Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pavel Snytnikov | -- | 2674 | 2023-02-03 09:04:30 | | | |
| 2 | Camila Xu | Meta information modification | 2674 | 2023-02-03 09:13:50 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Shilov, V.; Potemkin, D.; Rogozhnikov, V.; Snytnikov, P. Catalytic Materials Development for Fuel Cell Power Generators. Encyclopedia. Available online: https://encyclopedia.pub/entry/40797 (accessed on 24 July 2026).
Shilov V, Potemkin D, Rogozhnikov V, Snytnikov P. Catalytic Materials Development for Fuel Cell Power Generators. Encyclopedia. Available at: https://encyclopedia.pub/entry/40797. Accessed July 24, 2026.
Shilov, Vladislav, Dmitriy Potemkin, Vladimir Rogozhnikov, Pavel Snytnikov. "Catalytic Materials Development for Fuel Cell Power Generators" Encyclopedia, https://encyclopedia.pub/entry/40797 (accessed July 24, 2026).
Shilov, V., Potemkin, D., Rogozhnikov, V., & Snytnikov, P. (2023, February 03). Catalytic Materials Development for Fuel Cell Power Generators. In Encyclopedia. https://encyclopedia.pub/entry/40797
Shilov, Vladislav, et al. "Catalytic Materials Development for Fuel Cell Power Generators." Encyclopedia. Web. 03 February, 2023.
Copy Citation
Many research teams around the world persistently undertake attempts to create active and stable catalysts for the pre-reforming and steam reforming of diesel and kerosene fuels. The most active and stable catalysts for diesel fuel conversion are Rh- and other precious metal systems supported on oxide carriers containing mobile lattice oxygen, mainly zirconium and cerium oxides.
fuel cell
reforming
fuel processing
liquid fuel
1. Introduction
It was expected in most of the research work carried out over the past 20–25 years that by the time of mass use of fuel cells, the infrastructure for their supply with fuel (hydrogen) would already be created, including efficient logistics, a fully developed hydrogen refueling network and sufficient hydrogen long-time storing capacities. However, these forecasts appeared overly optimistic. Even regarding widely used natural gas, the existing and actively developing infrastructure for its transportation and consumption turns to be insufficient to cover completely the current level of mass demand.
That is why all the world’s research centers involved in R&D of fuel-cell-based power units show interest in using fuel of common types, such as natural gas, liquefied petroleum gas, gasoline, aviation kerosene, diesel fuel, methanol, ethanol, etc. Besides, compared to other currently available hydrogen storage technologies, hydrocarbon fuels demonstrate the highest hydrogen content per unit volume [1].
These fuels can hardly be oxidized directly in the anode space of a fuel cell (FC), since they are inert at low temperatures, and can initiate electrode coking and FC failure at high temperatures. Therefore, the fuel is first converted into a hydrogen-rich gas, which is then oxidized in a fuel cell. Depending on the FC type, hydrogen for FC fueling can contain some amounts of CO and CO2. Figure 1 presents a typical scheme of catalytic processes for the production and cleanup of hydrogen-rich gas mixtures from various fuels for fuel cell feeding. Note that sulfur-containing compounds are poisonous for FC of all types; therefore, any fuel must be desulfurized either at the stage of production or prior to using it for hydrogen production. The processes for liquid and gaseous fuels desulfurization are well-developed and widely used in industry, but remain beyond consideration in this research.
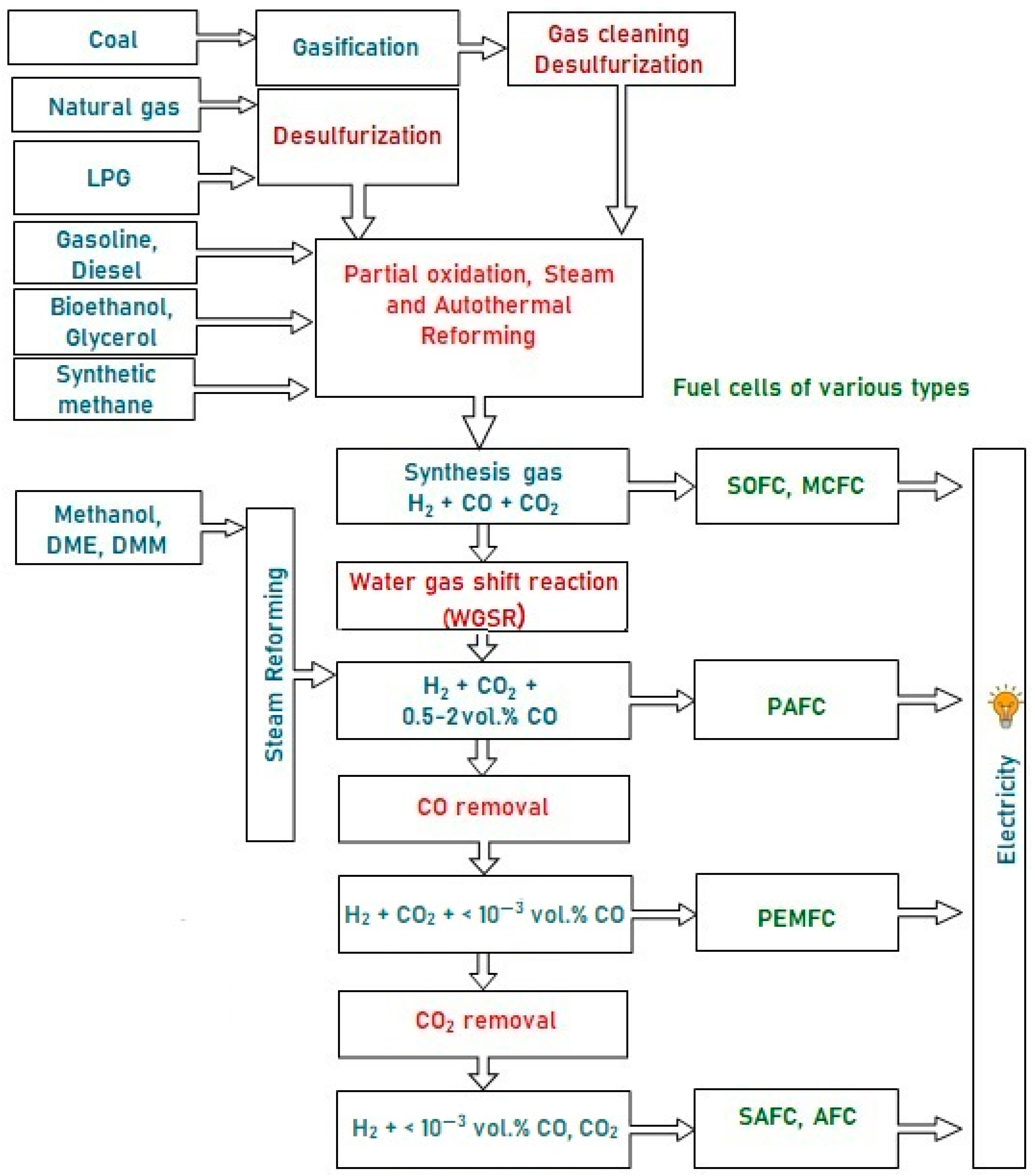
Figure 1. A typical scheme of hydrogen production from various fuels for feeding fuel cells of different types.
2. Active Component of Catalysts for the Conversion of Diesel Fuel into Synthesis Gas
At present, the main amount of commercial diesel fuel (DF) produced at refineries by the process of diesel fraction hydrotreating meets the Euro 5 standard in terms of fractional composition and the content of polycyclic aromatics and sulfur. Table 1 presents a typical average composition of diesel fuel.
Table 1. A typical average composition of commercial diesel fuel.
| Component | Content (vol.%) |
|---|---|
| n-Paraffins | 20 |
| iso-paraffins | 15–20 |
| Cycloparaffins | 35 |
| Alkylbenzenes | 20–23 |
| Diaromatic hydrocarbons | 5 |
| Polycyclic aromatics | <2 |
| Sulfur compounds | 0.0005–0.0008 (5–8 ppm) |
As noted above, a key factor for providing efficient catalytic conversion of diesel fuel into synthesis gas is to ensure the stable operation and coking resistance of the catalyst. The most active and stable catalysts for diesel fuel conversion are Rh- and other precious metal systems supported on oxide carriers containing mobile lattice oxygen, mainly zirconium and cerium oxides. The mobile lattice oxygen participates in the oxidation of incipient carbon deposits and thus significantly improves the catalyst stability. The most active carrier in this regard is cerium oxide, but at temperatures above 600 °C it is not strongly sintered and therefore cannot be used in its pure form. So, mixed oxides of composition CexZr1-xO2-δ were chosen as the support, as they possess both high mobility of lattice oxygen and thermal stability.
The main efforts were aimed at developing a procedure for depositing nanoparticles of platinum group metals (Ru, Rh, Pd and Pt) onto oxide supports, which would provide a high particle dispersion and adhesion to the support and be quite simple and adaptable for coating the structured substrates.
As a result of the research, a method of sorption–hydrolytic precipitation was proposed [2]. The method is based on the slow kinetics of ligand exchange in alkaline solutions of chloride complexes of platinum metals. This approach allowed the selection of appropriate concentrations of metal chlorides and precipitant (Na2CO3) and a temperature to obtain a metastable solution, in which homogeneous precipitation of platinum metal hydroxides is impeded for kinetic reasons. After immersing the carrier into the solution, the precipitation of metal hydroxide particles in its pores proceeds by the heterogeneous nucleation mechanism.
According to CO chemisorption data, the average size of the Rh, Ru and Pt particles deposited by the sorption–hydrolytic procedure on a commercial support of composition Ce0.75Zr0.25O2 (hereinafter CZ) was 1.1, 1.2 and 1.8 nm, respectively [2]. Transmission electron microscopy (TEM) data showed that Rh particles exist on the support surface predominantly in the form of 1–2 nm clusters. (Figure 2a). After HD ATR experiments, the support crystallites increased in size to 20–30 nm, while the aggregate size remained the same (Figure 2b). Additionally, carbon species in the form of 1–2 graphite layers appeared occasionally on the surface (Figure 2c). The size of Rh particles increased slightly to 2–4 nm. After oxidative treatment of the catalyst, the carbon deposits disappeared, while the size of support crystallites and Rh particles remained unchanged.

Figure 2. TEM images of 1 wt.% Rh/CZ: (a) as-prepared and (b,c) used in HD ATR [2].
Note that the DF ATR catalysts must be highly active and stable under DF SR conditions, since oxygen is rapidly consumed under ATR conditions and, in fact, most of the catalyst layer operates under SR conditions. The resulting Rh/CZ, Ru/CZ and Pt/CZ catalysts were studied in SR of n-hexadecane (HD), which served as a model DF compound. The fuel conversion was evaluated gravimetrically by collecting unreacted fuel into a condensate vessel and calculated using the following equation:
where X (%) is fuel conversion, V0—fuel flow rate (g/h), t—sampling time (h) and m—sample weight (g).
Figure 3 shows the time dependences of HD conversion, and product distribution for the Pt/CZ, Ru/CZ and Rh/CZ catalysts at HD SR. It is seen that Pt/CZ showed the worst catalytic properties: at 550 °C, it failed to achieve complete HD conversion and demonstrated its decrease from 59 to 27% within 3 h. The Ru/CZ catalyst rapidly lost activity after 5 h on stream and respective HD conversion decreased to 46%. The Rh/CZ catalyst demonstrated stable operation at 550 °C for 8 h, provided 100% HD conversion and the product concentrations (vol.%) of 54 H2, 18 CO2, 5 CO and ~6% CH4, which were close to the thermodynamically equilibrium ones. Then the catalyst was regenerated with hydrogen and retested at 650 °C. The HD conversion was 100%. The reaction product distribution was similar to the thermodynamically equilibrium one calculated for a temperature of 650 °C.
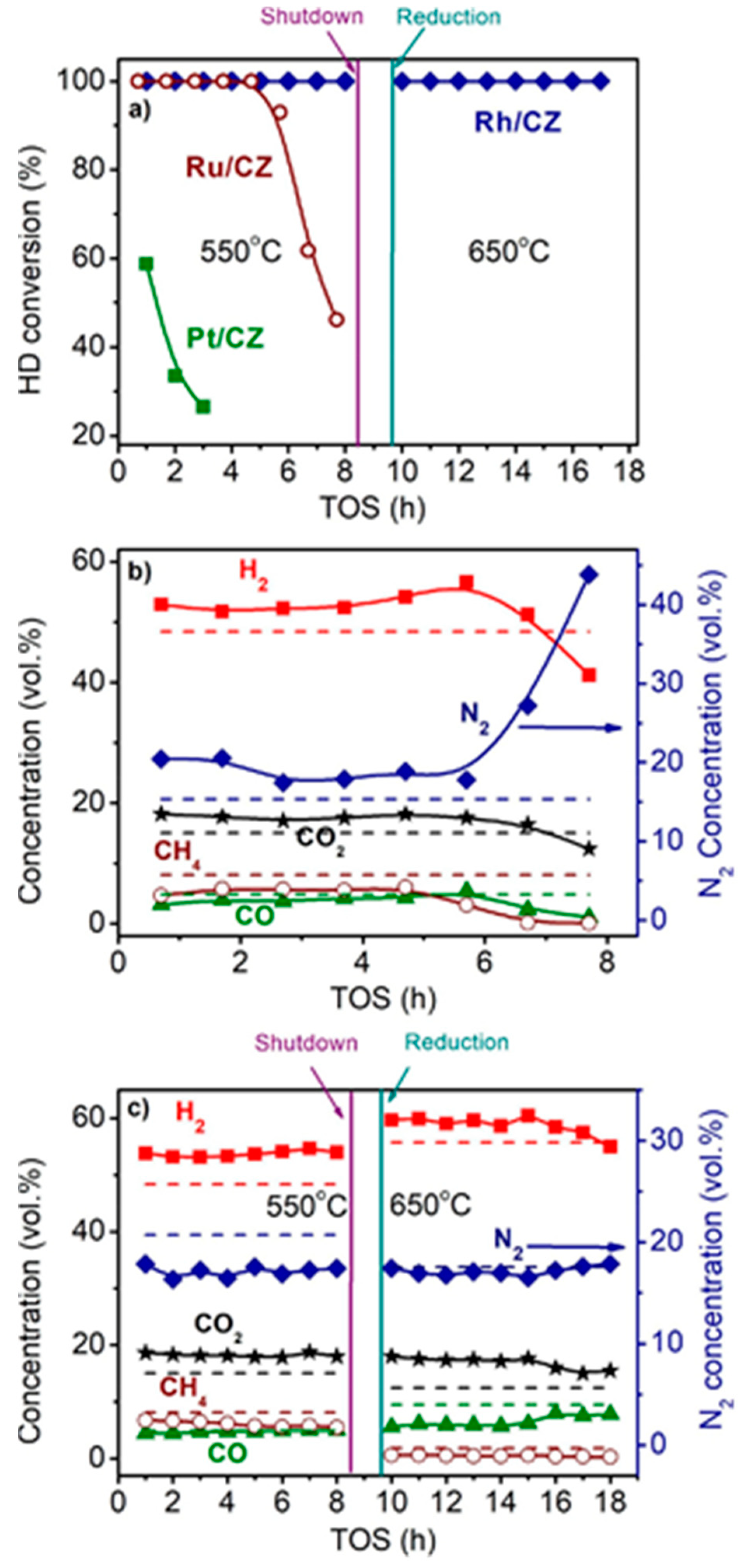
Figure 3. The HD conversion (a) and product distribution on dry basis (b,c) over 1.9 wt.% Pt/CZ (a), 1 wt.% Ru/CZ (a,b) and 1 wt.% Rh/CZ (a,c) in the HD SR as a function of time on stream at H2O/C = 3.0, T = 550–650 °C and GHSV = 23,000 h−1. Points—experiment, dashed line—equilibrium [2].
Thus, the activity and stability of the prepared noble-metal-based catalysts decreased in the following order: Rh/CZ > Ru/CZ >> Pt/CZ [2]—in good agreement with the results of other studies discussed above.
Note that catalysts supported on mixed cerium–zirconium oxides are usually inappropriate for practical use in granular form owing to insufficient mechanical strength and poor formability. Therefore, the feasibility of using a mixed Al2O3-Ce0.75Zr0.25O2 oxide as a support (commercial product 50 wt.% Al2O3 and 50 wt.% Ce0.75Zr0.25O2, hereinafter CZA) and alumina as a binding agent was investigated. The alumina additive is also intended to improve the catalyst thermal stability.
Besides Rh/CZ and Rh/CZA, Figure 4 presents the test results for Rh/CZA doped with MgO (Rh/CZA-Mg) and Rh-based catalyst supported on CZ containing 20 wt.% pseudoboehmite (Rh/CZB) as a structural promoter. Clearly, the Rh/CZ and Rh/CZB catalysts had the highest activity and coking resistance. The higher the aluminum content in the catalysts, the more rapidly they lost activity owing to acceleration of the side process of carbon deposition. Most likely, the catalyst deactivation is associated with the presence of acid sites on the alumina surface, while support doping with basic oxide MgO facilitated a significant increase in catalyst stability.
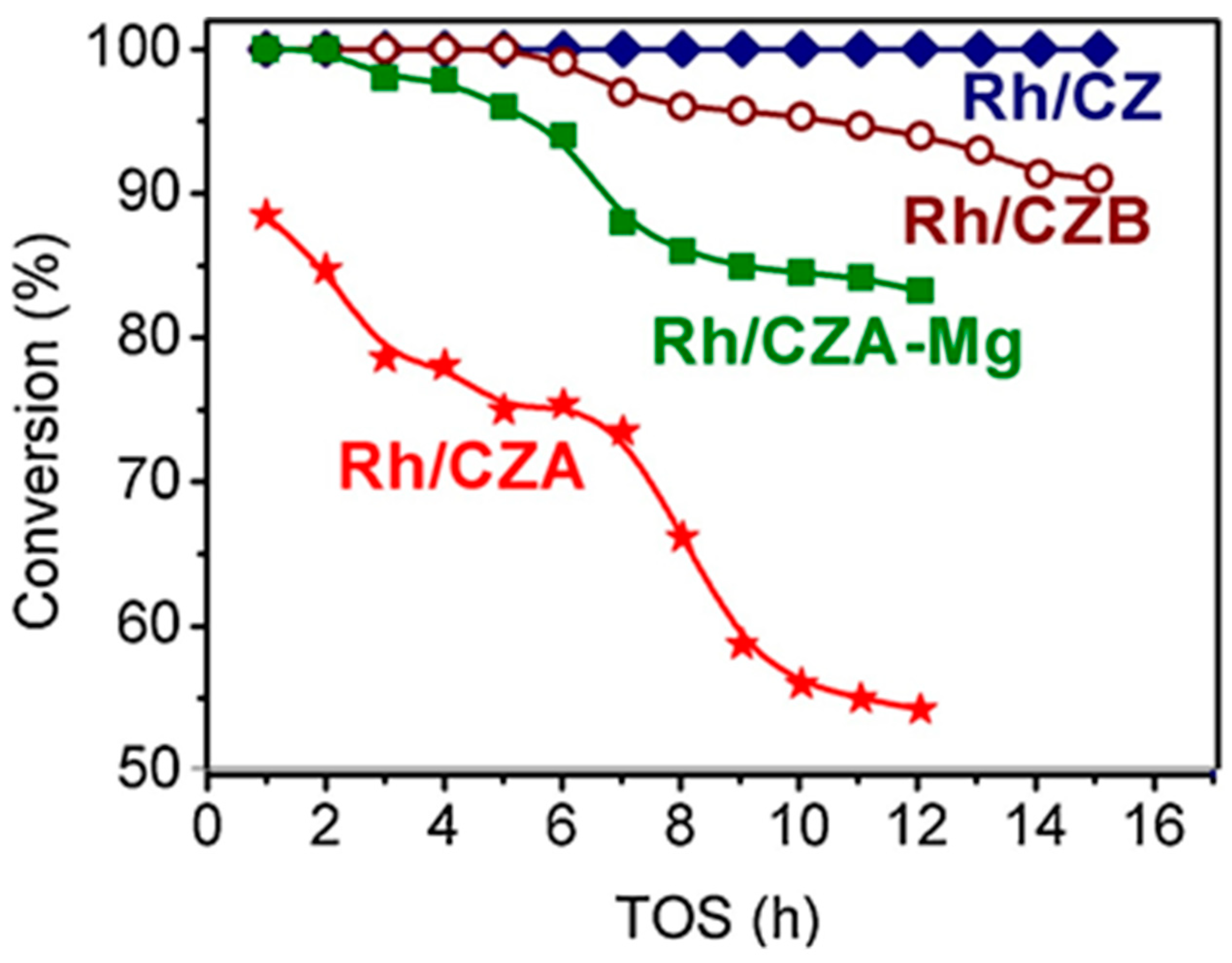
Figure 4. The time dependence of HD conversion under the following SR conditions: GHSV = 23,000 h−1, H2O/C = 3, T = 550 °C [3].
Rh/CZA was the most susceptible to coking: it accumulated 3.7 wt.% of carbon in 12 h of HD SR. Since the size of Rh particles in Rh/CZA after annealing remained unchanged (Figure 5f), rapid deactivation of the catalyst is explained by coke formation. As known, γ-Al2O3 contains acid sites, which are responsible for coke formation [4][5]. As proved by the results of Rh/CZA-Mg catalytic activity tests (Figure 4) and TPO data [3], the blocking of acid sites by Mg cations causes a threefold decrease in the amount of carbon formed. Compared to Rh/CZA-Mg and Rh/CZA, the use of 20 wt.% pseudoboehmite as a binder in Rh/CZB improved the catalyst performance in HD SR (Figure 4), but the carbon productivity exceeded that of Rh/CZA-Mg. The same amount of carbon was observed in Rh/CZB after HD ATR. Among the studied catalysts, Rh/CZ accumulated the lowest amount of carbon: 1.2 wt.% for 15 h of HD SR and 1.5 wt.% for 12 h of HD ATR.
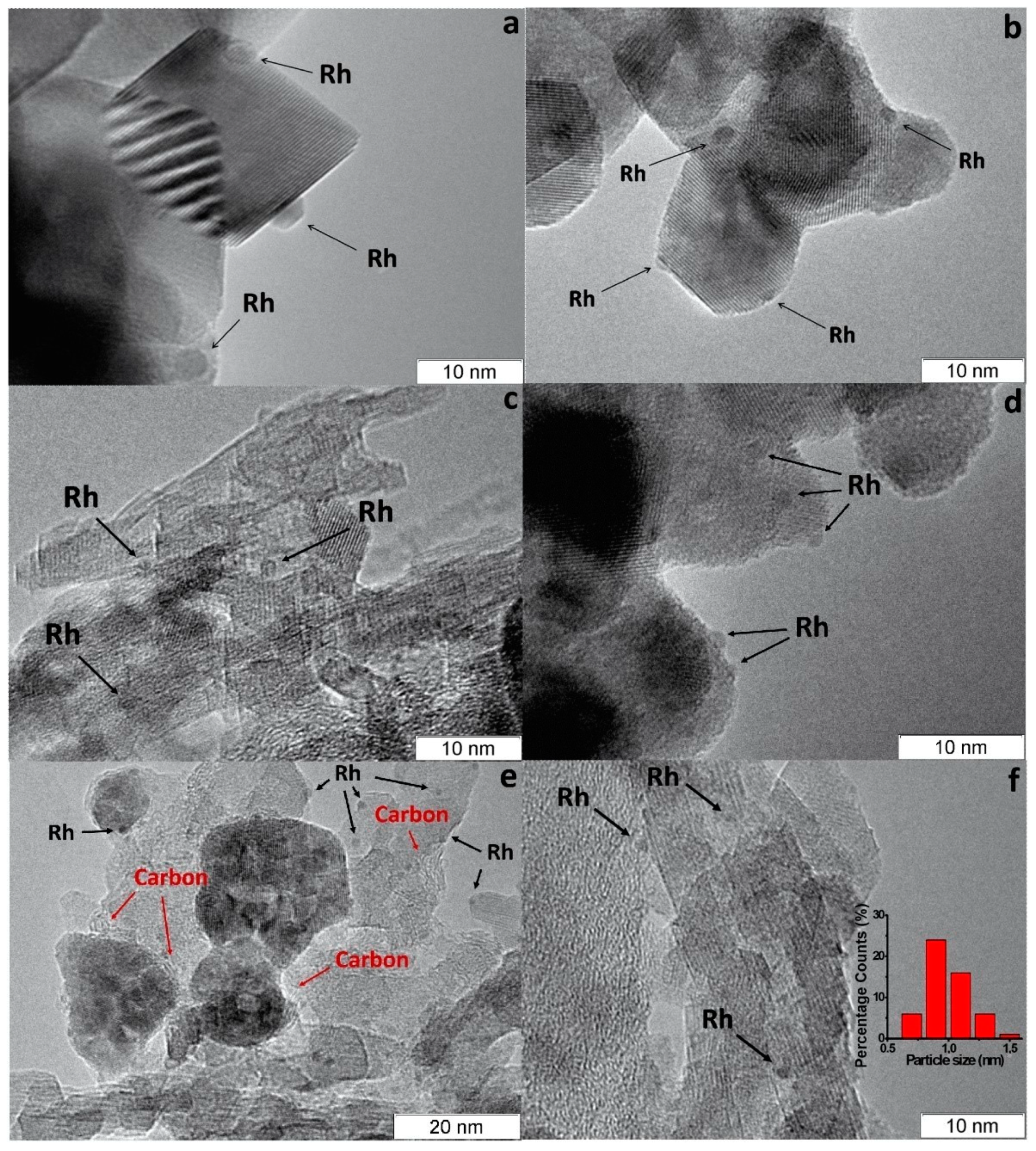
Figure 5. TEM images of as-prepared Rh/CZ (a), Rh/CZB (c) and used in HD SR Rh/CZ (b), Rh/CZB (d), Rh/CZA (e) and annealed Rh/CZA (f) catalysts [3].
Thus, to ensure the stable operation in DF SR and ATR of Rh-based catalysts supported on alumina-containing carriers, it is necessary to “neutralize” completely the acidity of the alumina surface [3].
3. Structured Composite Catalysts Supported on FeCrAl Alloy Wire Mesh
The DF ATR process is characterized by a combination of exo- and endothermic reactions. As discussed below in Section 8, modern understanding of the process mechanism assumes that fast complete oxidation reactions occur in the frontal part of the reactor with the release of heat, which is then consumed along the catalyst bed during the endothermic processes of steam and carbon-dioxide reforming of hydrocarbons. Therefore, when carrying out the ATR process, to prevent overheating in the frontal section and overcooling in the tail section, the structure of the catalyst bed must ensure efficient heat transfer between these zones. Conventional granular ceramic catalysts have low thermal conductivity and are hardly appropriate for this purpose. Besides, to reduce the pressure drop in the reactor, it is necessary to use large-size catalyst grains (1 cm and larger), which inevitably leads to a low utilization factor of the catalyst grain under conditions of fast ATR reactions.
For ensuring efficient implementation of the ATR process, it was proposed to use composite catalytic systems of the “metal nanoparticles/active oxide nanoparticles/structural oxide component/structured metal substrate” type. A structured metal substrate made of heat-resistant FeCrAl alloy facilitates fast heat removal/supply for exo-/endothermic reactions, has sufficient hydrodynamic characteristics, allows manufacturing products of various geometric shape and easy process scaling. The structural oxide component (alumina) provides thermal stability and high specific surface area and increases mechanical strength for the supported catalytic coating. The active oxide component (cerium oxide and mixed cerium–zirconium oxides with a fluorite structure) participates in the activation of water and oxygen molecules, improves the coke resistance because of high oxygen mobility and keeps the active component in a fine dispersed state owing to strong metal–carrier interaction. Metal nanoparticles of 1–2 nm size are involved in the activation of hydrocarbon molecules.
Based on this concept, the structured catalysts of composition 0.24 wt.% Rh/Ce0.75Zr0.25O2/Al2O3/FeCrAl (Rh/CZB/FCA) were prepared and tested. The structured metal substrate was made of FeCrAl alloy wire mesh (wire thickness of 0.25 mm, cell size of 0.5 × 0.5 mm). Structural oxide component, a layer of ƞ-Al2O3 in the amount of 6 wt.%, was supported on the metal substrate to ensure reliable adhesion of CZ active oxide nanoparticles (Figure 6). By calcining in air, α-Al2O3 layer was pre-formed on FeCrAl wire mesh; then a modified Bayer method (using aluminum hydroxide) was used to deposit an η-Al2O3 coating with a flexible (“breathing”) needle-shaped morphology [6]. According to SEM data (Figure 6), aluminum oxide consisted of tubular or acicular crystals (5–10 µm thick, up to 50 µm long).
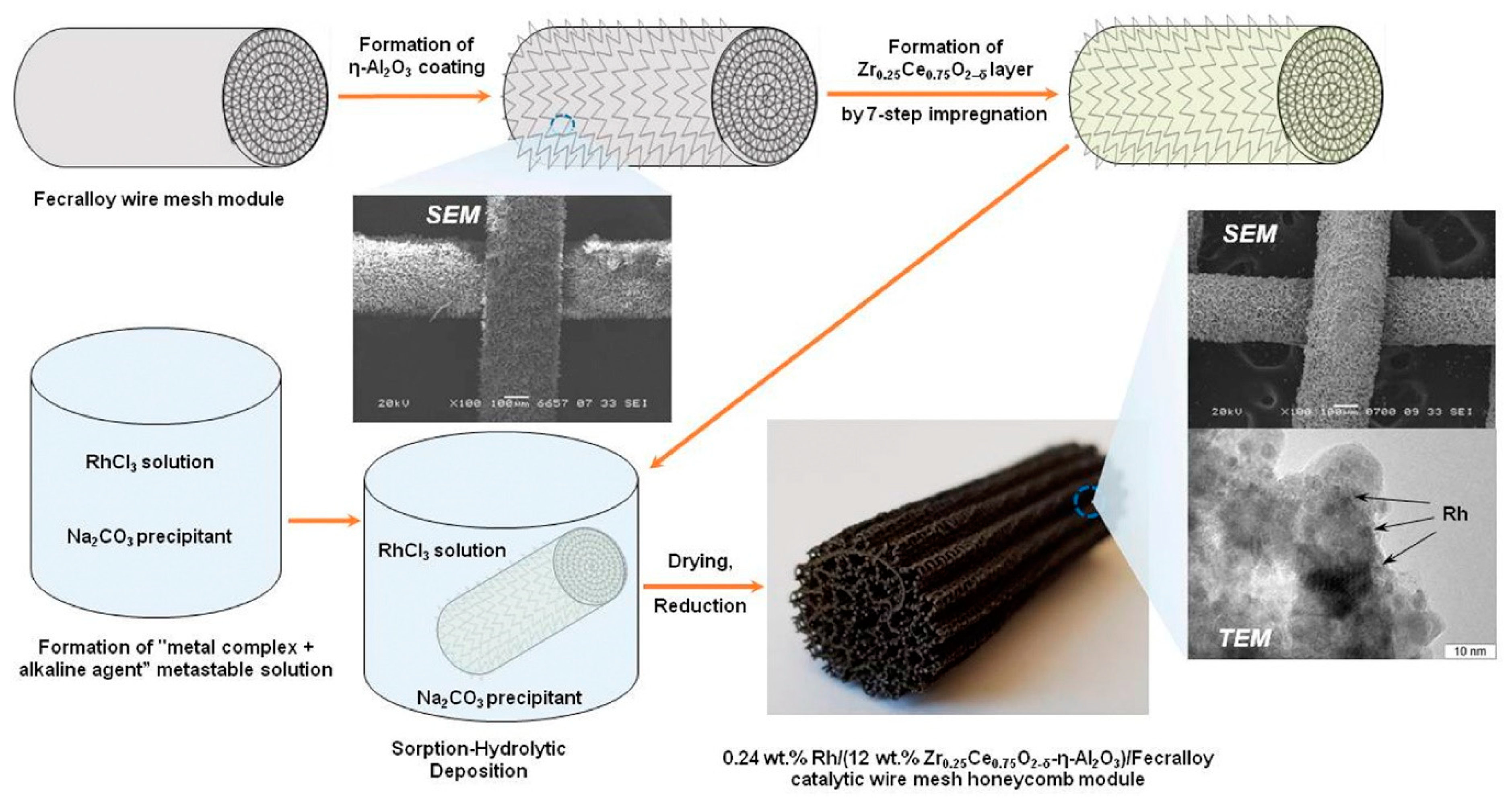
Figure 6. Schematic preparation procedure of Rh/CZB/FCA catalytic block, its general view, SEM and TEM images [7].
The obtained ƞ-Al2O3/FeCrAl sample was repeatedly impregnated with a solution of Ce(NO3)3·6H2O and ZrO(NO3)2·2H2O (Ce/Zr = 3) and calcined at 800 °C [7]. Thus, the 12 wt.% Ce0.75Zr0.25O2/Al2O3/FeCrAl (CZB/FCA) composite support was obtained. Rh nanoparticles in the amount of 0.24 wt.% were deposited on the CZB/FCA by sorption–hydrolytic precipitation method. The RhCl3 solution was mixed with Na2CO3 in the ratio Na/Cl = 1. At room temperature, the resulting solution is metastable with regard to the homogeneous precipitation of rhodium hydroxide. The solution was brought into contact with CZB/FCA at T = 75 °C to initiate the hydrolysis that facilitated uniform deposition of rhodium particles throughout the structured support surface. At the final stage, the structured Rh/CZB/FCA catalyst was dried in air and reduced in hydrogen flow at 250 °C for 30 min.
The TEM images of as-prepared Rh/CZB/FCA catalyst (Figure 7) [3] show that the support consisted of ~1 µm-sized Al2O3 particles containing 5–20 nm Ce1−xZrxO2−δ crystallites on their surface and aggregated into large porous species. Rh particles on the support surface were predominantly in the form of 1–2 nm clusters, though 3–4 nm particles were observed as well (Figure 7b).

Figure 7. TEM images of as-prepared (a,b) Rh/Ce0.75Zr0.25O2-Al2O3/FeCrAl and Rh particles size distribution (b) [3].
The SEM micrographs of Al2O3/FeCrAl (Figure 8a) and Rh/CZB/FCA (Figure 8b,c) clearly show that the ƞ-Al2O3 layer consisting of tubular or acicular crystals (5–10 µm thick, up to 50 µm long) evenly covers the surface of the FeCrAl wire mesh. The thickness of the ƞ-Al2O3 layer was 30–50 μm. After depositing Rh/Ce1−xZrxO2−δ onto the ƞ-Al2O3 layer, the surface microstructure changed (Figure 8b): the crystal surface became rougher, though the thickness of the final layer remained the same (30–50 µm).

Figure 8. SEM images of ƞ-Al2O3/FeCrAl (a) and as-prepared Rh/Ce0.75Zr0.25O2-Al2O3/FeCrAl (b,c) catalyst [3].
According to EDX data for several 100 × 100 µm areas, the surface of the Rh/CZB/FCA catalyst contained Rh, Ce, Zr, Al and O. The element concentrations in all regions were the same, which proves a uniform distribution of Rh and Ce1−xZrxO2−δ over the surface of alumina crystals and agrees well with the TEM data.
Thus, it was shown [3] that the structured catalyst Rh/Ce0.75Zr0.25O2-Al2O3/FeCrAl is a composite in which aluminum oxide, chemically bonded to the metal substrate, provides the mechanical strength of the catalytic layer and keeps Rh and Ce1−xZrxO2−δ particles in a highly dispersed state.
In comparative studies of the catalytic properties of granular Rh/CZB and composite Rh/CZB/FCA in HD ATR, the Rh/CZB catalyst showed stable operation for 6 h on stream and product distribution close to the thermodynamically equilibrium one. Furthermore, the HD conversion and concentrations of the main products decreased, while the outlet concentrations of C2–C5 components increased (Figure 9a). Rh/CZB/FCA under HD ATR conditions demonstrated a 100% HD conversion for 12 h on stream (Figure 9b) even at a higher space velocity compared to that in the experiment with the granular catalyst. The outlet product concentrations were close to the thermodynamically equilibrium values. Thus, the structured Rh/CZB/FCA catalyst showed high activity in HD ATR and provided hydrogen productivity of 2.5 kgH2kgcat−1h−1; it obviously possesses a high potential for the ATR of commercial diesel fuel.
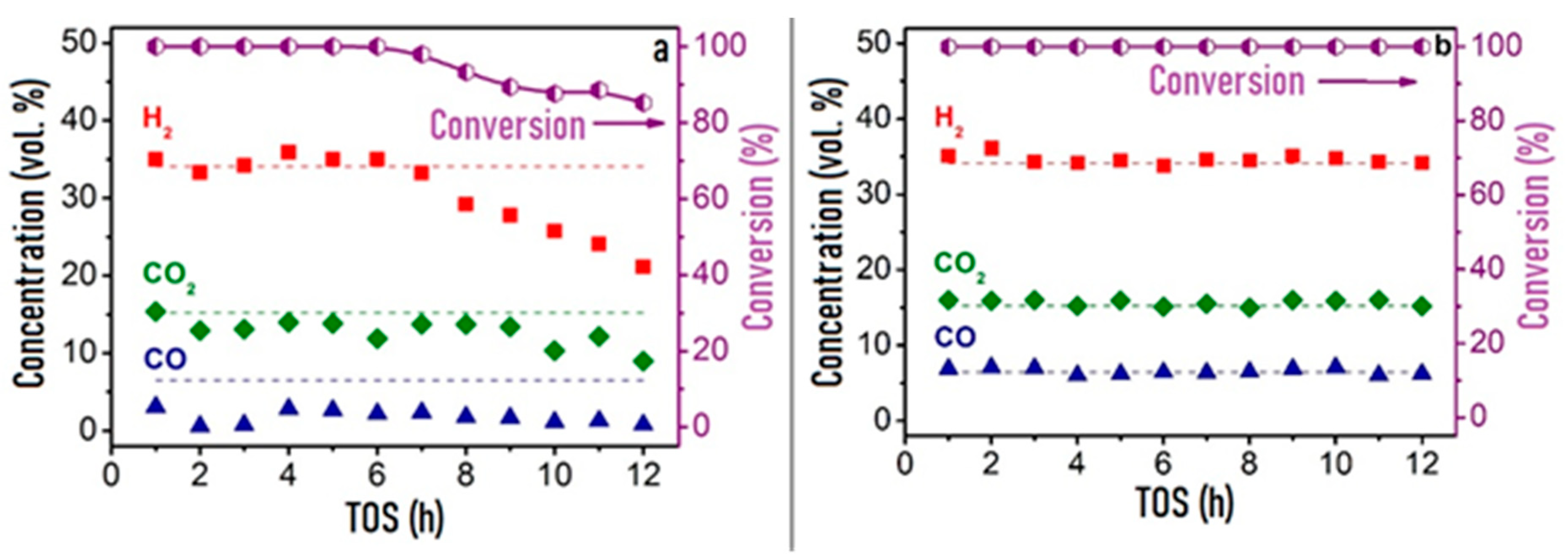
Figure 9. The time-on-steam dependence of HD conversion under ATR conditions: WHSV 30,000 cm3g−1h−1 (a) and 90,000 cm3g−1h−1 (b), T = 650 °C, H2O/C = 2.5, O2/C= 0.5 over Rh/CZB (a) and Rh/CZB/FCA (b) catalysts.Adapted from [3].
References
- Speight, J.G. Fuels for Fuel Cells. In Fuel Cells: Technologies for Fuel Processing; Elsevier: Amsterdam, The Netherlands, 2011; pp. 29–48.
- Shoynkhorova, T.B.; Simonov, P.A.; Potemkin, D.I.; Snytnikov, P.V.; Belyaev, V.D.; Ishchenko, A.V.; Svintsitskiy, D.A.; Sobyanin, V.A. Highly Dispersed Rh-, Pt-, Ru/Ce0.75Zr0.25O2–δ Catalysts Prepared by Sorption-Hydrolytic Deposition for Diesel Fuel Reforming to Syngas. Appl. Catal. B 2018, 237, 237–244.
- Shoynkhorova, T.B.; Snytnikov, P.V.; Simonov, P.A.; Potemkin, D.I.; Rogozhnikov, V.N.; Gerasimov, E.Y.; Salanov, A.N.; Belyaev, V.D.; Sobyanin, V.A. From Alumina Modified Rh/Ce0.75Zr0.25O2-δ Catalyst towards Composite Rh/Ce0.75Zr0.25O2-δ-ƞ-Al2O3/FeCrAl Catalytic System for Diesel Conversion to Syngas. Appl Catal B 2019, 245, 40–48.
- Samsun, R.C.; Pasel, J.; Janßen, H.; Lehnert, W.; Peters, R.; Stolten, D. Design and Test of a 5 KW High-Temperature Polymer Electrolyte Fuel Cell System Operated with Diesel and Kerosene. Appl. Energy 2014, 114, 238–249.
- Pasel, J.; Samsun, R.C.; Tschauder, A.; Peters, R.; Stolten, D. A Novel Reactor Type for Autothermal Reforming of Diesel Fuel and Kerosene. Appl. Energy 2015, 150, 176–184.
- Porsin, A.V.; Rogoznikov, V.N.; Kulikov, A.V.; Salanov, A.N.; Serkova, A.N. Crystallization of Aluminum Hydroxide in a Sodium Aluminate Solution on a Heterogeneous Surface. Cryst. Growth Des. 2017, 17, 4730–4738.
- Shoynkhorova, T.B.; Rogozhnikov, V.N.; Ruban, N.V.; Shilov, V.A.; Potemkin, D.I.; Simonov, P.A.; Belyaev, V.D.; Snytnikov, P.V.; Sobyanin, V.A. Composite Rh/Ce0.75Zr0.25O2-δ-ƞ-Al2O3/Fecralloy Wire Mesh Honeycomb Module for Natural Gas, LPG and Diesel Catalytic Conversion to Syngas. Int. J. Hydrogen Energy 2019, 44, 9941–9948.
More
Information
Subjects:
Chemistry, Applied
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
612
Revisions:
2 times
(View History)
Update Date:
03 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No