylates the transcription factors interferon regulatory factor 3 (IRF3) and nuclear factor ‘kappa-light-chain-enhancer’ (NFκB), causing its nuclear translocation and the further activation of the SASP gene expression (Figure 2) [71].
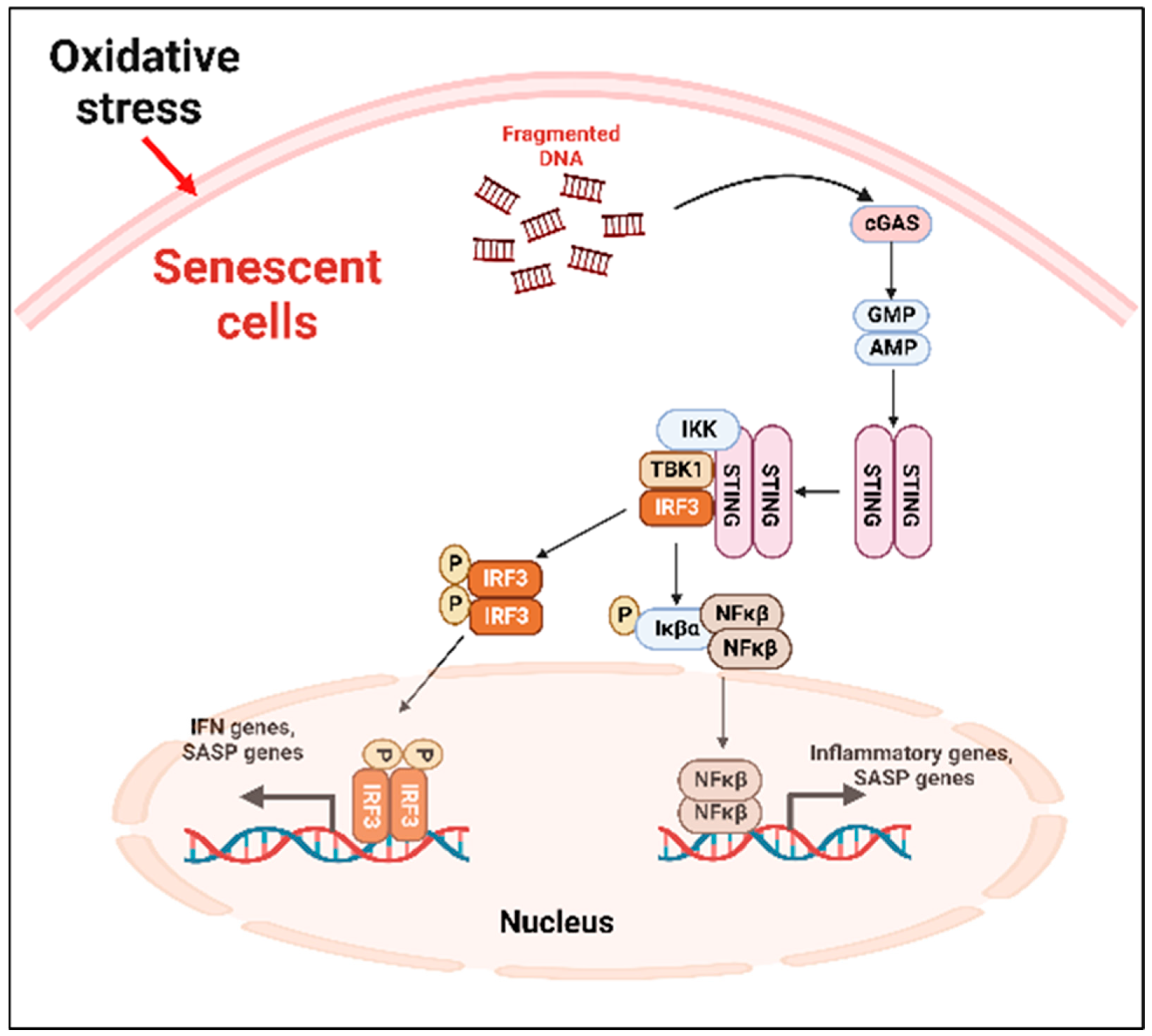
Figure 2. cGAS-STING-mediated cellular senescence. In response to oxidative stress, DNA fragments are released from the nuclei to the cytoplasm which are recognized by the cGAS, activating STING. The activated STING complex with TBK1 phosphorylates the transcription factor IRF3 and NFκβ. Activated NFκβ induces the transcription of proinflammatory SASP genes. cGAS: cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase; STING: stimulator of interferon genes; TBK1: TANK-binding kinase 1; NFκβ: nuclear factor ‘kappa-light-chain-enhancer.
- B.
-
p53 pathway
p53 plays an important role in the onset of cellular senescence. The telomeric erosion and the DNA damage response pathway leads to the activation of p53 [72][73]. In response to the DDR, the stress sensors’ telangiectasia-muted (ATM) or ataxia telangiectasia and Rad3-related (ATR) kinases are activated, which in turn activate the p53/p21cip1 with p53 stabilization [72]. The p21cip1 is one of the founding members of the mammalian CDK inhibitor family and, upon activation, p21cip1 binds with many apoptotic genes, including caspases; as a result, it inhibits apoptosis and induces senescence [74]. p53 also plays as a molecular switch in the insulin-like growth factor-1 (IGF-1)-induced cellular premature senescence. Increased IGF-1 levels are associated with cancer progression [75]. Tran et al. have shown that long-term IGF-1 exposure increased p53 acetylation, leading to p53 stabilization which ultimately induced premature senescence (Figure 3) [76].
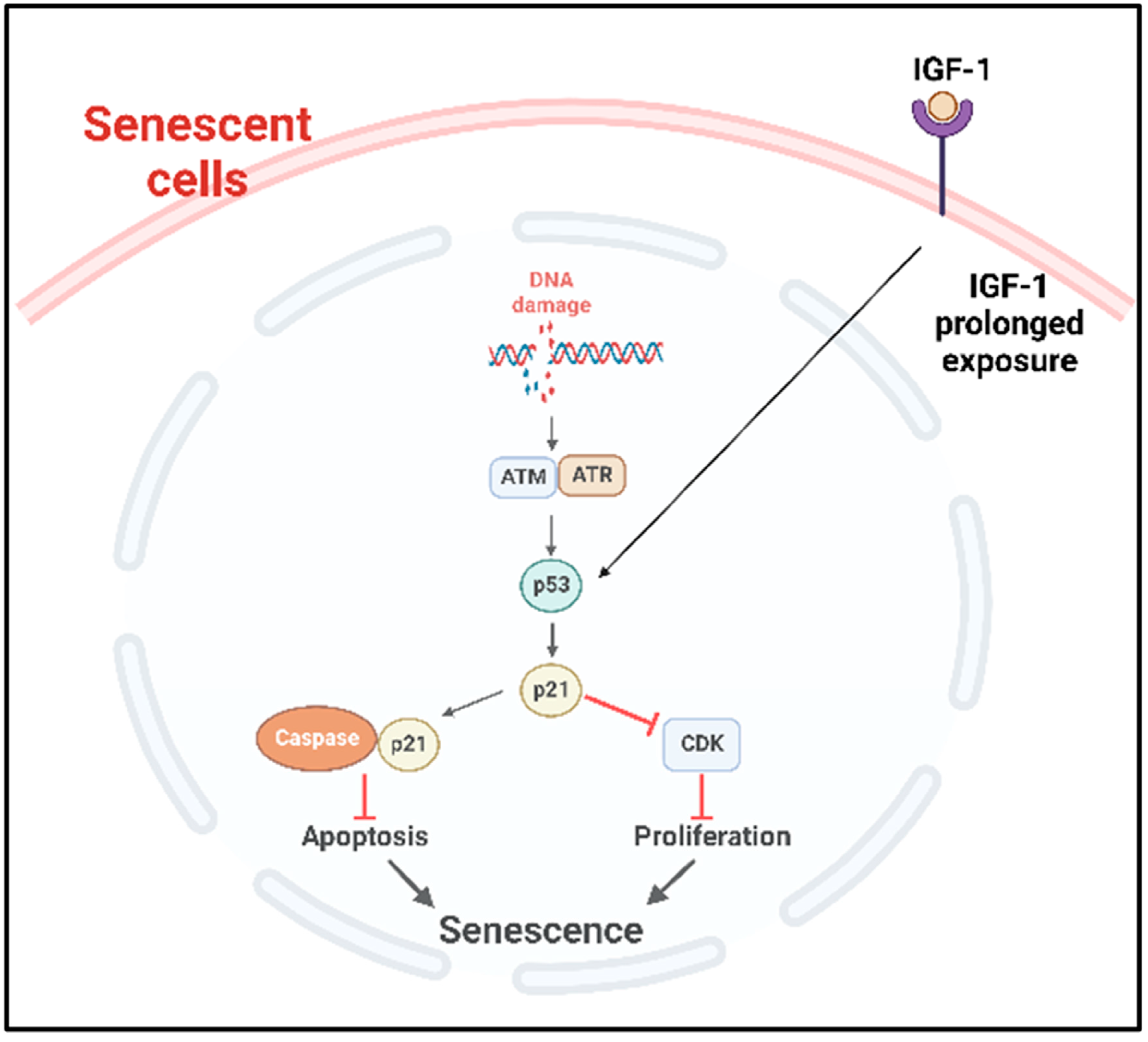
Figure 3. p53 mediated cellular senescence. In response to DNA damage caused by DNA damaging factors, the DDR pathway is activated with the concomitant activation of ATR or ATM which in turn stabilize p53. Prolonged exposure of the cancer cells to IGF-1 also causes the stabilization and activation of p53 pathway. Consequently, the downstream p53/p21cip1 are activated, which can then inhibit the apoptosis by binding with apoptotic genes including caspases and induce senescence. Activated p21cip1, which is a member of the CDK inhibitor, inhibits the cellular proliferation. DDR: DNA damage response, ATM: ataxia telangiectasia mutated, ATR: ataxia telangiectasia and Rad3-related kinase, IGF-1: insulin-like growth factor-1, CDK: cyclin-dependent kinases.
- C.
-
NFκβ pathway
In solid tumors, the noncanonical NFκβ pathway activation leads to senescence via the regulation of the enhancer of Zeste homologue 2 (EZH2) [77]. EZH2 is significantly increased in hematopoietic and solid tumors [78]. The overexpression of EZH2 suppresses the senescence by inhibiting p21Cip1 (CDKN1A) in a p53-independent manner [79]. Another important regulator of the canonical NFκβ pathway that induced premature senescence in cancer is the DNA damage which activates the NFκβ pathway with NFκβ essential modulator (NEMO) protein. NEMO, the regulatory subunit of the Iκβ Kinase Complex (IKK) complex protein, regulates NFκβ signaling via the regulation of the IKK complex [80]. The genotoxic stress-induced SUMOylation of the NFκβ essential modulator (NEMO) can also be promoted by p53-induced protein with a death domain (PIDD) and receptor-interacting protein kinase 1 (RIP1). The SUMOylation of NEMO induces its nuclear export. Additionally, the stress-induced double-stranded break (DSB) of DNA activates the ATM, which in turn phosphorylates NEMO, inducing its monoubiquitination and nuclear export. As a consequence, the IKK complex is activated, and the activated IKK phosphorylates Iκβα and its proteasomal degradation. Finally, the p65/p50 heterodimer is released and translocated to the nucleus to activate the NFκβ signaling cascade [81][82]. A study by Dong et al. reported that radiation-induced endothelial cell senescence is caused by the activation of the DSB/NEMO/NFκβ signal pathway (Figure 4) [83].
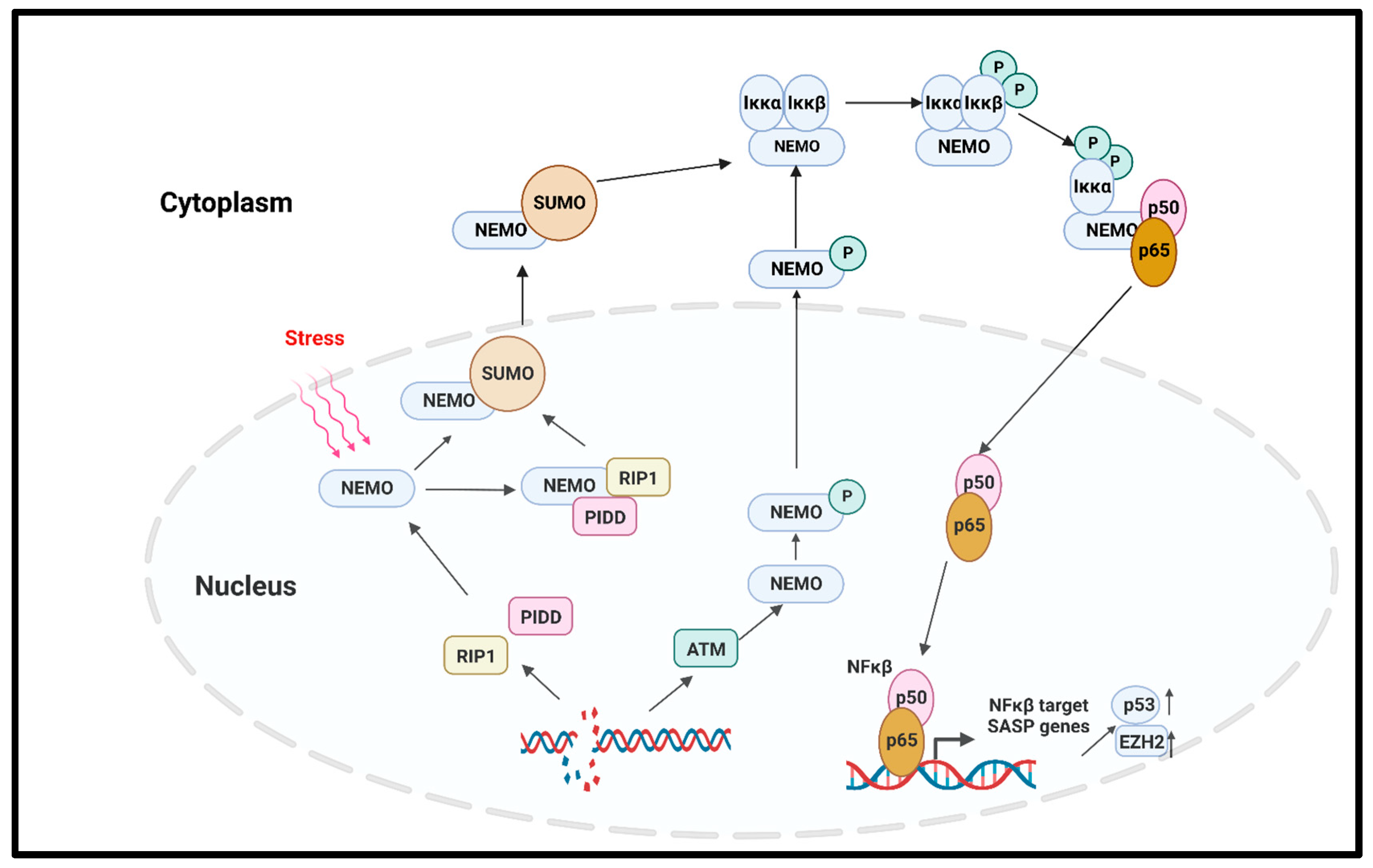
Figure 4. NFκβ-mediated cellular senescence. Extracellular stress or DNA damage caused by extra- or intracellular factors activate the NEMO, which is the regulatory subunit of IKK complex. In response to genotoxic stress, the p53-induced protein with death domain (PIDD) and receptor-interacting protein kinase 1 (RIP1) SUMOylate NEMO, and SUMOylated NEMO is exported to the cytoplasm. The DNA-damage-induced activation of ATM also phosphorylates the NEMO and causes its nuclear export. In the cytoplasm, NEMO activates IKK complex, phosphorylates the Iκβα, and induces its proteasomal degradation. p65/p50 heterodimer is released and transported to the nucleus to activate the NFκβ signaling, and as the downstream effect, SASP genes are expressed. NEMO: NFκβ essential modulator; IKK: Iκβ kinase; PIDD: p53-induced protein with death domain; RIP1: receptor interacting protein kinase 1.
An essential regulator of the TNF-induced NFkβ pathway is ataxia-telangiectasia mutated (ATM), a master regulator of the DNA double-strand break (DSB) repair pathway after genotoxic stress [84]. The downstream target of damage ATM for the cell cycle checkpoint is p53, which is regulated by ATM-dependent phosphorylation [85]. Cells defective in ATM function have defective telomere metabolism [86][87][88] as well as a higher frequency of SA-β-gal [89], which are both phenotypes associated with senescence. The agents causing DSBs lead to p16INK4a enrichment and the premature senescence of normal fibroblasts [90]. A transient increase in p21 is followed by a delayed induction of p16INK4a, which also happens with the permanent arrest that is observed with cellular senescence. These observations have indicated that damage-induced cells are very similar to senescent cells and have additional factor(s) beside p21 and p53 that maintain cell cycle arrest [90].
- D.
-
Mammalian target of rapamycin (mTOR) pathway
mTOR is the intracellular target of the pharmacological drug rapamycin, which is widely used in many cancers. The mTOR pathway positively regulates the protein synthesis pathway and inhibits autophagy, thereby playing an important role in senescence [91][92]. As discussed earlier, cellular senescence is associated with mitochondrial dysfunction, resulting in impaired ATP generation and increased reactive oxygen species (ROS). Mitochondrial metabolism and biogenesis are regulated by the master regulators Peroxisome proliferator-activated receptor gamma (PPARγ) and its coactivator 1(Peroxisome proliferator-activated receptor-γ coactivator, PGC1-α). Importantly, mTORC1, one of the TOR complexes (mTROC1 and mTORC2), regulates the transcriptional activity of PGC1-α [93]. Thus, rapamycin treatment to inhibit mTOR signaling reduces radiation-induced ROS production, inhibits senescence, and increases cellular life span [94][95].
- E.
-
Transforming growth factor-β (TGFβ) pathway
The role of TGFβ in premature senescence has been reported in several studies in multiple cell types, which include bronchial epithelial cells and hepatocellular carcinoma cells [96][97][98]. TGFβ induced the cyclin-dependent kinase inhibitors p15Ink4b, p21, and p27 and suppressed cellular proliferation [99][100]. TGFβ was also reported to induce ROS production in mitochondria in different cell types [101][102], and ROS are one of the inducers of premature senescence. Another TGFβ-targeted gene which is an important regulator of senescence and age-related diseases is plasminogen activator inhibitor-1 (PAI-1) [103]. Interestingly, TGFβ is also considered an important SASP factor, and it causes senescence in cells by autocrine and paracrine manners. In senescence, the polycomb protein Chromobox 7, CBX7, affects the upregulation of integrin β3 (ITGB3), which in turn activates the TGFβ signaling in an autocrine and paracrine manner in human fibroblast [104].
- F.
-
Mitogen-activated protein kinase (MAPK) pathway
Genotoxic stress in senescence activates the p38 MAPK pathway, which is independent of the DDR [105]. In senescent cells, p38 MAPK regulates the NFκβ activity; the role of NFκβ in senescence was discussed in a previous section [105]. In senescent T-cells, the intracellular metabolic sensor AMPK activates p38 via its autophosphorylation via the scaffold protein TAB1 [106]. The activation of this pathway leads to the inhibition of telomerase activity, T-cell proliferation, and senescence [106].
4. Telomerase Activity Suppresses Senescence and Its Inhibition Enhances Senescence
Telomerase consists of an RNA component (hTR), which serves as a telomeric template and a catalytic protein component (hTERT), which has a reverse transcriptase activity [107][108][109]. The ectopic expression of hTERT prevents replicative senescence in several cell types, including fibroblasts and epithelial cells, by exerting antiapoptotic action in early stages of the cell death prior to caspase activation and mitochondrial dysfunction [110][111]. Immortalization in human cells has been achieved by the expression of hTERT [89], which results in the loss of p16-dependent cell cycle control [112][113]. The inhibition of telomerase activity via treatment with GRN163L (human telomerase RNA-targeted antisense agents) inhibits cell growth [113], supporting the argument that telomerase regulates senescence.
References
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77.
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100.
- Harley, C.B.; Vaziri, H.; Counter, C.M.; Allsopp, R.C. The telomere hypothesis of cellular aging. Exp. Gerontol. 1992, 27, 375–382.
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621.
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Invest. 2018, 128, 1238–1246.
- Campisi, J. Cellular senescence and apoptosis: How cellular responses might influence aging phenotypes. Exp. Gerontol. 2003, 38, 5–11.
- Banerjee, P.; Kotla, S.; Reddy Velatooru, L.; Abe, R.J.; Davis, E.A.; Cooke, J.P.; Schadler, K.; Deswal, A.; Herrmann, J.; Lin, S.H.; et al. Senescence-Associated Secretory Phenotype as a Hinge Between Cardiovascular Diseases and Cancer. Front. Cardiovasc. Med 2021, 8, 763930.
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453.
- Lawrenson, K.; Grun, B.; Benjamin, E.; Jacobs, I.J.; Dafou, D.; Gayther, S.A. Senescent fibroblasts promote neoplastic transformation of partially transformed ovarian epithelial cells in a three-dimensional model of early stage ovarian cancer. Neoplasia 2010, 12, 317–325.
- Rojas, A.; Liu, G.; Coleman, I.; Nelson, P.S.; Zhang, M.; Dash, R.; Fisher, P.B.; Plymate, S.R.; Wu, J.D. IL-6 promotes prostate tumorigenesis and progression through autocrine cross-activation of IGF-IR. Oncogene 2011, 30, 2345–2355.
- Hartman, Z.C.; Poage, G.M.; den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480.
- Di, G.H.; Liu, Y.; Lu, Y.; Liu, J.; Wu, C.; Duan, H.F. IL-6 secreted from senescent mesenchymal stem cells promotes proliferation and migration of breast cancer cells. PLoS ONE 2014, 9, e113572.
- Song, L.; Rawal, B.; Nemeth, J.A.; Haura, E.B. JAK1 activates STAT3 activity in non-small-cell lung cancer cells and IL-6 neutralizing antibodies can suppress JAK1-STAT3 signaling. Mol. Cancer Ther. 2011, 10, 481–494.
- Kim, E.K.; Moon, S.; Kim, D.K.; Zhang, X.; Kim, J. CXCL1 induces senescence of cancer-associated fibroblasts via autocrine loops in oral squamous cell carcinoma. PLoS ONE 2018, 13, e0188847.
- Li, H.; Qiu, L.; Liu, Q.; Ma, Z.; Xie, X.; Luo, Y.; Wu, X. Senescent Fibroblasts Generate a CAF Phenotype through the Stat3 Pathway. Genes 2022, 13, 1579.
- Schosserer, M.; Grillari, J.; Breitenbach, M. The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Front. Oncol. 2017, 7, 278.
- Freitas-Rodriguez, S.; Folgueras, A.R.; Lopez-Otin, C. The role of matrix metalloproteinases in aging: Tissue remodeling and beyond. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2015–2025.
- Hassona, Y.; Cirillo, N.; Heesom, K.; Parkinson, E.K.; Prime, S.S. Senescent cancer-associated fibroblasts secrete active MMP-2 that promotes keratinocyte dis-cohesion and invasion. Br. J. Cancer 2014, 111, 1230–1237.
- Liu, D.; Hornsby, P.J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007, 67, 3117–3126.
- Canino, C.; Mori, F.; Cambria, A.; Diamantini, A.; Germoni, S.; Alessandrini, G.; Borsellino, G.; Galati, R.; Battistini, L.; Blandino, R.; et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 2012, 31, 3148–3163.
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547.
- Yang, F.; Tuxhorn, J.A.; Ressler, S.J.; McAlhany, S.J.; Dang, T.D.; Rowley, D.R. Stromal expression of connective tissue growth factor promotes angiogenesis and prostate cancer tumorigenesis. Cancer Res. 2005, 65, 8887–8895.
- Coppe, J.P.; Kauser, K.; Campisi, J.; Beausejour, C.M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 2006, 281, 29568–29574.
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362ra144.
- Wu, S.G.; He, Z.Y.; Li, Q.; Sun, J.Y.; Li, F.Y.; Lin, Q.; Lin, H.X.; Guan, X.X. Prognostic value of metastatic axillary lymph node ratio for Chinese breast cancer patients. PLoS ONE 2013, 8, e61410.
- Wang, L.; Dou, X.; Liu, T.; Lu, W.; Ma, Y.; Yang, Y. Tumor size and lymph node metastasis are prognostic markers of small cell lung cancer in a Chinese population. Medicine 2018, 97, e11712.
- Taghizadeh-Kermani, A.; Yahouiyan, S.Z.; AliAkbarian, M.; Seilanian Toussi, M. Prognostic significance of metastatic lymph node ratio in patients with gastric cancer: An evaluation in north-East of iran. Iran. J. Cancer Prev. 2014, 7, 73–79.
- Kim, Y.H.; Choi, Y.W.; Lee, J.; Soh, E.Y.; Kim, J.H.; Park, T.J. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat. Commun. 2017, 8, 15208.
- Milanovic, M.; Yu, Y.; Schmitt, C.A. The Senescence-Stemness Alliance—A Cancer-Hijacked Regeneration Principle. Trends Cell Biol. 2018, 28, 1049–1061.
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011, 21, 440–446.
- Khadka, D.B.; Cho, W.J. Topoisomerase inhibitors as anticancer agents: A patent update. Expert. Opin. Ther. Pat. 2013, 23, 1033–1056.
- Rothenberg, M.L. Topoisomerase I inhibitors: Review and update. Ann. Oncol. 1997, 8, 837–855.
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anticancer Agents 2005, 5, 363–372.
- Karabicici, M.; Alptekin, S.; Firtina Karagonlar, Z.; Erdal, E. Doxorubicin-induced senescence promotes stemness and tumorigenicity in EpCAM-/CD133- nonstem cell population in hepatocellular carcinoma cell line, HuH-7. Mol. Oncol. 2021, 15, 2185–2202.
- Mitry, M.A.; Laurent, D.; Keith, B.L.; Sira, E.; Eisenberg, C.A.; Eisenberg, L.M.; Joshi, S.; Gupte, S.; Edwards, J.G. Accelerated cardiomyocyte senescence contributes to late-onset doxorubicin-induced cardiotoxicity. Am. J. Physiol. Cell Physiol. 2020, 318, C380–C391.
- Beltzig, L.; Schwarzenbach, C.; Leukel, P.; Frauenknecht, K.B.M.; Sommer, C.; Tancredi, A.; Hegi, M.E.; Christmann, M.; Kaina, B. Senescence Is the Main Trait Induced by Temozolomide in Glioblastoma Cells. Cancers 2022, 14, 2233.
- Fang, K.; Chiu, C.C.; Li, C.H.; Chang, Y.T.; Hwang, H.T. Cisplatin-induced senescence and growth inhibition in human non-small cell lung cancer cells with ectopic transfer of p16INK4a. Oncol. Res. 2007, 16, 479–488.
- Zhao, W.; Lin, Z.X.; Zhang, Z.Q. Cisplatin-induced premature senescence with concomitant reduction of gap junctions in human fibroblasts. Cell Res. 2004, 14, 60–66.
- Sun, X.; Shi, B.; Zheng, H.; Min, L.; Yang, J.; Li, X.; Liao, X.; Huang, W.; Zhang, M.; Xu, S.; et al. Senescence-associated secretory factors induced by cisplatin in melanoma cells promote non-senescent melanoma cell growth through activation of the ERK1/2-RSK1 pathway. Cell Death Dis. 2018, 9, 260.
- Li, W.; Wang, W.; Dong, H.; Li, Y.; Li, L.; Han, L.; Han, Z.; Wang, S.; Ma, D.; Wang, H. Cisplatin-induced senescence in ovarian cancer cells is mediated by GRP78. Oncol. Rep. 2014, 31, 2525–2534.
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284.
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176.
- Hu, X.; Zhang, H. Doxorubicin-Induced Cancer Cell Senescence Shows a Time Delay Effect and Is Inhibited by Epithelial-Mesenchymal Transition (EMT). Med. Sci. Monit 2019, 25, 3617–3623.
- Tamamori-Adachi, M.; Koga, A.; Susa, T.; Fujii, H.; Tsuchiya, M.; Okinaga, H.; Hisaki, H.; Iizuka, M.; Kitajima, S.; Okazaki, T. DNA damage response induced by Etoposide promotes steroidogenesis via GADD45A in cultured adrenal cells. Sci. Rep. 2018, 8, 9636.
- Teng, Y.N.; Chang, H.C.; Chao, Y.Y.; Cheng, H.L.; Lien, W.C.; Wang, C.Y. Etoposide Triggers Cellular Senescence by Inducing Multiple Centrosomes and Primary Cilia in Adrenocortical Tumor Cells. Cells 2021, 10, 1466.
- Kasper, M.; Barth, K. Bleomycin and its role in inducing apoptosis and senescence in lung cells-modulating effects of caveolin-1. Curr. Cancer Drug Targets 2009, 9, 341–353.
- Wang, X.; Wong, S.C.; Pan, J.; Tsao, S.W.; Fung, K.H.; Kwong, D.L.; Sham, J.S.; Nicholls, J.M. Evidence of cisplatin-induced senescent-like growth arrest in nasopharyngeal carcinoma cells. Cancer Res. 1998, 58, 5019–5022.
- Seifrtova, M.; Havelek, R.; Soukup, T.; Filipova, A.; Mokry, J.; Rezacova, M. Mitoxantrone ability to induce premature senescence in human dental pulp stem cells and human dermal fibroblasts. J. Physiol. Pharmacol. 2013, 64, 255–266.
- Han, L.; Long, Q.; Li, S.; Xu, Q.; Zhang, B.; Dou, X.; Qian, M.; Jiramongkol, Y.; Guo, J.; Cao, L.; et al. Senescent Stromal Cells Promote Cancer Resistance through SIRT1 Loss-Potentiated Overproduction of Small Extracellular Vesicles. Cancer Res. 2020, 80, 3383–3398.
- Aasland, D.; Gotzinger, L.; Hauck, L.; Berte, N.; Meyer, J.; Effenberger, M.; Schneider, S.; Reuber, E.E.; Roos, W.P.; Tomicic, M.T.; et al. Temozolomide Induces Senescence and Repression of DNA Repair Pathways in Glioblastoma Cells via Activation of ATR-CHK1, p21, and NF-kappaB. Cancer Res. 2019, 79, 99–113.
- Mhaidat, N.M.; Zhang, X.D.; Allen, J.; Avery-Kiejda, K.A.; Scott, R.J.; Hersey, P. Temozolomide induces senescence but not apoptosis in human melanoma cells. Br. J. Cancer 2007, 97, 1225–1233.
- Mohiuddin, M.; Kasahara, K. The Mechanisms of the Growth Inhibitory Effects of Paclitaxel on Gefitinib-resistant Non-small Cell Lung Cancer Cells. Cancer Genom. Proteom. 2021, 18, 661–673.
- Milczarek, M. The Premature Senescence in Breast Cancer Treatment Strategy. Cancers 2020, 12, 1815.
- Dabrowska, M.; Mosieniak, G.; Skierski, J.; Sikora, E.; Rode, W. Methotrexate-induced senescence in human adenocarcinoma cells is accompanied by induction of p21(waf1/cip1) expression and lack of polyploidy. Cancer Lett. 2009, 284, 95–101.
- Zhang, J.W.; Zhang, S.S.; Song, J.R.; Sun, K.; Zong, C.; Zhao, Q.D.; Liu, W.T.; Li, R.; Wu, M.C.; Wei, L.X. Autophagy inhibition switches low-dose camptothecin-induced premature senescence to apoptosis in human colorectal cancer cells. Biochem. Pharmacol. 2014, 90, 265–275.
- Aguado-Flor, E.; Fuentes-Raspall, M.J.; Gonzalo, R.; Alonso, C.; Ramon, Y.C.T.; Fisas, D.; Seoane, A.; Sanchez-Pla, A.; Giralt, J.; Diez, O.; et al. Cell Senescence-Related Pathways Are Enriched in Breast Cancer Patients With Late Toxicity After Radiotherapy and Low Radiation-Induced Lymphocyte Apoptosis. Front. Oncol. 2022, 12, 825703.
- Meng, J.; Li, Y.; Wan, C.; Sun, Y.; Dai, X.; Huang, J.; Hu, Y.; Gao, Y.; Wu, B.; Zhang, Z.; et al. Targeting senescence-like fibroblasts radiosensitizes non-small cell lung cancer and reduces radiation-induced pulmonary fibrosis. JCI Insight 2021, 6, e146334.
- Dabritz, J.H.; Yu, Y.; Milanovic, M.; Schonlein, M.; Rosenfeldt, M.T.; Dorr, J.R.; Kaufmann, A.M.; Dorken, B.; Schmitt, C.A. CD20-Targeting Immunotherapy Promotes Cellular Senescence in B-Cell Lymphoma. Mol. Cancer Ther. 2016, 15, 1074–1081.
- Chibaya, L.; Snyder, J.; Ruscetti, M. Senescence and the tumor-immune landscape: Implications for cancer immunotherapy. Semin. Cancer Biol. 2022, 86, 827–845.
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199.
- Chen, Z.; Cao, K.; Xia, Y.; Li, Y.; Hou, Y.; Wang, L.; Li, L.; Chang, L.; Li, W. Cellular senescence in ionizing radiation (Review). Oncol. Rep. 2019, 42, 883–894.
- Wang, Y.; Boerma, M.; Zhou, D. Ionizing Radiation-Induced Endothelial Cell Senescence and Cardiovascular Diseases. Radiat. Res. 2016, 186, 153–161.
- Nishimura, T.; Iwakabe, K.; Sekimoto, M.; Ohmi, Y.; Yahata, T.; Nakui, M.; Sato, T.; Habu, S.; Tashiro, H.; Sato, M.; et al. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J. Exp. Med. 1999, 190, 617–627.
- Qin, S.; Schulte, B.A.; Wang, G.Y. Role of senescence induction in cancer treatment. World J. Clin. Oncol. 2018, 9, 180–187.
- Muller-Hermelink, N.; Braumuller, H.; Pichler, B.; Wieder, T.; Mailhammer, R.; Schaak, K.; Ghoreschi, K.; Yazdi, A.; Haubner, R.; Sander, C.A.; et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell 2008, 13, 507–518.
- Braumuller, H.; Wieder, T.; Brenner, E.; Assmann, S.; Hahn, M.; Alkhaled, M.; Schilbach, K.; Essmann, F.; Kneilling, M.; Griessinger, C.; et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013, 494, 361–365.
- Rosemblit, C.; Datta, J.; Lowenfeld, L.; Xu, S.; Basu, A.; Kodumudi, K.; Wiener, D.; Czerniecki, B.J. Oncodriver inhibition and CD4(+) Th1 cytokines cooperate through Stat1 activation to induce tumor senescence and apoptosis in HER2+ and triple negative breast cancer: Implications for combining immune and targeted therapies. Oncotarget 2018, 9, 23058–23077.
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314.
- Chen, Q.; Fischer, A.; Reagan, J.D.; Yan, L.J.; Ames, B.N. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc. Natl. Acad. Sci. USA 1995, 92, 4337–4341.
- Kotla, S.; Zhang, A.; Imanishi, M.; Ko, K.A.; Lin, S.H.; Gi, Y.J.; Moczygemba, M.; Isgandarova, S.; Schadler, K.L.; Chung, C.; et al. Nucleus-mitochondria positive feedback loop formed by ERK5 S496 phosphorylation-mediated poly (ADP-ribose) polymerase activation provokes persistent pro-inflammatory senescent phenotype and accelerates coronary atherosclerosis after chemo-radiation. Redox. Biol. 2021, 47, 102132.
- Ernst, P.; Heidel, F.H. Molecular Mechanisms of Senescence and Implications for the Treatment of Myeloid Malignancies. Cancers 2021, 13, 612.
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420.
- Benslimane, Y.; Sanchez-Osuna, M.; Coulombe-Huntington, J.; Bertomeu, T.; Henry, D.; Huard, C.; Bonneil, E.; Thibault, P.; Tyers, M.; Harrington, L. A novel p53 regulator, C16ORF72/TAPR1, buffers against telomerase inhibition. Aging Cell 2021, 20, e13331.
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295.
- de Ostrovich, K.K.; Lambertz, I.; Colby, J.K.; Tian, J.; Rundhaug, J.E.; Johnston, D.; Conti, C.J.; DiGiovanni, J.; Fuchs-Young, R. Paracrine overexpression of insulin-like growth factor-1 enhances mammary tumorigenesis in vivo. Am. J. Pathol. 2008, 173, 824–834.
- Tran, D.; Bergholz, J.; Zhang, H.; He, H.; Wang, Y.; Zhang, Y.; Li, Q.; Kirkland, J.L.; Xiao, Z.X. Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell 2014, 13, 669–678.
- De Donatis, G.M.; Le Pape, E.; Pierron, A.; Cheli, Y.; Hofman, V.; Hofman, P.; Allegra, M.; Zahaf, K.; Bahadoran, P.; Rocchi, S.; et al. NF-kB2 induces senescence bypass in melanoma via a direct transcriptional activation of EZH2. Oncogene 2016, 35, 2735–2745.
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104.
- Fan, T.; Jiang, S.; Chung, N.; Alikhan, A.; Ni, C.; Lee, C.C.; Hornyak, T.J. EZH2-dependent suppression of a cellular senescence phenotype in melanoma cells by inhibition of p21/CDKN1A expression. Mol. Cancer Res. 2011, 9, 418–429.
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal. 2012, 24, 835–845.
- Wu, Z.H.; Miyamoto, S. Many faces of NF-kappaB signaling induced by genotoxic stress. J. Mol. Med. 2007, 85, 1187–1202.
- McCool, K.W.; Miyamoto, S. DNA damage-dependent NF-kappaB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012, 246, 311–326.
- Dong, X.; Tong, F.; Qian, C.; Zhang, R.; Dong, J.; Wu, G.; Hu, Y. NEMO modulates radiation-induced endothelial senescence of human umbilical veins through NF-kappaB signal pathway. Radiat. Res. 2015, 183, 82–93.
- Fang, L.; Choudhary, S.; Zhao, Y.; Edeh, C.B.; Yang, C.; Boldogh, I.; Brasier, A.R. ATM regulates NF-kappaB-dependent immediate-early genes via RelA Ser 276 phosphorylation coupled to CDK9 promoter recruitment. Nucleic Acids Res. 2014, 42, 8416–8432.
- Pandita, T.K. ATM function and telomere stability. Oncogene 2002, 21, 611–618.
- Smilenov, L.B.; Morgan, S.E.; Mellado, W.; Sawant, S.G.; Kastan, M.B.; Pandita, T.K. Influence of ATM function on telomere metabolism. Oncogene 1997, 15, 2659–2665.
- Pandita, T.K.; Pathak, S.; Geard, C.R. Chromosome end associations, telomeres and telomerase activity in ataxia telangiectasia cells. Cytogenet. Cell Genet. 1995, 71, 86–93.
- Smilenov, L.B.; Dhar, S.; Pandita, T.K. Altered telomere nuclear matrix interactions and nucleosomal periodicity in ataxia telangiectasia cells before and after ionizing radiation treatment. Mol. Cell Biol. 1999, 19, 6963–6971.
- Wood, L.D.; Halvorsen, T.L.; Dhar, S.; Baur, J.A.; Pandita, R.K.; Wright, W.E.; Hande, M.P.; Calaf, G.; Hei, T.K.; Levine, F.; et al. Characterization of ataxia telangiectasia fibroblasts with extended life-span through telomerase expression. Oncogene 2001, 20, 278–288.
- Robles, S.J.; Adami, G.R. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene 1998, 16, 1113–1123.
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293.
- Xu, S.; Cai, Y.; Wei, Y. mTOR Signaling from Cellular Senescence to Organismal Aging. Aging Dis. 2014, 5, 263–273.
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007, 450, 736–740.
- Iglesias-Bartolome, R.; Patel, V.; Cotrim, A.; Leelahavanichkul, K.; Molinolo, A.A.; Mitchell, J.B.; Gutkind, J.S. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell 2012, 11, 401–414.
- Lerner, C.; Bitto, A.; Pulliam, D.; Nacarelli, T.; Konigsberg, M.; Van Remmen, H.; Torres, C.; Sell, C. Reduced mammalian target of rapamycin activity facilitates mitochondrial retrograde signaling and increases life span in normal human fibroblasts. Aging Cell 2013, 12, 966–977.
- Senturk, S.; Mumcuoglu, M.; Gursoy-Yuzugullu, O.; Cingoz, B.; Akcali, K.C.; Ozturk, M. Transforming growth factor-beta induces senescence in hepatocellular carcinoma cells and inhibits tumor growth. Hepatology 2010, 52, 966–974.
- Minagawa, S.; Araya, J.; Numata, T.; Nojiri, S.; Hara, H.; Yumino, Y.; Kawaishi, M.; Odaka, M.; Morikawa, T.; Nishimura, S.L.; et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-beta-induced senescence of human bronchial epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L391–L401.
- Debacq-Chainiaux, F.; Borlon, C.; Pascal, T.; Royer, V.; Eliaers, F.; Ninane, N.; Carrard, G.; Friguet, B.; de Longueville, F.; Boffe, S.; et al. Repeated exposure of human skin fibroblasts to UVB at subcytotoxic level triggers premature senescence through the TGF-beta1 signaling pathway. J. Cell Sci. 2005, 118, 743–758.
- Papageorgis, P. Complex Interplay Between Aging and Cancer: Role of TGF-beta Signaling. Crit. Rev. Oncog. 2017, 22, 313–321.
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-beta Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb Perspect. Biol. 2017, 9, a022145.
- Yoon, Y.S.; Lee, J.H.; Hwang, S.C.; Choi, K.S.; Yoon, G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene 2005, 24, 1895–1903.
- Albright, C.D.; Salganik, R.I.; Craciunescu, C.N.; Mar, M.H.; Zeisel, S.H. Mitochondrial and microsomal derived reactive oxygen species mediate apoptosis induced by transforming growth factor-beta1 in immortalized rat hepatocytes. J. Cell Biochem. 2003, 89, 254–261.
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. Plasminogen Activator Inhibitor-1 Is a Marker and a Mediator of Senescence. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1446–1452.
- Rapisarda, V.; Borghesan, M.; Miguela, V.; Encheva, V.; Snijders, A.P.; Lujambio, A.; O’Loghlen, A. Integrin Beta 3 Regulates Cellular Senescence by Activating the TGF-beta Pathway. Cell Rep. 2017, 18, 2480–2493.
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548.
- Lanna, A.; Henson, S.M.; Escors, D.; Akbar, A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014, 15, 965–972.
- Harley, C.B.; Sherwood, S.W. Telomerase, checkpoints and cancer. Cancer Surv. 1997, 29, 263–284.
- Greider, C.W. Telomere length regulation. Annu. Rev. Biochem. 1996, 65, 337–365.
- Harley, C.B.; Sherwood, S.W. Aging of cultured human skin fibroblasts. Methods Mol. Biol. 1997, 75, 23–30.
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352.
- Fu, W.; Killen, M.; Culmsee, C.; Dhar, S.; Pandita, T.K.; Mattson, M.P. The catalytic subunit of telomerase is expressed in developing brain neurons and serves a cell survival-promoting function. J. Mol. Neurosci. 2000, 14, 3–15.
- Dickson, M.A.; Hahn, W.C.; Ino, Y.; Ronfard, V.; Wu, J.Y.; Weinberg, R.A.; Louis, D.N.; Li, F.P.; Rheinwald, J.G. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell Biol. 2000, 20, 1436–1447.
- Kiyono, T.; Foster, S.A.; Koop, J.I.; McDougall, J.K.; Galloway, D.A.; Klingelhutz, A.J. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 1998, 396, 84–88.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
08 Feb 2023
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No