 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | BING LI | -- | 1822 | 2023-01-30 16:12:44 | | | |
| 2 | Jessie Wu | + 12 word(s) | 1834 | 2023-01-31 03:05:36 | | | | |
| 3 | Jessie Wu | + 9 word(s) | 1843 | 2023-01-31 03:07:40 | | | | |
| 4 | Jessie Wu | + 4 word(s) | 1847 | 2023-01-31 03:08:52 | | | | |
| 5 | Jessie Wu | Meta information modification | 1847 | 2023-01-31 03:13:00 | | |
Video Upload Options
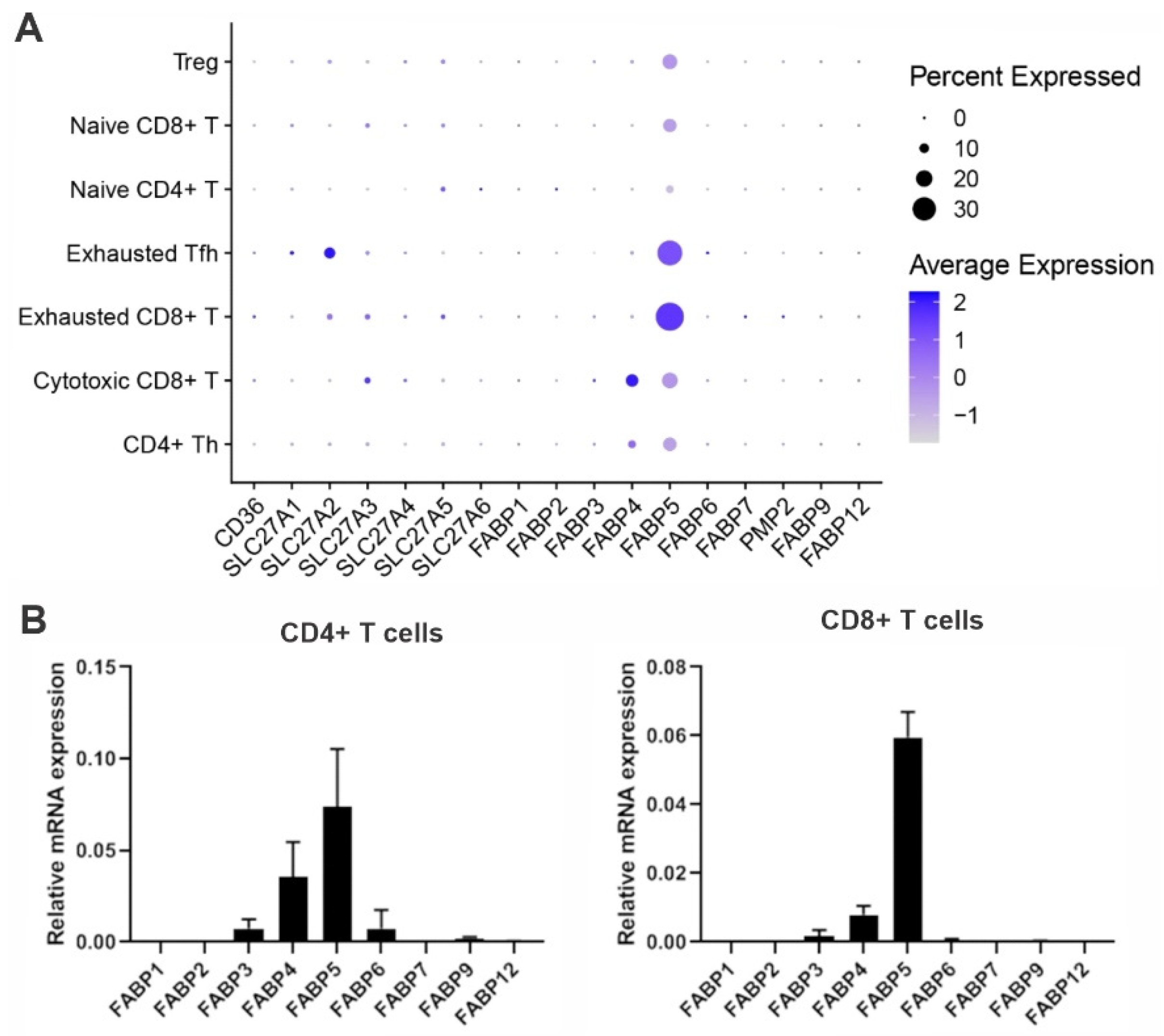
To evade immune surveillance, tumors develop a hostile microenvironment that inhibits anti-tumor immunity. Recent immunotherapy breakthroughs that target the reinvigoration of tumor-infiltrating T lymphocytes (TIL) have led to unprecedented success in treating some cancers that are resistant to conventional therapy, suggesting that T cells play a pivotal role in anti-tumor immunity. In the hostile tumor microenvironment (TME), activated T cells are known to mainly rely on aerobic glycolysis to facilitate their proliferation and anti-tumor function. However, TILs usually exhibit an exhausted phenotype and impaired anti-tumor activity due to the limited availability of key nutrients (e.g., glucose) in the TME. Emerging evidence suggests that fatty acid binding protein 5 (FABP5) expression in T cells regulates T cell lipid metabolism and function.
1. Introduction
2. Fatty Acid Binding Protein 5 in Naïve T Cells
3. Fatty Acid Binding Protein 5 in Tregs
4. Fatty Acid Binding Protein 5 in Memory T Cells
5. Fatty Acid Binding Protein 5 in Other T Cell Subsets
References
- Rosenberg, S.A. Raising the bar: The curative potential of human cancer immunotherapy. Sci. Transl. Med. 2012, 4, 127ps8.
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465.
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517.
- Powles, T.; Eder, J.P.; Fine, G.D.; Braiteh, F.S.; Loriot, Y.; Cruz, C.; Bellmunt, J.; Burris, H.A.; Petrylak, D.P.; Teng, S.-L.; et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014, 515, 558–562.
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients with Platinum-Resistant Ovarian Cancer. J. Clin. Oncol. 2015, 33, 4015–4022.
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924.
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740.
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262.
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867.
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264.
- Kim, J.M.; Chen, D.S. Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure). Ann. Oncol. 2016, 27, 1492–1504.
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10.
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571.
- Lochner, M.; Berod, L.; Sparwasser, T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015, 36, 81–91.
- Bonnefont, J.P.; Djouadi, F.; Prip-Buus, C.; Gobin, S.; Munnich, A.; Bastin, J. Carnitine palmitoyltransferases 1 and 2: Biochemical, molecular and medical aspects. Mol. Aspects Med. 2004, 25, 495–520.
- Pan, Y.; Tian, T.; Park, C.O.; Lofftus, S.Y.; Mei, S.; Liu, X.; Luo, C.; O’Malley, J.T.; Gehad, A.; Teague, J.E.; et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 2017, 543, 252–256.
- Jin, R.; Hao, J.; Yi, Y.; Yin, D.; Hua, Y.; Li, X.; Bao, H.; Han, X.; Egilmez, N.K.; Sauter, E.R.; et al. Dietary fats high in linoleic acids impair anti-tumor T cell responses by inducing E-FABP-mediated mitochondrial dysfunction. Cancer Res. 2021, 81, 5296–5310.
- Nasi, M.; Troiano, L.; Lugli, E.; Pinti, M.; Ferraresi, R.; Monterastelli, E.; Mussi, C.; Salvioli, G.; Franceschi, C.; Cossarizza, A. Thymic output and functionality of the IL-7/IL-7 receptor system in centenarians: Implications for the neolymphogenesis at the limit of human life. Aging Cell 2006, 5, 167–175.
- de Jong, A.J.; Kloppenburg, M.; Toes, R.E.; Ioan-Facsinay, A. Fatty acids, lipid mediators, and T-cell function. Front. Immunol. 2014, 5, 483.
- Ellulu, M.S.; Patimah, I.; KhazáAi, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863.
- Imes, C.C.; Burke, L.E. The Obesity Epidemic: The United States as a Cautionary Tale for the Rest of the World. Curr. Epidemiol. Rep. 2014, 1, 82–88.
- Naik, A.; Monjazeb, A.M.; Decock, J. The Obesity Paradox in Cancer, Tumor Immunology, and Immunotherapy: Potential Therapeutic Implications in Triple Negative Breast Cancer. Front. Immunol. 2019, 10, 1940.
- Ma, C.; Kesarwala, A.H.; Eggert, T.; Medina-Echeverz, J.; Kleiner, D.E.; Jin, P.; Stroncek, D.F.; Terabe, M.; Kapoor, V.; ElGindi, M.; et al. NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016, 531, 253–257.
- Ma, X.; Xiao, L.; Liu, L.; Ye, L.; Su, P.; Bi, E.; Wang, Q.; Yang, M.; Qian, J.; Yi, Q. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab. 2021, 33, 1001–1012.e5.
- He, N.; Fan, W.; Henriquez, B.; Yu, R.T.; Atkins, A.R.; Liddle, C.; Zheng, Y.; Downes, M.; Evans, R.M. Metabolic control of regulatory T cell (Treg) survival and function by Lkb1. Proc. Natl. Acad. Sci. USA 2017, 114, 12542–12547.
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J. Immunol. 2011, 186, 3299–3303.
- Gualdoni, G.A.; Mayer, K.; Göschl, L.; Boucheron, N.; Ellmeier, W.; Zlabinger, G. The AMP analog AICAR modulates the Treg/Th17 axis through enhancement of fatty acid oxidation. FASEB J. 2016, 30, 3800–3809.
- Galgani, M.; De Rosa, V.; La Cava, A.; Matarese, G. Role of Metabolism in the Immunobiology of Regulatory T Cells. J. Immunol. 2016, 197, 2567–2575.
- Wang, H.; Franco, F.; Tsui, Y.-C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernández-García, J.; Tsai, C.-H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308.
- Field, C.S.; Baixauli, F.; Kyle, R.L.; Puleston, D.J.; Cameron, A.M.; Sanin, D.E.; Hippen, K.L.; Loschi, M.; Thangavelu, G.; Corrado, M.; et al. Mitochondrial Integrity Regulated by Lipid Metabolism Is a Cell-Intrinsic Checkpoint for Treg Suppressive Function. Cell Metab. 2019, 31, 422–437.e5.
- Li, B.; Reynolds, J.M.; Stout, R.D.; Bernlohr, D.A.; Suttles, J. Regulation of Th17 differentiation by epidermal fatty acid-binding protein. J. Immunol. 2009, 182, 7625–7633.
- Zhang, Y.; Sun, Y.; Rao, E.; Yan, F.; Li, Q.; Zhang, Y.; Silverstein, K.; Liu, S.; Sauter, E.; Cleary, M.; et al. Fatty acid-binding protein E-FABP restricts tumor growth by promoting IFN-beta responses in tumor-associated macrophages. Cancer Res. 2014, 74, 2986–2998.
- Kumar, B.V.; Ma, W.; Miron, M.; Granot, T.; Guyer, R.S.; Carpenter, D.J.; Senda, T.; Sun, X.; Ho, S.-H.; Lerner, H.; et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep. 2017, 20, 2921–2934.
- Park, C.O.; Kupper, T.S. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat. Med. 2015, 21, 688–697.
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.-S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107.
- Webb, J.R.; Milne, K.; Watson, P.; Deleeuw, R.J.; Nelson, B.H. Tumor-Infiltrating Lymphocytes Expressing the Tissue Resident Memory Marker CD103 Are Associated with Increased Survival in High-Grade Serous Ovarian Cancer. Clin. Cancer Res. 2014, 20, 434–444.
- Ganesan, A.-P.; Clarke, J.; Wood, O.; Garrido-Martin, E.M.; Chee, S.J.; Mellows, T.; Samaniego-Castruita, D.; Singh, D.; Seumois, G.; Alzetani, A.; et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat. Immunol. 2017, 18, 940–950.
- Savas, P.; Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; Salgado, R.; Byrne, D.J.; Teo, Z.L.; Dushyanthen, S.; et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018, 24, 986–993.
- Lin, R.; Zhang, H.; Yuan, Y.; He, Q.; Zhou, J.; Li, S.; Sun, Y.; Li, D.Y.; Qiu, H.-B.; Wang, W.; et al. Fatty acid oxidation controls CD8+ tissue-resident memory T-cell survival in gastric adenocarcinoma. Cancer Immunol. Res. 2020, 8, 479–492.
- O’Sullivan, D.; van der Windt, G.J.; Huang, S.C.-C.; Curtis, J.D.; Chang, C.-H.; Buck, M.D.; Qiu, J.; Smith, A.M.; Lam, W.Y.; DiPlato, L.M.; et al. Memory CD8+ T Cells Use Cell-Intrinsic Lipolysis to Support the Metabolic Programming Necessary for Development. Immunity 2014, 41, 75–88.
- Liu, F.; Liu, W.; Zhou, S.; Yang, C.; Tian, M.; Jia, G.; Wang, H.; Zhu, B.; Feng, M.; Lu, Y.; et al. Identification of FABP5 as an immunometabolic marker in human hepatocellular carcinoma. J. Immunother. Cancer 2020, 8, e000501.
- Barbi, J.; Pardoll, D.; Pan, F. Metabolic control of the Treg/Th17 axis. Immunol. Rev. 2013, 252, 52–77.
- Waickman, A.T.; Powell, J.D. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol. Rev. 2012, 249, 43–58.
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The Biochemistry and Physiology of Mitochondrial Fatty Acid beta-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44.
- Berod, L.; Friedrich, C.; Nandan, A.; Freitag, J.; Hagemann, S.; Harmrolfs, K.; Sandouk, A.; Hesse, C.; Castro, C.N.; Bähre, H.; et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 2014, 20, 1327–1333.
- Lee, J.; Walsh, M.; Hoehn, K.; James, D.; Wherry, J.; Choi, Y. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J. Immunol. 2014, 192, 3190–3199.
- Endo, Y.; Asou, H.K.; Matsugae, N.; Hirahara, K.; Shinoda, K.; Tumes, D.J.; Tokuyama, H.; Yokote, K.; Nakayama, T. Obesity Drives Th17 Cell Differentiation by Inducing the Lipid Metabolic Kinase, ACC1. Cell Rep. 2015, 12, 1042–1055.
- Lee, J.; Kim, B.; Chu, H.; Zhang, K.; Kim, H.; Kim, J.H.; Kim, S.H.; Pan, Y.; Noh, J.Y.; Sun, Z.; et al. FABP5 as a possible biomarker in atopic march: FABP5-induced Th17 polarization, both in mouse model and human samples. EBioMedicine 2020, 58, 102879.
- Li, B.; Schmidt, N.W. Epidermal Fatty Acid Binding Protein (E-FABP) Is Not Required for the Generation or Maintenance of Effector and Memory T Cells following Infection with Listeria monocytogenes. PLoS ONE 2016, 11, e0162427.
- Vitiello, G.A.; Miller, G. Targeting the interleukin-17 immune axis for cancer immunotherapy. J. Exp. Med. 2019, 217, e20190456.
- Asadzadeh, Z.; Mohammadi, H.; Safarzadeh, E.; Hemmatzadeh, M.; Mahdian-Shakib, A.; Jadidi-Niaragh, F.; Azizi, G.; Baradaran, B. The paradox of Th17 cell functions in tumor immunity. Cell. Immunol. 2017, 322, 15–25.


