Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hiroaki Kume | -- | 2112 | 2023-01-31 08:22:30 | | | |
| 2 | Conner Chen | + 4 word(s) | 2116 | 2023-01-31 09:27:35 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kume, H.; Yamada, R.; Sato, Y.; Togawa, R. Oxidative Stress in Chronic Obstructive Pulmonary Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/40645 (accessed on 06 August 2026).
Kume H, Yamada R, Sato Y, Togawa R. Oxidative Stress in Chronic Obstructive Pulmonary Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/40645. Accessed August 06, 2026.
Kume, Hiroaki, Ryuki Yamada, Yuki Sato, Ryuichi Togawa. "Oxidative Stress in Chronic Obstructive Pulmonary Disease" Encyclopedia, https://encyclopedia.pub/entry/40645 (accessed August 06, 2026).
Kume, H., Yamada, R., Sato, Y., & Togawa, R. (2023, January 31). Oxidative Stress in Chronic Obstructive Pulmonary Disease. In Encyclopedia. https://encyclopedia.pub/entry/40645
Kume, Hiroaki, et al. "Oxidative Stress in Chronic Obstructive Pulmonary Disease." Encyclopedia. Web. 31 January, 2023.
Copy Citation
Since chronic obstructive pulmonary disease (COPD) is a heterogeneous disease, a specific anti-inflammatory therapy for this disease has not been established yet. Oxidative stress is recognized as a major predisposing factor to COPD related inflammatory responses, resulting in pathological features of small airway fibrosis and emphysema.
oxidants
antioxidants
Ca2+ dynamics
1. Introduction
Although chronic obstructive pulmonary disease (COPD) is simply diagnosed based on persistent air flow limitation that will not return to the normal range, using spirometric measurements, this disease is heterogenous and complex in symptoms, disease progression, lung function and response to therapies [1]. The pathogenesis of this disease results from chronic lung inflammation due to cigarette smoke and other environmental exposures (biomass fuel etc.); and this chronic inflammation is associated with activation not only of neutrophils and macrophages but also of eosinophils. While these responses to lung inflammation are normal in many healthy subjects, in contrast, the response is potentiated in patients who develop COPD. This chronic lung inflammation affects distal airways, leading to emphysema and small airway fibrosis (pathological characteristics of this disease) [2][3]; and these pathological alterations in COPD are progressive in most cases [1]. The mechanisms of this modified inflammation are not understood well. Moreover, a wide variety of inflammatory mediators are related to this chronic lung inflammation. For this reason, specific treatment for inflammation is not well established in this disease. Oxidative stress is defined as a state in which oxidation exceeds the capacity of antioxidant systems in the body secondary to a loss of the balance between them. Disturbances in the normal redox state of cells can cause toxic effects through the production of peroxides and free radicals that damage all components of the cell, including proteins, lipids, and DNA. Oxidative stress from oxidative metabolism causes base damage, as well as strand breaks in DNA. Base damage is mostly indirect and caused by reactive oxygen species (ROS) generation, e.g., O2•− (superoxide anion radical), •OH (hydroxyl radical), H2O2 (hydrogen peroxide) and O3 (ozone) (Figure 1) [4]. Inflammatory cells as described above are recruited into the lungs in COPD [5]; and structural cells in the respiratory system (airway epithelial cells, fibroblasts, and endothelial cells) also contribute to the lung inflammation. These cells generate multiple mediators, including cytokines that perpetuate and amplify the inflammation in the lungs. These cells are also important sources of ROS, leading to oxidative stress in the lungs (Figure 1). Oxidative stress in the lungs due to exogenous oxidants (cigarette smoke, biomass fuel, air pollution) and endogenous oxidants (ROS generated by inflammatory cells, epithelium) are associated with clinical and pathophysiological characteristics of COPD (Figure 1) [6]. Mitochondrial respiration is an important source of ROS, and cigarette smoke produces excessive ROS via mitochondrial dysfunction (Figure 1) [7]. It is now generally considered that COPD results from an acceleration of lung ageing with the accumulation of senescent cells [8][9][10]. Senescent cells secrete high levels of inflammatory cytokines, immune modulators, growth factors, and proteases, referred to as senescence-associated secretory phenotype (SASP) [10]. This phenotype change is perhaps an essential mechanism in the chronic lung inflammation of COPD [11]. Since senescent cells also release ROS more than intact cells, this chronic lung inflammation potentiates oxidative stress in COPD (Figure 2). Therefore, oxidative stress is probably a major driving mechanism of many of the pathophysiological changes in COPD [12].
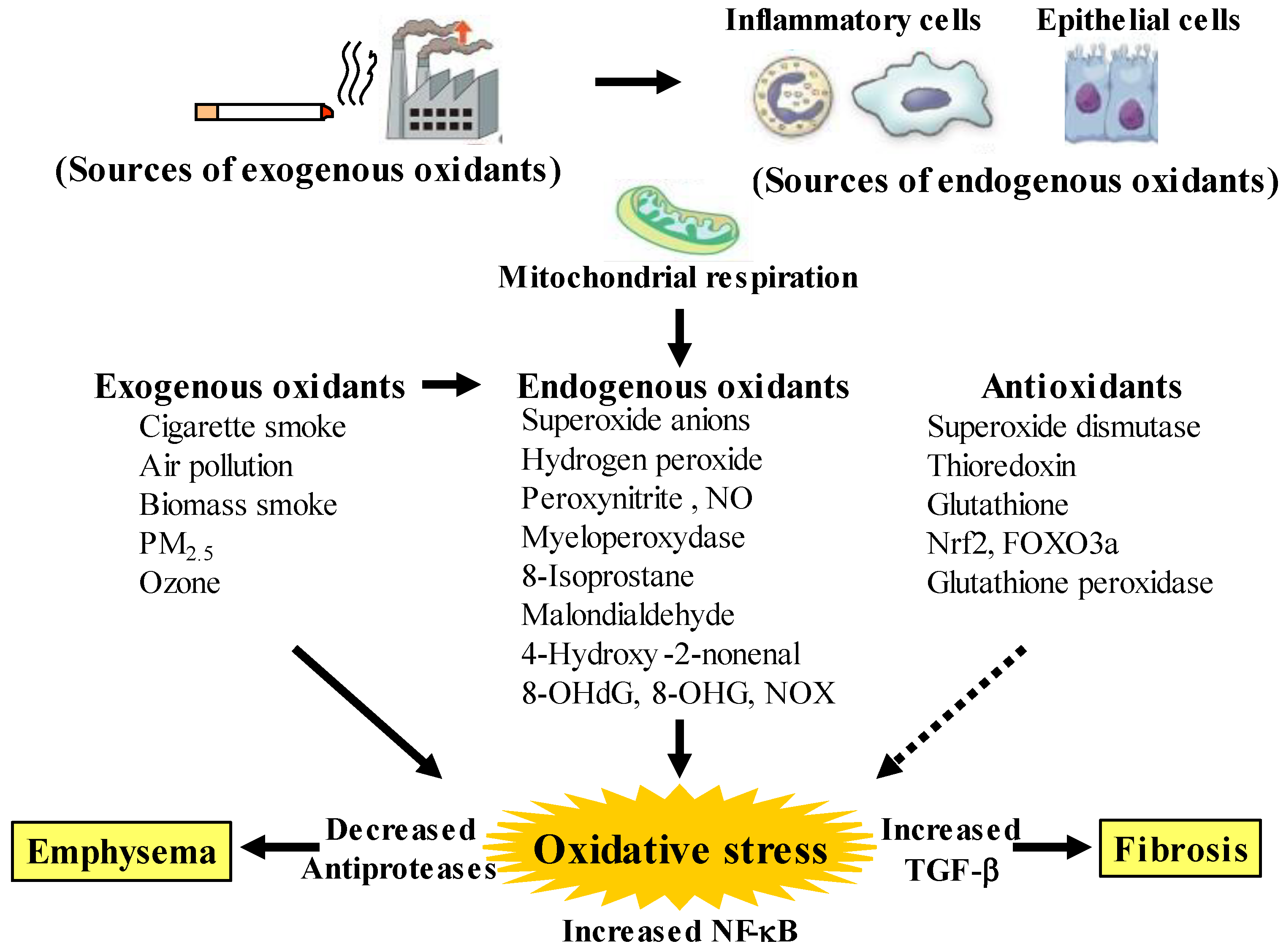
Figure 1. Oxidants and antioxidants involved in COPD, and relationships between oxidative stress and the pathology related to this disease. Oxidative stress in the lungs results from increased exogenous and endogenous oxidants, and from reduced antioxidants. Endogenous oxidants are generated by mitochondrial respiration. Elevated production of endogenous oxidants continues after stopping smoking. Increased oxidative stress is caused by a lack of balance between oxidants and antioxidants. Exogenous oxidants are derived from cigarette smoke, air pollution and biomass smoke, etc.; endogenous oxidants are derived from inflammatory cells (macrophages, neutrophils) and airway epithelial cells. Oxidative stress results in emphysema in alveolar areas with decreased antiproteases, and in fibrosis in the small airways with increased transforming growth factor (TGF)-β. PM: small particulate matter, 8-OHdG: 8-hydroxy-2′-deoxyguanosine, 8-OHG: 8-oxo-7,8-dihydroguanosine, NOX: membrane-bound NADPH oxidases, Nrf2: nuclear erythroid-2 related factor 2, FOXO3a: forkhead box O3a. Arrows: activation, dotted arrows: inactivation.
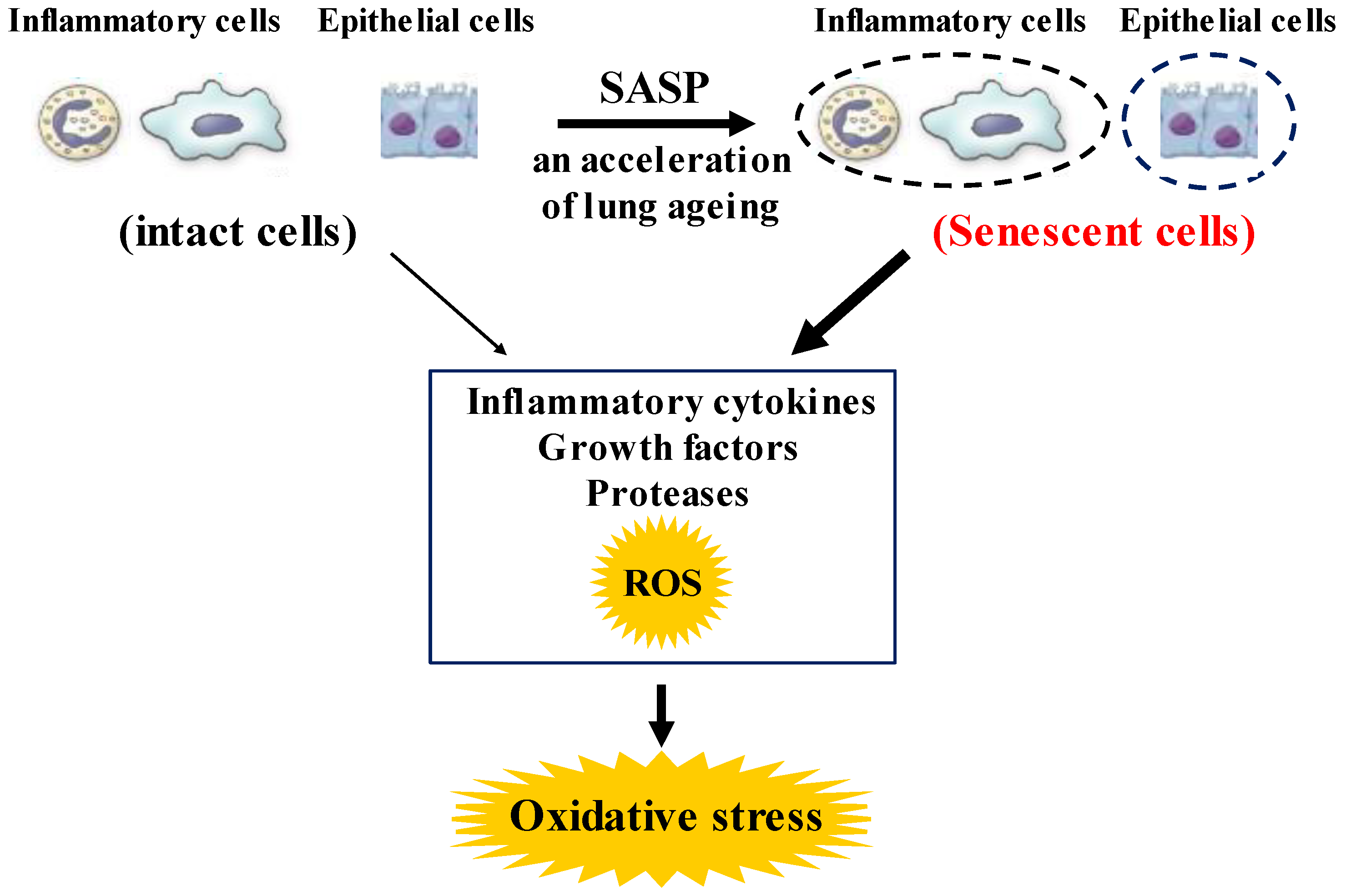
Figure 2. Roles of senescence in inflammatory and airway epithelial cells to enhance oxidative stress in COPD. These senescent cells in the lungs synthesize inflammatory cytokines, growth factors proteases, and ROS more than intact cells in them, referred to as senescence-associated secretary phenotype (SASP). These phenotype changes in these cells perhaps potentiate not only the lung inflammation but also oxidative stress in COPD. ROS: Reactive oxygen species.
To improve the management and treatment for COPD, patients with COPD should be classified by grouping according to distinct clinical phenotypes. These groupings, based on multiple dimensions (clinical, physiological, imaging, and endotyping) determine clusters of patients with common characteristics, which are associated with clinically meaningful outcomes such as symptoms, exacerbations, response to therapy, and disease progression (stratified medicine). Moreover, since several phenotypes can coexist in individual patients with COPD, an approach due to therapeutic target identified phenotypes and endotypes (treatable traits) has been proposed as an advanced therapy recently (precision medicine) [13]. Although oxidative stress perhaps plays an important role in amplifying the chronic lung inflammation in COPD [14], little is currently known about the involvement of oxidative stress in the pathogenesis of COPD. Therefore, research for clinical phenotype classification focused on oxidative stress is needed to establish precision medicine for development of the therapeutic management for COPD.
2. Oxidative Stress in COPD
2.1. Pathological Features
Oxidative stress occurs in the lungs during COPD, leading to characteristic pathological changes in this disease (Figure 1). It is well proven by data derived from bronchial biopsy [15], sputum examination [16], and in vitro studies [17] that inflammatory cells such as neutrophils, macrophages and T lymphocytes infiltrate and various proinflammatory molecules are present at increased levels in smokers’ lungs. Inflammatory cells, particularly neutrophils and macrophages that are recruited into the lungs, as well as structural cells, such as airway epithelial cells and fibroblasts, generate endogenous oxidants (ROS) in the lungs, leading to destruction of peripheral airways and alveoli. Mitochondrial respiration in these related cells is a key source of ROS, and cigarette smoke enhances generation of ROS through mitochondrial dysfunction, supporting the pathophysiological characteristics in COPD [18][19]. These destructive processes overcome the local protective mechanisms, and cause tissue damage without manifestations. The inflammatory tissue damage may be perpetuated for a long time after smoking cessation in patients with COPD [20]. Cigarette smoke causes the chronic lung inflammation; however, only about 20% of smokers develop COPD, indicating that there are factors that increase susceptibility and amplify the normal inflammatory response to cigarette smoke. Although these mechanisms are still unknown in detail, this phenomenon is probably involved in oxidative stress due to synthesis of ROS and imbalance of local proteolysis/antiproteolysis states that are related to oxidative stress (imbalance of oxidants/antioxidants).
2.2. Oxidants Related to COPD
Oxidative stress is recognized as a major predisposing factor of the inflammatory response related to COPD. Oxidative stress is probably associated with the pathology and severity of COPD. Oxidative stress is potentiated in patients with COPD, especially when acute exacerbations occur. Cigarette smoke, air pollution and biomass smoke are major exogenous oxidants related to COPD in the lungs, referred to as exogenous oxidative stress, but oxidative stress also arises from endogenous processes due to endogenous oxidants, after stop smoking, referred to as endogenous oxidative stress (Figure 1). The number of activated alveolar macrophages is markedly increased in the lungs of patients with COPD, compared to healthy subjects; and a large amount of ROS is released from these activated macrophages as superoxide anions and hydrogen peroxide (H2O2) [21]. This phenomenon is more potentiated during COPD exacerbations. Activated neutrophils also infiltrate to the lungs in patients with COPD, and activated neutrophils release a large amount of ROS, especially during COPD exacerbations [22]. In patients with COPD, generation of 4-hydroxy-2-nonenal (4HNE) is increased in the lungs, indicating that lipid peroxidation, a marker of oxidative stress, occurs on endogenous lipids [23]. Clinical studies have demonstrated that H2O2, 8-isoprostane, 4HNE, myeloperoxidase (MPO) and malondialdehyde (MDA) (endogenous oxidants as biomarkers of oxidative stress) are increased in exhaled breath condensate in patients with COPD [13][24][25][26][27], compared to healthy individuals; and these makers are more elevated during exacerbations [28]. These markers, such as MDA, 8-isoprostane, 8-hydroxy-2′-deoxyguanosine (8-OHdG) and MPO, are also elevated in sputum from patients with COPD [29][30]. Furthermore, nucleic acid oxidation, 8-oxo-7,8-dihydroguanosine (8-OHG) in RNA and 8-OHdG in DNA are elevated in alveolar lung fibroblasts from patients with emphysematous COPD [18][31]. These augmented biomarkers of oxidative stress do not decrease, and remain elevated in ex-smokers after the cessation of smoking, suggesting that persistent lung inflammation is caused by endogenous oxidative stress [25].
The respiratory system is constantly exposed to oxidative stress due to sources of endogenous ROS generated by mitochondrial respiration and inflammatory responses to bacterial and viral infections. The persistent oxidative stress in COPD results not only from activated neutrophils and macrophages but also from epithelial cells in the respiratory system. Oxidative stress is associated with mitochondrial respiration in these structural cells [32]. Other sources of intracellular ROS include the cytoplasmic ROS generating enzymes, such as membrane-bound nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) and the xanthine/xanthine oxidase system, as well as neutrophil derived MPO [33]. Superoxide anions, relatively weak oxidizing agents, are mainly produced endogenously by NOX; and are rapidly converted to more damaging ROS, such as the hydroxyl radical and H2O2, or the powerful and damaging peroxynitrite radical in the presence of nitric oxide (NO) [34]. MPO is released from activated neutrophils which are recruited into the lungs of patients with COPD; MPO also produces very destructive hypochlorous acid such as 3-chlorotyrosine [35]. However, in healthy adults, intracellular antioxidant defenses can overcome these damaging ROS, thus limiting their cellular effects; on the other hand, in patients with COPD these antioxidant defenses are overwhelmed. Increased oxidative stress has great effects on driving the pathophysiology of COPD as described below [33]. Oxidative stress causes activation of the proinflammatory transcription factor nuclear factor-KB (NF-κB) pathways; expression of NF-κB is augmented in COPD in airway epithelium and macrophages in patients with COPD. Oxidative stress also causes activation of the transforming growth factor (TGF)-β1 pathways, which acts on the lung epithelium, and induces fibrotic repair via driving epithelial-to-mesenchymal transition (EMT) [36], leading to small airway fibrosis. The inhibitory effects of TGF-β1 on nuclear erythroid-2 related factor 2 (Nrf2) bring about reduced expression of endogenous antioxidants [37]. Oxidative stress increases the expression of matrix metallopeptidase 9 (MMP9), an elastolytic enzyme, related to the development of emphysema.
2.3. Antioxidants Related to COPD
Increased oxidative stress may be potentiated with a reduction in endogenous anti-oxidant-induced defenses in patients with COPD (Figure 1). A clinical trial has demonstrated that concentrations of glutathione are lower in bronchoalveolar lavage fluid from unstable COPD patients with frequent exacerbations than in that from stable COPD [38]. Extracellular superoxide dismutase (SOD3) is decreased around small airways in patients with COPD [39]. Thioredoxin, which is an important regulator of redox balance, is decreased in COPD [38]. Nrf2 and forkhead box O3a (FOXO3a) are decreased in the lungs of patients with COPD [40][41]. Nrf2 and FOXO3a, which are key transcription factors that regulate multiple antioxidant genes, protect the respiratory system against oxidative damage [42]. Nrf2 is activated in healthy smokers, but its activation is impaired by oxidative stress in patients with COPD, resulting in reduced antioxidant gene expression [42]. Glutathione peroxidase is decreased in the lungs of COPD patients [43]. Glutathione peroxidase transgenic mice are protected against the development of inflammation and emphysema after cigarette smoke exposure, whereas glutathione peroxidase gene knockout increases the tissue destruction in the lung’s response to cigarette smoke [44]. Imbalance between oxidants and antioxidants probably plays an essential role in the chronic inflammation related to the pathology of COPD (Figure 1 and Figure 2).
2.4. Reduced Responsiveness to Corticosteroids Caused by Oxidative Stress
Oxidative stress probably causes corticosteroid resistance in COPD. Oxidative stress reduces activity and expression of histone deacetylase-2 (HDAC2), which is required for inflammatory gene suppression [45], by activation of phosphoinositide-3-kinase (PI3K)-δ [46]. This phenomenon prevents the acetylation of glucocorticoid receptors, which is necessary for the inhibition of NF-κB that mediates the anti-inflammatory effects of corticosteroids, leading to reduced responsiveness to corticosteroids. Therefore, chronic lung inflammation is not fully inhibited by corticosteroids in COPD, different from mild asthma [6][33]. Recent preclinical studies have indicated that improvement of the redox balance by the administration of antioxidants or the stimulation of endogenous antioxidant response may overcome the corticosteroid resistance in COPD [47][48]. Nrf2 is known to act as an antioxidant. Sulforaphate, an activator of Nrf2, improves reduced responsiveness to corticosteroids mediated by upregulation of Nrf2 and enhancement of HDAC2 expression and activity in the allergen challenged mice that were exposed to cigarette smoke [49]. Nrf2 may be a potential molecular target for cigarette smoke-related resistance to corticosteroids in COPD.
References
- Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for Prevention, Diagnosis and Management of Chronic Obstructive Pulmonary Disease: 2023 Report. Available online: https://goldcopd.org/ (accessed on 1 December 2022).
- Hogg, J.C.; Timens, W. The pathology of chronic obstructive pulmonary disease. Annu. Rev. Pathol. 2009, 4, 435–459.
- Barnes, P.J.; Burney, P.G.J.; Silverman, E.K.; Celli, B.R.; Vestbo, J.; Wedzicha, J.A.; Wouters, E.F.M. Chronic obstructive pulmonary disease. Nat. Rev. Prim. 2015, 1, 15076.
- Birnboim, H.C. DNA strand breaks in human leukocytes induced by superoxide anion, hydrogen peroxide and tumor promoters are repaired slowly compared to breaks induced by ionizing radiation. Carcinogenesis 1986, 7, 1511–1517.
- Agustí, A.; Hogg, J.C. Update on the pathogenesis of chronic obstructive pulmonary disease. N. Engl. J. Med. 2019, 381, 1248–1256.
- Barnes, P.J. Oxidative stress in chronic obstructive pulmonary disease. Antioxidants 2022, 11, 965.
- Aravamudan, B.; Kiel, A.; Freeman, M.; Delmotte, P.; Thompson, M.; Vassallo, R.; Sieck, G.C.; Pabelick, C.M.; Prakash, Y.S. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L840–L854.
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular senescence as a mechanism and target in chronic lung diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564.
- Tsuji, T.; Aoshiba, K.; Nagai, A. Cigarette smoke induces senescence in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 643–649.
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735.
- Kumar, M.; Seeger, W.; Voswinckel, R. Senescence-associated secretory phenotype and its possible role in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2014, 51, 323–333.
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273.
- Agusti, A.; Bel, E.; Thomas, M.; Vogelmeier, C.; Brusselle, G.; Holgate, S.; Humbert, M.; Jones, P.; Gibson, P.G.; Vestbo, J.; et al. Treatable traits: Toward precision medicine of chronic airway diseases. Eur. Respir. J. 2016, 47, 410–419.
- Domej, W.; Oettl, K.; Renner, W. Oxidative stress and free radicals in COPD–Implications and relevance for treatment. Int. J. Chron. Obs. Pulmon. Dis. 2014, 9, 1207–1224.
- Di Stefano, A.; Caramori, G.; Oates, T.; Capelli, A.; Lusuardi, M.; Gnemmi, I.; Ioli, F.; Chung, K.F.; Donner, C.F.; Barnes, P.J.; et al. Increased expression of nuclear factor-kappaB in bronchial biopsies from smokers and patients with COPD. Eur. Respir. J. 2002, 20, 556–563.
- Ravi, A.K.; Khurana, S.; Lemon, J.; Plumb, J.; Booth, G.; Healy, L.; Catley, M.; Vestbo, J.; Singh, D. Increased levels of soluble interleu-kin-6 receptor and CCL3 in COPD sputum. Respir. Res. 2014, 15, 103.
- Shao, M.X.; Nakanaga, T.; Nadel, J.A. Cigarette smoke induces MUC5AC mucin overproduction via tumor necrosis factor-α-converting enzyme in human airway epithelial (NCI-H292) cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L420–L427.
- van der Toorn, M.; Rezayat, D.; Kauffman, H.F.; Bakker, S.J.; Gans, R.O.; Koëter, G.H.; Choi, A.M.; van Oosterhout, A.J.; Slebos, D.J. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L109–L114.
- Schumacker, P.T.; Gillespie, M.N.; Nakahira, K.; Choi, A.M.; Crouser, E.D.; Piantadosi, C.A.; Bhattacharya, J. Mitochondria in lung biology and pathology: More than just a powerhouse. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L962–L974.
- Shapiro, S.D. End-stage chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2001, 164, 339–340.
- Schaberg, T.; Klein, U.; Rau, M.; Eller, J.; Lode, H. Subpopulations of alveolar macrophages in smokers and nonsmokers: Relation to the expression of CD11/CD18 molecules and superoxide anion production. Am. J. Respir. Crit. Care Med. 1995, 151, 1551–1558.
- Noguera, A.; Batle, S.; Miralles, C.; Iglesias, J.; Busquets, X.; MacNee, W.; Agustí, A.G. Enhanced neutrophil response in chronic obstructive pulmonary disease. Thorax 2001, 56, 432–437.
- Rahman, I.; van Schadewijk, A.A.; Crowther, A.J.; Hiemstra, P.S.; Stolk, J.; MacNee, W.; De Boer, W.I. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2002, 166, 490–495.
- Dekhuijzen, P.N.; Aben, K.K.; Dekker, I.; Aarts, L.P.; Wielders, P.L.; van Herwaarden, C.L.; Bast, A. Increased exhalation of hydro-gen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1996, 154, 813–816.
- Montuschi, P.; Collins, J.V.; Ciabattoni, G.; Lazzeri, N.; Corradi, M.; Kharitonov, S.A.; Barnes, P.J. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am. J. Respir. Crit. Care Med. 2000, 162, 1175–1177.
- Corradi, M.; Pignatti, P.; Manini, P.; Andreoli, R.; Goldoni, M.; Poppa, M.; Moscato, G.; Balbi, B.; Mutti, A. Comparison between exhaled and sputum oxidative stress biomarkers in chronic airway inflammation. Eur. Respir. J. 2004, 24, 1011–1017.
- Bartoli, M.L.; Novelli, F.; Costa, F.; Malagrinò, L.; Melosini, L.; Bacci, E.; Cianchetti, S.; Dente, F.L.; Di Franco, A.; Vagaggini, B.; et al. Malondialdehyde in exhaled breath condensate as a marker of oxidative stress in different pulmonary diseases. Mediat. Inflamm. 2011, 2011, 891752.
- Biernacki, W.A.; Kharitonov, S.A.; Barnes, P.J. Increased leukotriene B4 and 8-isoprostane in exhaled breath condensate of patients with exacerbations of COPD. Thorax 2003, 58, 294–298.
- Zhu, A.; Ge, D.; Zhang, J.; Teng, Y.; Yuan, C.; Huang, M.; Adcock, I.M.; Barnes, P.J.; Yao, X. Sputum myeloperoxidase in chronic obstructive pulmonary disease. Eur. J. Med. Res. 2014, 19, 12.
- Antus, B. Oxidative Stress Markers in Sputum. Oxid. Med. Cell Longev. 2016, 2016, 2930434.
- Deslee, G.; Adair-Kirk, T.L.; Betsuyaku, T.; Woods, J.C.; Moore, C.H.; Gierada, D.S.; Conradi, S.H.; Atkinson, J.J.; Toennies, H.M.; Battaile, J.T.; et al. Cigarette smoke induces nucleic-acid oxidation in lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 2010, 43, 576–584.
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol Physiol. 2000, 279, L1005–L1028.
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020, 33, 101544.
- Osoata, G.O.; Hanazawa, T.; Brindicci, C.; Ito, M.; Barnes, P.J.; Kharitonov, S.; Ito, K. Peroxynitrite elevation in exhaled breath condensate of COPD and its inhibition by fudosteine. Chest 2009, 135, 1513–1520.
- O’Donnell, C.; Newbold, P.; White, P.; Thong, B.; Stone, H.; Stockley, R.A. 3-Chlorotyrosine in sputum of COPD patients: Relation-ship with airway inflammation. COPD. 2010, 7, 411–417.
- Gorowiec, M.R.; Borthwick, L.A.; Parker, S.M.; Kirby, J.A.; Saretzki, G.C.; Fisher, A.J. Free radical generation induces epithelial-to-mesenchymal transition in lung epithelium via a TGF-β1-dependent mechanism. Free Radic. Biol. Med. 2012, 52, 1024–1032.
- Michaeloudes, C.; Sukkar, M.B.; Khorasani, N.M.; Bhavsar, P.K.; Chung, K.F. TGF-β regulates Nox4, MnSOD and catalase expres-sion, and IL-6 release in airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L295–L304.
- Drost, E.M.; Skwarski, K.M.; Sauleda, J.; Soler, N.; Roca, J.; Agusti, A.; MacNee, W. Oxidative stress and airway inflammation in severe exacerbations of COPD. Thorax 2005, 60, 293–300.
- Yao, H.; Arunachalam, G.; Hwang, J.W.; Chung, S.; Sundar, I.K.; Kinnula, V.L.; Crapo, J.D.; Rahman, I. Extracellular superoxide dis-mutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc. Natl. Acad. Sci. USA 2010, 107, 15571–15576.
- Malhotra, D.; Thimmulappa, R.; Vij, N.; Navas-Acien, A.; Sussan, T.; Merali, S.; Zhang, L.; Kelsen, S.G.; Myers, A.; Wise, R.; et al. Heightened endoplasmic reticulum stress in the lungs of patients with chronic obstructive pulmonary disease: The role of Nrf2-regulated proteasomal activity. Am. J. Respir. Crit. Care Med. 2009, 180, 1196–1207.
- Hwang, J.W.; Rajendrasozhan, S.; Yao, H.; Chung, S.; Sundar, I.K.; Huyck, H.L.; Pryhuber, G.S.; Kinnula, V.L.; Rahman, I. FOXO3 deficiency leads to increased susceptibility to cigarette smoke-induced inflammation, airspace enlargement, and chronic obstructive pulmonary disease. J. Immunol. 2011, 187, 987–998.
- Liu, Q.; Gao, Y.; Ci, X. Role of Nrf2 and Its Activators in Respiratory Diseases. Oxid. Med. Cell Longev. 2019, 2019, 7090534.
- Vlahos, R.; Bozinovski, S. Glutathione peroxidase-1 as a novel therapeutic target for COPD. Redox Rep. 2013, 18, 142–149.
- Geraghty, P.; Hardigan, A.A.; Wallace, A.M.; Mirochnitchenko, O.; Thankachen, J.; Arellanos, L.; Thompson, V.; D’Armiento, J.M.; Foronjy, R.F. The glutathione peroxidase 1-protein tyrosine phosphatase 1B-protein phosphatase 2A axis. A key determinant of airway inflammation and alveolar destruction. Am. J. Respir. Cell Mol. Biol. 2013, 49, 721–730.
- Barnes, P.J. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2013, 131, 636–645.
- To, Y.; Ito, K.; Kizawa, Y.; Failla, M.; Ito, M.; Kusama, T.; Elliott, W.M.; Hogg, J.C.; Adcock, I.M.; Barnes, P.J. Targeting phosphoinositide-3-kinase-d with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 897–904.
- Lewis, B.W.; Ford, M.L.; Rogers, L.K.; Britt, R.D., Jr. Oxidative stress promotes corticosteroid insensitivity in asthma and COPD. Antioxidants 2021, 10, 1335.
- Mei, D.; Tan, W.S.D.; Wong, W.S.F. Pharmacological strategies to regain steroid sensitivity in severe asthma and COPD. Curr. Opin. Pharmacol. 2019, 46, 73–81.
- Sakurai, H.; Morishima, Y.; Ishii, Y.; Yoshida, K.; Nakajima, M.; Tsunoda, Y.; Hayashi, S.Y.; Kiwamoto, T.; Matsuno, Y.; Kawaguchi, M.; et al. Sulforaphane ameliorates steroid insensitivity through an Nrf2-dependent pathway in cigarette smoke-exposed asthmatic mice. Free Radic. Biol. Med. 2018, 129, 473–485.
More
Information
Subjects:
Respiratory System
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Revisions:
2 times
(View History)
Update Date:
06 Feb 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No