Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marialuisa Zedde | -- | 2125 | 2023-02-06 04:24:40 | | | |
| 2 | Catherine Yang | Meta information modification | 2125 | 2023-02-06 04:28:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Paolucci, M.; Vincenzi, C.; Romoli, M.; Amico, G.; Ceccherini, I.; Lattanzi, S.; Bersano, A.; Longoni, M.; Sacco, S.; Vernieri, F.; et al. The Genetic Landscape of Patent Foramen Ovale. Encyclopedia. Available online: https://encyclopedia.pub/entry/40846 (accessed on 25 July 2026).
Paolucci M, Vincenzi C, Romoli M, Amico G, Ceccherini I, Lattanzi S, et al. The Genetic Landscape of Patent Foramen Ovale. Encyclopedia. Available at: https://encyclopedia.pub/entry/40846. Accessed July 25, 2026.
Paolucci, Matteo, Chiara Vincenzi, Michele Romoli, Giulia Amico, Isabella Ceccherini, Simona Lattanzi, Anna Bersano, Marco Longoni, Simona Sacco, Fabrizio Vernieri, et al. "The Genetic Landscape of Patent Foramen Ovale" Encyclopedia, https://encyclopedia.pub/entry/40846 (accessed July 25, 2026).
Paolucci, M., Vincenzi, C., Romoli, M., Amico, G., Ceccherini, I., Lattanzi, S., Bersano, A., Longoni, M., Sacco, S., Vernieri, F., Pascarella, R., Valzania, F., & Zedde, M. (2023, February 06). The Genetic Landscape of Patent Foramen Ovale. In Encyclopedia. https://encyclopedia.pub/entry/40846
Paolucci, Matteo, et al. "The Genetic Landscape of Patent Foramen Ovale." Encyclopedia. Web. 06 February, 2023.
Copy Citation
Patent Foramen Ovale (PFO) is a common postnatal defect of cardiac atrial septation. A certain degree of familial aggregation has been reported. Animal studies suggest the involvement of the Notch pathway and other cardiac transcription factors (GATA4, TBX20, NKX2-5) in Foramen Ovale closure.
Congenital Heart Defects
Atrial Septal Defects
paradoxical embolism
1. Introduction
Paradoxical embolism through Patent Foramen Ovale (PFO) is an important cause of cryptogenic ischemic stroke, especially in younger patients [1]. Its diagnosis may be suspected based on Transcranial Doppler (TCD), a screening test achieving high sensitivity and specificity values (96.1% and 92.4%, respectively) in detecting right-to-left shunts [2]. PFO confirmation requires transesophageal echocardiography (TEE), the gold standard diagnostic test, or transthoracic echocardiography (TTE) with intravenous contrast agent. However, the latter is affected by a lower sensitivity value (47.5% for TTE with intravenous contrast) [2]. PFO is highly prevalent in the general population [3], ranging from 35% in younger subjects to 20% in the elderly [4]. Migraine with aura carries an even higher rate of PFO [5], particularly large ones [6]. In this condition, PFO shows a strong familial aggregation with an autosomal dominant inheritance pattern [7]. Consistent with this observation, the rate of PFO in siblings of young patients with ischemic stroke and PFO is three times higher than siblings of patients without PFO [8].
PFO (Figure 1) is the result of an incomplete postnatal merger of primary and secondary atrial septum. The requirement for two different atrial septa and the high patency rate indicate how atrial septation is a protracted and complex process [9]. In week 4 of human gestation, a mesenchymal structure (the primary septum) arises from the atrial roof, shaping a primary foramen that gradually closes. Then, in week 5, a secondary foramen is formed by the coalescence of perforations of the primary septum. Around week 12, the right atrial roof folds downward and gradually become the secondary septum. After birth, the decreased pulmonary resistance and the increased pressure in the left atrium force the primary septum against the secondary septum: the structures merge, the foramen closes, and the remnant on the right atrium is called fossa ovalis [9]. The need for right-to-left shunt throughout fetal life, particularly during the prolonged gestations of mammals, must be balanced with the progressive septation process. It has been proposed that a mechanism unbalanced towards an incomplete septation could provide greater reproductive success, at the expense of negligible impact in post-natal life [9].
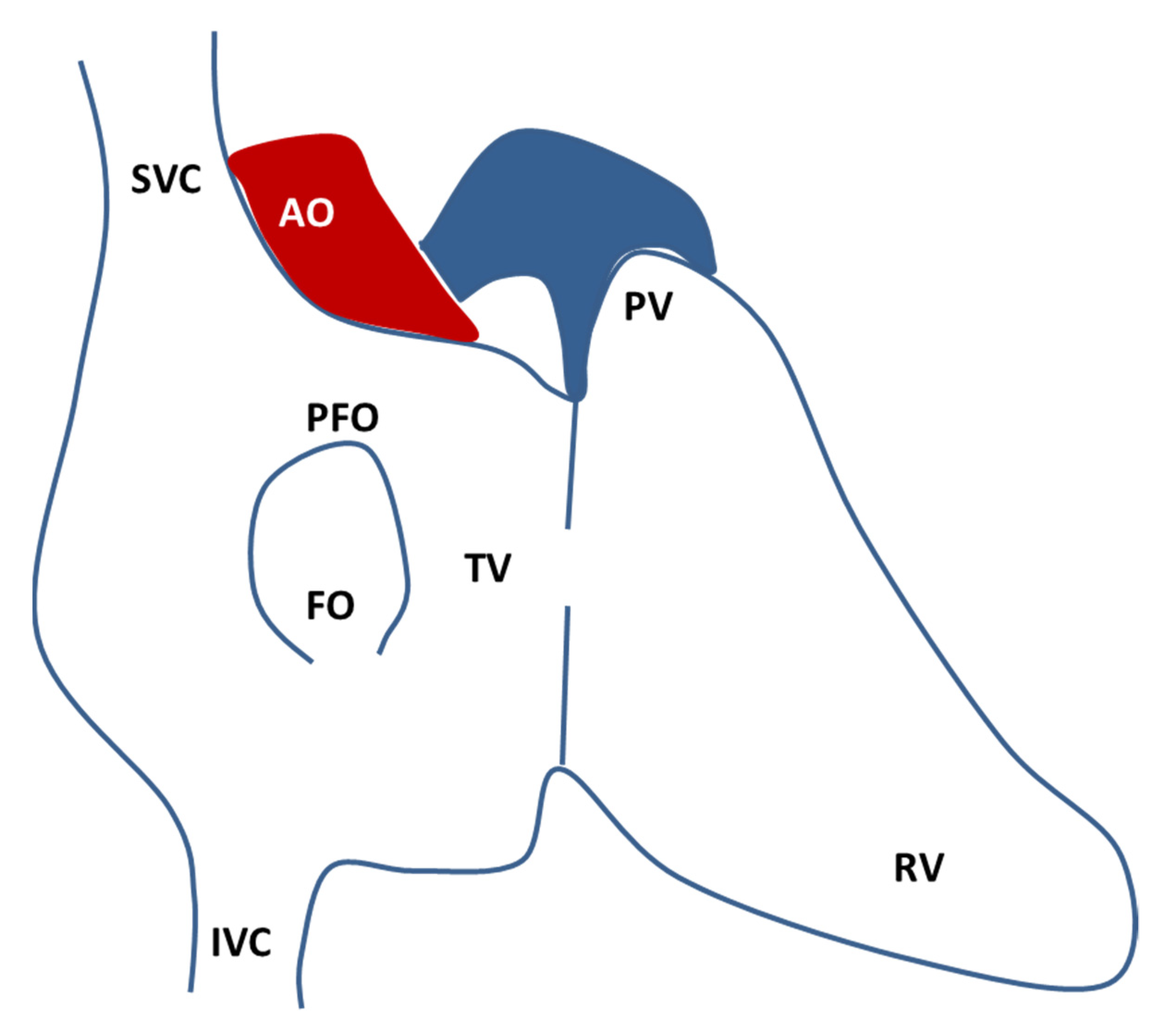
Figure 1. Schematic drawing of the atrial septal anatomy seen from the right atrium (redrawn from Calvert, P., Rana, B., Kydd, A. et al. Patent foramen ovale: anatomy, outcomes, and closure. Nat Rev Cardiol 8, 148–160 (2011). https://doi.org/10.1038/nrcardio.2010.224 accessed date 28 October 2021.). The fossa ovalis (FO) is composed by the septum primum and the septum secundum and the usual location of the PFO is on the anterosuperior border of the FO. AO, aorta; FO, fossa ovalis; IVC, inferior vena cava; PFO, patent foramen ovale; PV, pulmonary valve; RV, right ventricle; SVC, superior vena cava; TV, tricuspid valve.
The complexity of the atrial septation process is reflected by the intricacy of the involved genetic pathways. The highly conserved Notch signaling pathway is implicated in heart development, with a clear role in Foramen Ovale closure [10][11]. Other potentially involved genes (GATA Binding Protein 4, GATA4; T-Box Transcription Factor 20, TBX20; NK2 Homeobox 5, NKX2-5; Zic Family Member 3, ZIC3) were initially recognized as pathogenic for Atrial Septal Defects (ASD) [12][13]. Co-existence of other atrial abnormalities, such as right atrial septal pouch, Eustachian valve and Chiari’s network, has been reported as added factors in increasing PFO-related stroke risk [14][15]. There are no data in the literature about the inheritance of these minor atrial abnormalities neither individually nor in association with PFO. Similarly, the inheritance of the atrial septal aneurysm (ASA) is unknown.
Given the high rate of PFO in the general population, the large majority of PFOs do not play any pathological role. For those PFO subjects that suffer a cryptogenic ischemic stroke, the role of PFO as a contributing factor should be considered along with clotting disorders that may increase stroke risk. The mechanisms through which PFO may contribute to the risk of stroke and systemic embolism are at least two [16]: acting as a channel for paradoxical embolism (from a travelling venous clot, particularly in presence of a prothrombotic state, or from a clot formed in situ within the PFO) and triggering atrial arrhythmias because of electrical signaling disruption (the latter has been hypothesized especially in the setting of PFO associated with ASA or a hypermobile atrial septum). In both situations, the pattern of ischemic lesion on neuroimaging studies is coherent with an embolic source [17][18] and the potential genetic link between the presence of ASD, including PFO, and atrial arrhythmias, including atrial fibrillation, has not been extensively explored to date. The increasing knowledge has led to the proposal of an updated nomenclature and classification of potential causative mechanisms of embolic stroke in patients with a PFO [19].
Despite PFO being intensively studied for therapeutic approaches, little is known about its pathophysiological role. Can PFO be considered as a benign anatomical variant, or does it gather a more variegated range of alterations, some of which at a greater risk of comorbidity? Genetic findings may elucidate the genesis and significance of PFO.
2. The Genetic Landscape of Patent Foramen Ovale
Researchers' systematic review only found four studies evaluating genetic anomalies in PFO subjects. All of them included patients with PFO and either paradoxical embolism, stroke or TIA, or AF. This last association deserves a dedicated reasoning. While isolated PFO is usually not associated with atrial arrhythmias, these can be triggered by ASA or hypermobile atrial septum, often in combination with a PFO [20]. The underlying hypothesis is that ASDs induce atrial vulnerability, i.e., the electrophysiological trend to induce AF. Indeed, the rate of AF or atrial flutter in patients with PFO and/or ASA is 20–42% [21] and inducible AF longer than 60 s was documented in 58% of patients with PFO and/or ASA, as compared to 25% of patients without. A proposed mechanism is the stretch or pressure on the atrial septum [22].
The included studies are quite heterogeneous in terms of population, intervention, and sample size. Two of them respectively evaluated 25 [23] and 100 subjects [24], and thus their sample size is probably inadequate. We did not find any genetic variant or mutation associated with PFO, except for the rs2200733 variation of chromosome 4q25 in AF patients. The rs2200733 variation, lying in an intergenic genomic region, is indeed a known risk factor for AF [25]. The closest gene to this variation is Paired Like Homeodomain 2 (PITX2), implicated in cardiac morphogenesis, particularly in the differential identity of left and right atria [26][27]. The AF risk of rs2200733 variant and PITX2 absence, as seen in knockout mice models [28][29], is probably due to alterations of the sinoatrial node formation. However, to the knowledge, no other studies investigated the role of the rs2200733 variant nor the PITX2 gene in foramen ovale closure. On the other hand, non-PFO studies have also been reported in the literature but none of them found a positive association between the variant and the diseases investigated [30][31]. The reported association between PFO and the rs2200733 variant in patients with AF requires further validation as it was described in a single study published as a conference abstract [32]. Therefore, the hypothesis of a genetic link between PFO and AF may be proposed, enriching the list of potential correlations between PFO and stroke risk from which derive considerations with important therapeutic implications. This consideration could be particularly of interest in terms of practical translation because the main reason for the search for a PFO is a clinical presentation with an ischemic stroke with an imaging pattern suggestive of embolism (Figure 2) [33], which is defined as cryptogenic mainly by virtue of the exclusion of causes (atherothrombotic and AF) and substantially falls within the concept of Embolic Stroke of Unknown Source (ESUS). The possibility of testing AF in patients with ESUS strictly depends on the modalities and duration of cardiac monitoring and remains a matter of debate despite the negative result of therapeutic trials with direct oral anticoagulants, including the subgroup of patients with PFO [34].
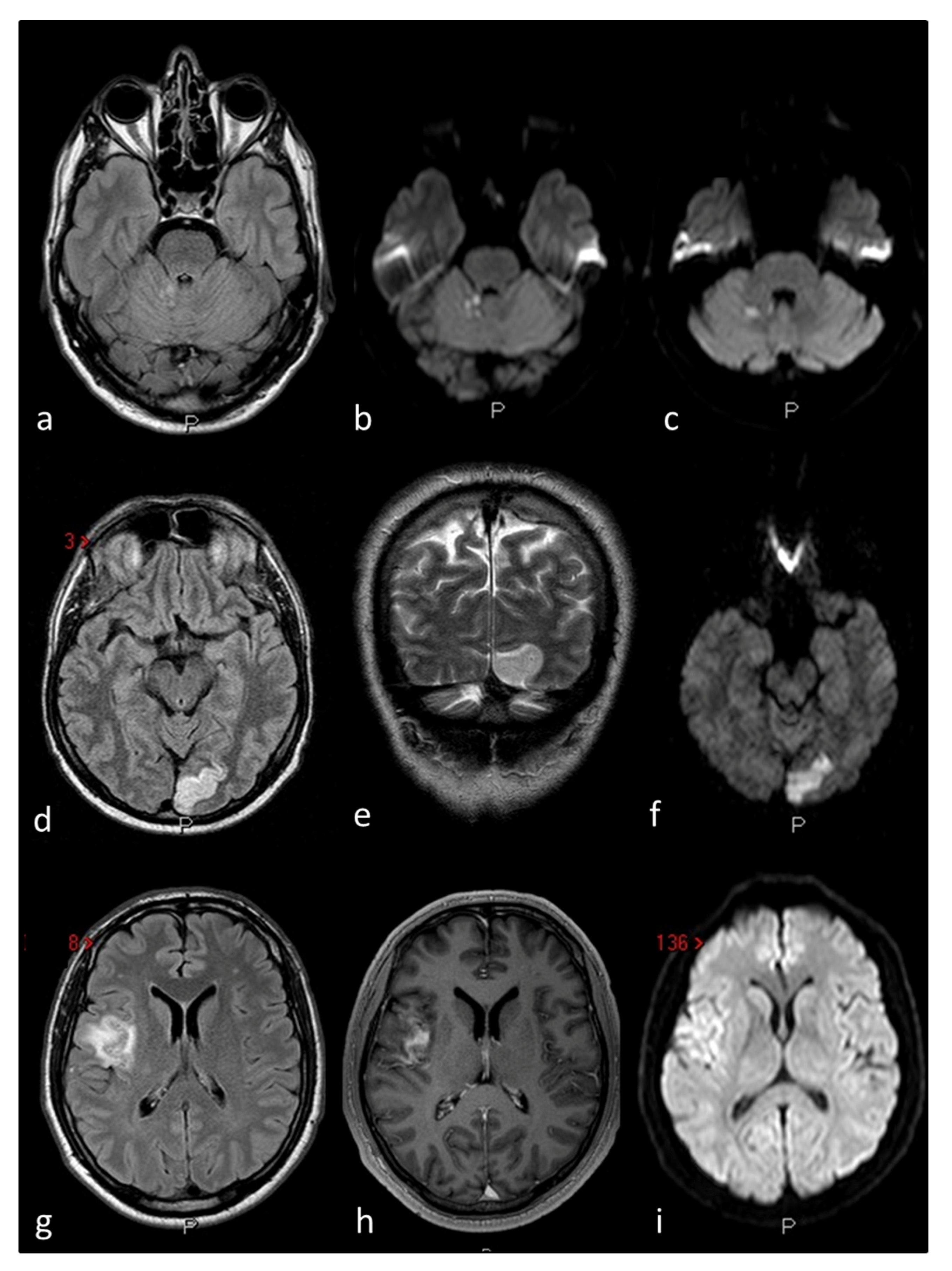
Figure 2. The figure outlines three examples of cryptogenic embolism in patients with PFO as imaged in Magnetic Resonance Imaging (MRI) of the brain: Patient 1 (a–c) multiple right cerebellar recent ischemic lesions on axial FLAIR (a) and DWI (b,c) MRI sequences. Patient 2 (d–f) left occipital ischemic stroke in a patient with migraine with aura and PFO on axial FLAIR (d) coronal T2W (e) and DWI MRI sequences. Patient 3 (g–i) right temporo-parietal ischemic stroke on axial FLAIR (g) contrast-enhanced axial T1W (h) and DWI (i) MRI sequences.
Despite the lack of epidemiological studies and the scarceness of evidence so far, animal studies and other studies that did not fulfil the inclusion criteria of the review indicate a robust genetic background in PFO.
A gain-of-function mutation in the TBX20 gene (p.I121M) was found in a patient with ostium secundum atrial septal defects (ASDII) and in his sister and mother, both affected by large PFOs [12]. Similarly, the mother of a proband with an ASD, both carrying such mutation, had a large PFO [35]. TBX20 is a T-box transcription factor, and its gain-of-function significantly enhanced transcriptional activity, which was further increased in the presence of its cardiac co-transcription factors GATA4/5 and NKX2-5. The homeobox transcription factor NKX2-5 mutations have been associated, among others, with ASD [36]. NKX2-5 heterozygous-null mice showed a significant 2.5–3.5 higher rate of PFO compared to wild type mice [37]. The synergistic role of TBX20 and NKX2-5 has been demonstrated in an animal study in which the rate of PFO was more than double in mice defective for TBX20 or NKX2-5 compared to wild type mice. Interestingly, mice defective for both genes showed a higher rate of complex PFO (PFO + atrial septal aneurysm) or even ASD [38].
GATA4 is a zinc finger transcription factor that, similarly, has a synergistic interaction with TBX20 and, likewise, mutations are responsible for Congenital Heart Defects (CHD), particularly ASD [39]. The interaction between the cardiac T-boxes (TBX20 and TBX5) and the transcription factors NKX2-5 and GATA4 suggest the complexity of the genetic regulation of cardiac septal formation.
Another signaling pathway to consider is Notch. Animal studies demonstrated its important role in the endothelial-to-mesenchymal transition (EndMT) occurring during the Foramen Ovale closure [10]. EndMT is an important mechanism involved in heart septation. The Notch receptors on the cell surface induce EndMT by activating the transcription factor Snail. In mammals, there are four types of Notch receptor (Notch 1–4) and five ligands (Delta-like-1, −3 and −4, and Jagged−1 and −2). Neurogenic locus notch homolog protein 3 (NOTCH3) is the leading gene in this process. Indeed, in rat hearts, increasingly high levels of Notch3 receptor expression, and to a lesser extent Notch1 and Jagged-1, were found in the foramen ovale region during its closure process [10]. The EndMT converted the cells to a fibroblast-like phenotype, leading to the merge of the tissues. Interestingly, NOTCH3 mutations are responsible for CADASIL (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy), the most common genetic cause of small vessel disease ischemic stroke, often associated with migraine with aura. The main pattern of cerebrovascular damage in CADASIL is small vessel disease, as seen in Figure 3. The association of PFO with CADASIL has been reported but remains controversial [40][41] and needs further insights. As of human studies, exome sequencing of members of a family with a dominant inheritance of ASD, including PFO, identified the pathogenic variant of the Notch homolog 1, translocation-associated (Drosophila) (NOTCH1) gene (c.3835C>T) [42]. However, only a minor decrease in signaling activity was found.
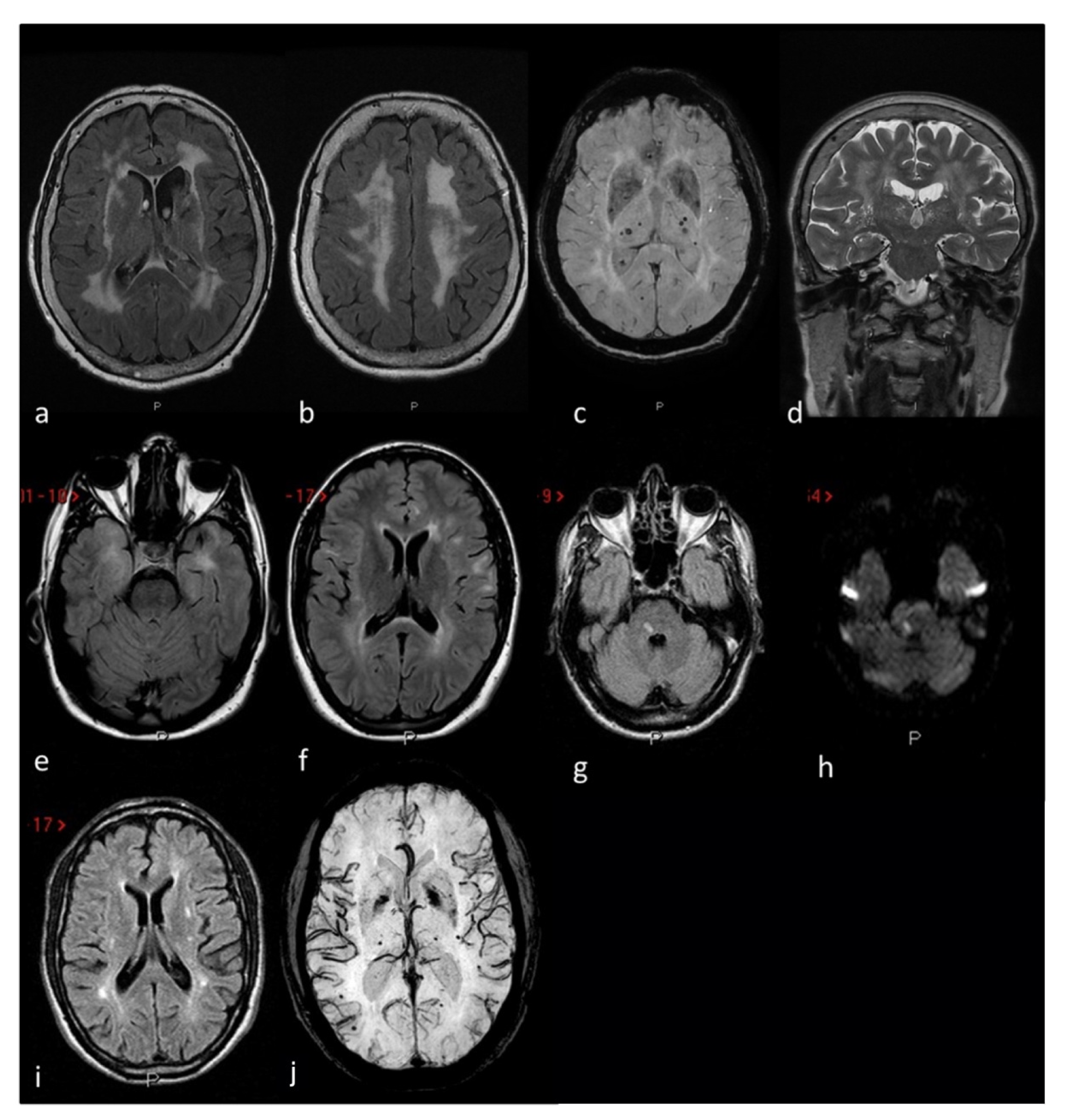
Figure 3. MRI markers of small vessel disease in three CADASIL patients: Patient 1 (a–d) severe leukoaraiosis involving subcortical white matter, external capsule and periventricular regions in a symmetrical pattern on axial FLAIR (a,b) MRI, associated with deel and lobar supratentorial microbleeds on SWI (axial MiP sequences) MRI (c) and enlarged perivascular spaces in the basal ganglia on coronal T2W (d) MRI. Patient 2 (e,f) anterior temporal lobe involvement (e) and periventricular and iuxtacortical white matter hyperintensities (f) on axial FLAIR MRI. Patient 3 (g–j) recent subcortical ischemic stroke on the right portion of the pons on FLAIR (g) and DWI (h) MRI sequences, supratentorial white matter hyperintensities with a dotted distribution involving the external capsule and the periventricular regions on axial FLAIR (i) MRI and multiple supratentorial deep and lobar microbleeds on SWI (MiP sequences) MRI (j).
So far, no single genetic alteration responsible for PFO has been found. Cardiac development, particularly atrial septation, may be influenced by the combination of different genetic factors rather than by a single gene variant. In this light, a recent genome-wide association study on Congenital Heart Defects (CHD) identified 20 risk-inducing SNPs [43]. Knockout mice for the related genes, however, did not show clinical CHD, reinforcing the concept of a multigenic etiology of the defects.
References
- Homma, S.; Sacco, R.L. Patent Foramen Ovale and Stroke. Circulation 2005, 112, 1063–1072.
- Katsanos, A.H.; Psaltopoulou, T.; Sergentanis, T.N.; Frogoudaki, A.; Vrettou, A.; Ikonomidis, I.; Paraskevaidis, I.; Parissis, J.; Bogiatzi, C.; Zompola, C.; et al. Transcranial Doppler versus Transthoracic Echocardiography for the Detection of Patent Foramen Ovale in Patients with Cryptogenic Cerebral Ischemia: A Systematic Review and Diagnostic Test Accuracy Meta-Analysis. Ann. Neurol. 2016, 79, 625–635.
- Homma, S.; Messé, S.R.; Rundek, T.; Sun, Y.-P.; Franke, J.; Davidson, K.; Sievert, H.; Sacco, R.L.; Tullio, M.R.D. Patent Foramen Ovale. Nat. Rev. Dis. Primers 2016, 66, 15086.
- Hagen, P.T.; Scholz, D.G.; Edwards, W.D. Incidence and Size of Patent Foramen Ovale during the First 10 Decades of Life: An Autopsy Study of 965 Normal Hearts. Mayo Clin. Proc. Mayo Clin. 1984, 59, 17–20.
- Del Sette, M.; Angeli, S.; Leandri, M.; Ferriero, G.; Bruzzone, G.L.; Finocchi, C.; Gandolfo, C. Migraine with Aura and Right-to-Left Shunt on Transcranial Doppler: A Case-Control Study. Cerebrovasc. Dis. 1998, 8, 327–330.
- Schwerzmann, M.; Nedeltchev, K.; Lagger, F.; Mattle, H.P.; Windecker, S.; Meier, B.; Seiler, C. Prevalence and Size of Directly Detected Patent Foramen Ovale in Migraine with Aura. Neurology 2005, 65, 1415–1418.
- Wilmshurst, P.T.; Pearson, M.J.; Nightingale, S.; Walsh, K.P.; Morrison, W.L. Inheritance of Persistent Foramen Ovale and Atrial Septal Defects and the Relation to Familial Migraine with Aura. Heart 2004, 90, 1315–1320.
- Arquizan, C.; Coste, J.; Touboul, P.-J.; Mas, J.-L. Is Patent Foramen Ovale a Family Trait? Stroke 2001, 32, 1563–1566.
- Jensen, B.; Wang, T.; Moorman, A.F.M. Evolution and Development of the Atrial Septum. Anat. Rec. 2019, 302, 32–48.
- Elliott, G.C.; Gurtu, R.; McCollum, C.; Newman, W.G.; Wang, T. Foramen Ovale Closure Is a Process of Endothelial-to-Mesenchymal Transition Leading to Fibrosis. PLoS ONE 2014, 9, e107175.
- High, F.A.; Epstein, J.A. The Multifaceted Role of Notch in Cardiac Development and Disease. Nat. Rev. Genet. 2008, 9, 49–61.
- Posch, M.G.; Gramlich, M.; Sunde, M.; Schmitt, K.R.; Lee, S.H.Y.; Richter, S.; Kersten, A.; Perrot, A.; Panek, A.N.; Khatib, I.H.A.; et al. A Gain-of-Function TBX20 Mutation Causes Congenital Atrial Septal Defects, Patent Foramen Ovale and Cardiac Valve Defects. J. Med. Genet. 2010, 47, 230–235.
- Clark, K.L.; Yutzey, K.E.; Benson, D.W. Transcription factors and congenital heart defects. Annu. Rev. Physiol. 2006, 68, 97–121.
- Schuchlenz, H.W.; Saurer, G.; Weihs, W.; Rehak, P. Persisting Eustachian Valve in Adults: Relation to Patent Foramen Ovale and Cerebrovascular Events. J. Am. Soc. Echocardiog. 2004, 17, 231–233.
- Goel, S.S.; Tuzcu, E.M.; Shishehbor, M.H.; de Oliveira, E.I.; Borek, P.P.; Krasuski, R.A.; Rodriguez, L.L.; Kapadia, S.R. Morphology of the Patent Foramen Ovale in Asymptomatic Versus Symptomatic (Stroke or Transient Ischemic Attack) Patients. Am. J. Cardiol. 2009, 103, 124–129.
- Ioannidis, S.G.; Mitsias, P.D. Patent Foramen Ovale in Cryptogenic Ischemic Stroke: Direct Cause, Risk Factor, or Incidental Finding? Front. Neurol. 2020, 11, 567.
- Sharobeam, A.; Churilov, L.; Parsons, M.; Donnan, G.A.; Davis, S.M.; Yan, B. Patterns of Infarction on MRI in Patients with Acute Ischemic Stroke and Cardio-Embolism: A Systematic Review and Meta-Analysis. Front. Neurol. 2020, 11, 606521.
- Wessels, T.; Wessels, C.; Ellsiepen, A.; Reuter, I.; Trittmacher, S.; Stolz, E.; Jauss, M. Contribution of Diffusion-Weighted Imaging in Determination of Stroke Etiology. AJNR Am. J. Neuroradiol. 2006, 27, 35–39.
- Elgendy, A.Y.; Saver, J.L.; Amin, Z.; Boudoulas, K.D.; Carroll, J.D.; Elgendy, I.Y.; Grunwald, I.Q.; Gertz, Z.M.; Hijazi, Z.M.; Horlick, E.M.; et al. Proposal for Updated Nomenclature and Classification of Potential Causative Mechanism in Patent Foramen Ovale–Associated Stroke. JAMA Neurol. 2020, 77, 878–886.
- Schernthaner, C.; Danmayr, F.; Daburger, A.; Eichinger, J.; Hammerer, M.; Strohmer, B. High Incidence of Echocardiographic Abnormalities of the Interatrial Septum in Patients Undergoing Ablation for Atrial Fibrillation. Echocardiography 2013, 30, 402–406.
- Berthet, K.; Lavergne, T.; Cohen, A.; Guize, L.; Bousser, M.-G.; Heuzey, J.-Y.L.; Amarenco, P. Significant Association of Atrial Vulnerability with Atrial Septal Abnormalities in Young Patients with Ischemic Stroke of Unknown Cause. Stroke 2000, 31, 398–403.
- Cotter, P.E.; Martin, P.J.; Pugh, P.J.; Warburton, E.A.; Cheriyan, J.; Belham, M. Increased Incidence of Interatrial Block in Younger Adults with Cryptogenic Stroke and Patent Foramen Ovale. Cereb. Dis. Extra 2011, 1, 36–43.
- Elliott, D.A.; Kirk, E.P.; Yeoh, T.; Chandar, S.; McKenzie, F.; Taylor, P.; Grossfeld, P.; Fatkin, D.; Jones, O.; Hayes, P.; et al. Cardiac Homeobox Gene NKX2-5 Mutations and Congenital Heart Disease Associations with Atrial Septal Defect and Hypoplastic Left Heart Syndrome. J. Am. Coll. Cardiol. 2003, 41, 2072–2076.
- Belvís, R.; Tizzano, E.F.; Martí-Fàbregas, J.; Leta, R.G.; Baena, M.; Carreras, F.; Pons-Lladó, G.; Baiget, M.; Martí-Vilalta, J.L. Mutations in the NKX2-5 Gene in Patients with Stroke and Patent Foramen Ovale. Clin. Neurol. Neurosur. 2009, 111, 574–578.
- Gudbjartsson, D.F.; Arnar, D.O.; Helgadottir, A.; Gretarsdottir, S.; Holm, H.; Sigurdsson, A.; Jonasdottir, A.; Baker, A.; Thorleifsson, G.; Kristjansson, K.; et al. Variants Conferring Risk of Atrial Fibrillation on Chromosome 4q25. Nature 2007, 448, 353–357.
- Franco, D.; Campione, M. The Role of Pitx2 during Cardiac Development Linking Left–Right Signaling and Congenital Heart Diseases. Trends Cardiovasc. Med. 2003, 13, 157–163.
- Tessari, A.; Pietrobon, M.; Notte, A.; Cifelli, G.; Gage, P.J.; Schneider, M.D.; Lembo, G.; Campione, M. Myocardial Pitx2 Differentially Regulates the Left Atrial Identity and Ventricular Asymmetric Remodeling Programs. Circ. Res. 2008, 102, 813–822.
- Faucourt, M.; Houliston, E.; Besnardeau, L.; Kimelman, D.; Lepage, T. The Pitx2 Homeobox Protein Is Required Early for Endoderm Formation and Nodal Signaling. Dev. Biol. 2001, 229, 287–306.
- Mommersteeg, M.T.M.; Hoogaars, W.M.H.; Prall, O.W.J.; Vries, C.d.G.; Wiese, C.; Clout, D.E.W.; Papaioannou, V.E.; Brown, N.A.; Harvey, R.P.; Moorman, A.F.M.; et al. Molecular Pathway for the Localized Formation of the Sinoatrial Node. Circ. Res. 2007, 100, 354–362.
- Kristjansson, R.P.; Benonisdottir, S.; Oddsson, A.; Galesloot, T.E.; Thorleifsson, G.; Aben, K.K.; Davidsson, O.B.; Jonsson, S.; Arnadottir, G.A.; Jensson, B.O.; et al. Sequence Variant at 4q25 near PITX2 Associates with Appendicitis. Sci. Rep. 2017, 7, 3119.
- Hauer, A.J.; Pulit, S.L.; van den Berg, L.H.; de Bakker, P.I.W.; Veldink, J.H.; Ruigrok, Y.M.; Klijn, C.J.M.; Algra, A.; van Dijk, E.J.; Koudstaal, P.J.; et al. A Replication Study of Genetic Risk Loci for Ischemic Stroke in a Dutch Population: A Case-Control Study. Sci. Rep. 2017, 7, 12175.
- Bollmann, A.; Kornej, J.; Adams, V.; Arya, A.; Piorkowski, C.; Hindricks, G.; Husser, D. Patent Foramen Ovale in Atrial Fibrillation: Relation with Chromosome 4q25 Variants and Rhythm Outcome of Catheter Ablation. Eur. Heart J. 2010, 31, 873–1071.
- McCarty, J.L.; Leung, L.Y.; Peterson, R.B.; Sitton, C.W.; Sarraj, A.; Riascos, R.F.; Brinjikji, W. Ischemic Infarction in Young Adults: A Review for Radiologists. Radiographics 2019, 39, 1629–1648.
- Diener, H.-C.; Chutinet, A.; Easton, J.D.; Granger, C.B.; Kleine, E.; Marquardt, L.; Meyerhoff, J.; Zini, A.; Sacco, R.L. Dabigatran or Aspirin After Embolic Stroke of Undetermined Source in Patients with Patent Foramen Ovale: Results from RE-SPECT ESUS. Stroke 2021, 52, 1065–1068.
- Kirk, E.P.; Sunde, M.; Costa, M.W.; Rankin, S.A.; Wolstein, O.; Castro, M.L.; Butler, T.L.; Hyun, C.; Guo, G.; Otway, R.; et al. Mutations in Cardiac T-Box Factor Gene TBX20 Are Associated with Diverse Cardiac Pathologies, Including Defects of Septation and Valvulogenesis and Cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 280–291.
- Schott, J.-J.; Benson, D.W.; Basson, C.T.; Pease, W.; Silberbach, G.M.; Moak, J.P.; Maron, B.J.; Seidman, C.E.; Seidman, J.G. Congenital Heart Disease Caused by Mutations in the Transcription Factor NKX2-5. Science 1998, 281, 108–111.
- Biben, C.; Weber, R.; Kesteven, S.; Stanley, E.; McDonald, L.; Elliott, D.A.; Barnett, L.; Köentgen, F.; Robb, L.; Feneley, M.; et al. Cardiac Septal and Valvular Dysmorphogenesis in Mice Heterozygous for Mutations in the Homeobox Gene Nkx2-5. Circ. Res. 2000, 87, 888–895.
- Stennard, F.A.; Costa, M.W.; Lai, D.; Biben, C.; Furtado, M.B.; Solloway, M.J.; McCulley, D.J.; Leimena, C.; Preis, J.I.; Dunwoodie, S.L.; et al. Murine T-Box Transcription Factor Tbx20 Acts as a Repressor during Heart Development, and Is Essential for Adult Heart Integrity, Function and Adaptation. Development 2005, 132, 2451–2462.
- Garg, V.; Kathiriya, I.S.; Barnes, R.; Schluterman, M.K.; King, I.N.; Butler, C.A.; Rothrock, C.R.; Eapen, R.S.; Hirayama-Yamada, K.; Joo, K.; et al. GATA4 Mutations Cause Human Congenital Heart Defects and Reveal an Interaction with TBX5. Nature 2003, 424, 443–447.
- Angeli, S.; Carrera, P.; Sette, M.D.; Assini, A.; Grandis, M.; Biancolini, D.; Ferrari, M.; Gandolfo, C. Very High Prevalence of Right-to-Left Shunt on Transcranial Doppler in an Italian Family with Cerebral Autosomal Dominant Angiopathy with Subcortical Infarcts and Leukoencephalopathy. Eur. Neurol. 2001, 46, 198–201.
- Mazzucco, S.; Anzola, G.P.; Rizzuto, N. Methodological Issues in Right-to-Left Shunt Detection in CADASIL Patients. Stroke 2009, 40, e509.
- Blue, G.M.; Humphreys, D.; Szot, J.; Major, J.; Chapman, G.; Bosman, A.; Kirk, E.P.; Sholler, G.F.; Harvey, R.P.; Dunwoodie, S.L.; et al. The Promises and Challenges of Exome Sequencing in Familial, Non-Syndromic Congenital Heart Disease. Int. J. Cardiol. 2017, 230, 155–163.
- Lahm, H.; Jia, M.; Dreßen, M.; Wirth, F.F.M.; Puluca, N.; Gilsbach, R.; Keavney, B.; Cleuziou, J.; Beck, N.; Bondareva, O.; et al. Congenital Heart Disease Risk Loci Identified by Genome-Wide Association Study in European Patients. J. Clin. Investig. 2020, 131, e141837.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
791
Revisions:
2 times
(View History)
Update Date:
06 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No