Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mario Mastrangelo | -- | 1750 | 2023-01-30 06:53:48 | | | |
| 2 | Conner Chen | Meta information modification | 1750 | 2023-01-30 08:45:05 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mastrangelo, M.; Tolve, M.; Artiola, C.; Bove, R.; Carducci, C.; Carducci, C.; Angeloni, A.; Pisani, F.; Leuzzi, V. Inherited Disorders of Biogenic Amine Neurotransmitter Metabolism. Encyclopedia. Available online: https://encyclopedia.pub/entry/40547 (accessed on 29 June 2026).
Mastrangelo M, Tolve M, Artiola C, Bove R, Carducci C, Carducci C, et al. Inherited Disorders of Biogenic Amine Neurotransmitter Metabolism. Encyclopedia. Available at: https://encyclopedia.pub/entry/40547. Accessed June 29, 2026.
Mastrangelo, Mario, Manuela Tolve, Cristiana Artiola, Rossella Bove, Claudia Carducci, Carla Carducci, Antonio Angeloni, Francesco Pisani, Vincenzo Leuzzi. "Inherited Disorders of Biogenic Amine Neurotransmitter Metabolism" Encyclopedia, https://encyclopedia.pub/entry/40547 (accessed June 29, 2026).
Mastrangelo, M., Tolve, M., Artiola, C., Bove, R., Carducci, C., Carducci, C., Angeloni, A., Pisani, F., & Leuzzi, V. (2023, January 30). Inherited Disorders of Biogenic Amine Neurotransmitter Metabolism. In Encyclopedia. https://encyclopedia.pub/entry/40547
Mastrangelo, Mario, et al. "Inherited Disorders of Biogenic Amine Neurotransmitter Metabolism." Encyclopedia. Web. 30 January, 2023.
Copy Citation
Inherited disorders of biogenic amine metabolism are genetically determined conditions resulting in dysfunctions or lack of enzymes involved in the synthesis, degradation, or transport of dopamine, serotonin, adrenaline/noradrenaline, and their metabolites or defects of their cofactor or chaperone biosynthesis. They represent a group of treatable diseases presenting with complex patterns of movement disorders (dystonia, oculogyric crises, severe/hypokinetic syndrome, myoclonic jerks, and tremors) associated with a delay in the emergence of postural reactions, global development delay, and autonomic dysregulation. The earlier the disease manifests, the more severe and widespread the impaired motor functions.
neurotransmitter disorders
movement disorders
encephalopathy
1. Background
Inherited defects of biogenic amine neurotransmitter metabolism are ultrarare genetically determined conditions resulting in dysfunctions/lack of enzymes involved in the synthesis, degradation, or transport of dopamine, serotonin, adrenaline/noradrenaline, and their metabolites or defects of their cofactor or chaperone biosynthesis (Table 1) [1]. All these conditions are inherited as autosomal recessive diseases, except for DYT/PARK-GCH1, including dominant and recessive forms and monoamine oxidase deficiency A and B, which are transmitted with an autosomal dominant inheritance [1].
Table 1. Clinical and biochemical features of disorders of biogenic amine metabolism.
| Disease | Gene | Clinical Features | Biochemical Markers | ||
|---|---|---|---|---|---|
| Plasma | Urine | CSF | |||
| AD-DYT/PARK-GCH1 (OMIM#128230) |
AD GCH1 | Parkinsonism, dystonia, motor delay, diurnal fluctuation, truncal hypotonia, hypertonia of extremities, tremors, and hypokinetic/rigid syndrome | Normal Phe Normal response to Phe loading |
↓BIO, ↓NEO | ↓NEO, ↓BIO, ↓HVA, ↓5-HIAA, Normal Sep and BH2 |
| AR-DYT/PARK-GCH1 (OMIM#233910) |
AR GCH1 | Truncal hypotonia, parkinsonism, feeding/swallowing difficulties, dystonia, excessive sweating, temperature instability, intellectual disability, motor delay, choreoathetosis, drooling, oculogyric crises, ptosis, and seizures | ↑Phe | ↓BIO, ↓NEO | ↓NEO, ↓BIO, ↓HVA, ↓5-HIAA, Normal Sep and BH2 |
| DYT/PARK-PTS (OMIM#261640) |
PTS | Dystonia, diurnal fluctuation, excessive sweating, temperature instability, hypo/hypertonia, parkinsonism, intellectual disability, motor delay, choreoathetosis, low birthweight, ptosis, and seizures | ↑Phe ↑Prolactin |
↓BIO, ↑NEO | ↑NEO, ↓BIO, ↓HVA ↓5-HIAA, Normal Sep and BH2 |
| DYT/PARK-SPR (OMIM#612716) |
SPR | Motor delay, dystonia, truncal hypotonia, diurnal fluctuation, intellectual disability, parkinsonism, hypertonia, drooling, oculogyric crises, and hypokinetic/rigid syndrome | Normal Phe | ↑Sep | Normal NEO, ↑BIO, ↓HVA, ↓5-HIAA ↑BH2, ↑Sep |
| DYT/PARK-QDPR (OMIM#261630) |
QDPR | Dystonia, diurnal fluctuation, sweating, temperature instability, hypo/hypertonia, parkinsonism, intellectual disability, motor delay, choreoathetosis, low birthweight, ptosis, epileptic encephalopathy, and basal ganglia calcifications | ↑Phe | ↑BIO, ↓NEO | N NEO, ↑BIO, ↓HVA ↓5-HIAA, ↓Folate, ↑BH2 Normal Sep |
| PCBD deficiency (OMIM#264070) |
PCBD1 | No severe neurological symptoms and transient and benign hyperphenylalaninemia | ↑Phe | ↑Primapterin | / |
| DYT/PARK-TH (OMIM#605407) |
TH | Hypokinetic/rigid syndrome, dystonia/parkinsonism, oculogyric crises, ptosis, autonomic dysfunctions, lethargy, irritability, sleep disturbances, pre-term birth, foetal distress, perinatal asphyxia, intellectual disability, growth retardation, microcephaly, motor delay, spasticity, and myoclonus | Normal Phe | / | ↓HVA, Normal NEO, BIO, 5HIAA, Sep, BH2 |
| DYT-DDC (OMIM#608643) |
DDC | Truncal hypotonia, developmental delay, intellectual disability, oculogyric crises, dystonia, dysarthria, ptosis, limb hypertonia, choreoathetosis, sleep disturbances, excessive sweating, temperature instability, orthostatic hypotension, diarrhea, nasal congestion, and hypoglycemia | Normal Phe ↑ Prolactin |
↑ Dopamine, ↓ VMA ↑ Vanillactic acid |
↓HVA, ↓5-HIAA, ↑3OMD ↓MHPG, Normal NEO, BIO, Sep, BH2 |
| MAOA/MAOB deficiency (OMIM#300615 MAOA) |
MAOA/ MAOB |
Behavioral disturbances, mild intellectual disability, hand stereotypes, flushing, and diarrhea | / | ↑normetanephrin ↑3-methoxytyramine, ↑tyramine ↓VMA, ↓HVA, ↓MHPG, ↓5-HIAA |
/ |
| DBH deficiency (OMIM#223360) |
DBH | Severe orthostatic hypotension, eyelid ptosis, sporadic dysmorphic features, rare reproductive dysfunctions, and normal cognitive development | ↓Norepinefrine ↑ Dopamine |
/ | / |
| DYT/PARK-SLC6A3 (OMIM#613135) |
SLC6A3 | Severe developmental delay or no acquisition of developmental milestones, anarthria, dystonia, parkinsonism, dyskinesia, oculogyric crises, swallowing difficulties, failure to thrive, and respiratory complications | Normal Phe ↑Prolactin ↓Norepinefrine |
↓Norepinefrine ↑3MT |
↑ HVA, Normal NEO, BIO,5-HIAA, Sep, BH2 |
| DYT/PARK-SLC18A2 (OMIM#618049) |
SLC18A2 | Severe developmental delay or no acquisition of developmental milestones, dysarthria, dystonia, parkinsonism, facial dyskinesia, oculogyric crises, vertical gaze palsy, and ptosis | / | ↑ HVA, ↑5-HIAA, ↓Dopamine, ↓Norepinefrine | ↑HVA/5-HIAA |
| DNAJC12 deficiency (OMIM#617384) |
DNAJC12 | Juvenile parkinsonism, dystonia, autism, intellectual disability, attention deficit hyperactivity disorder, psychiatric symptoms, and no symptoms in a quote of patients | ↑Phe | / | ↑BH4 ↓HVA, ↓5-HIAA |
LEGEND: 3MT, 3-methoxytyramine; 5 HIAA, 5-hydroxyindolacetic acid; GCH1, GTP Cyclohydrolase 1 gene; BIO, biopterin; BH2, dihydrobiopterin; DBH, dopamine β hydroxylase gene; DNAJC12, DNAJ/HSP40 homolog, subfamily c, member 12; DDC, DOPA decarboxylase gene; DYT/PARK-PTS, 6-pyruvoyltetrahydropterin synthase gene; QDPR, quinoid dihydropteridine reductase gene; SLC6A3, solute carrier family 6 member A3, dopamine transporter gene; SLC18A2, solute carrier family 18 member A2, vesicular monoamine transporter 2 gene; SPR, sepiapterin reductase gene; TH, tyrosine hydroxylase gene; HVA, homovanillic acid; MAO A/B, monoamine oxydase A/B gene; MHPG, 3-methoxy-4-hydroxyophenylglycol; NEO, neopterin; PCBD1, pterin-4a-carbinolamine dehydratase gene; Phe, phenylalanine; Sep, sepiapterin; VMA, vanillylmandelic acid. ↓ = decreased;↑ = increased.
Figure 1 summarizes the metabolic pathway and the sites of so-far identified proteins implied in clinically relevant alterations. Four enzymes (phenylalanine hydroxylase-PAH, tyrosine hydroxylase-TH, tryptophane hydroxylase-TPH, and aromatic aminoacidic decarboxylase-AADC) are directly implied in the synthesis of dopamine and serotonin. Five enzymes (guanosine triphosphate cyclohydrolase-GTPCH, 6-pyruvoyl tetrahydropterin synthase-PTPS, sepiapterin reductase-SR, dihydropteridine reductase-DHPR, and pterin-4a-carbinolamine dehydratase-PCD) are involved in the synthesis/regeneration of the PAH, TH, and TPH (tetrahydrobiopterin-BH4) cofactors. Three other disease-associated genes codify proteins acting as a chaperone of the three abovementioned hydroxylases (DNAJC12), an enzyme involved in biogenic amine catabolism (MAO), and a synaptic dopamine transporter (SLC6A3).
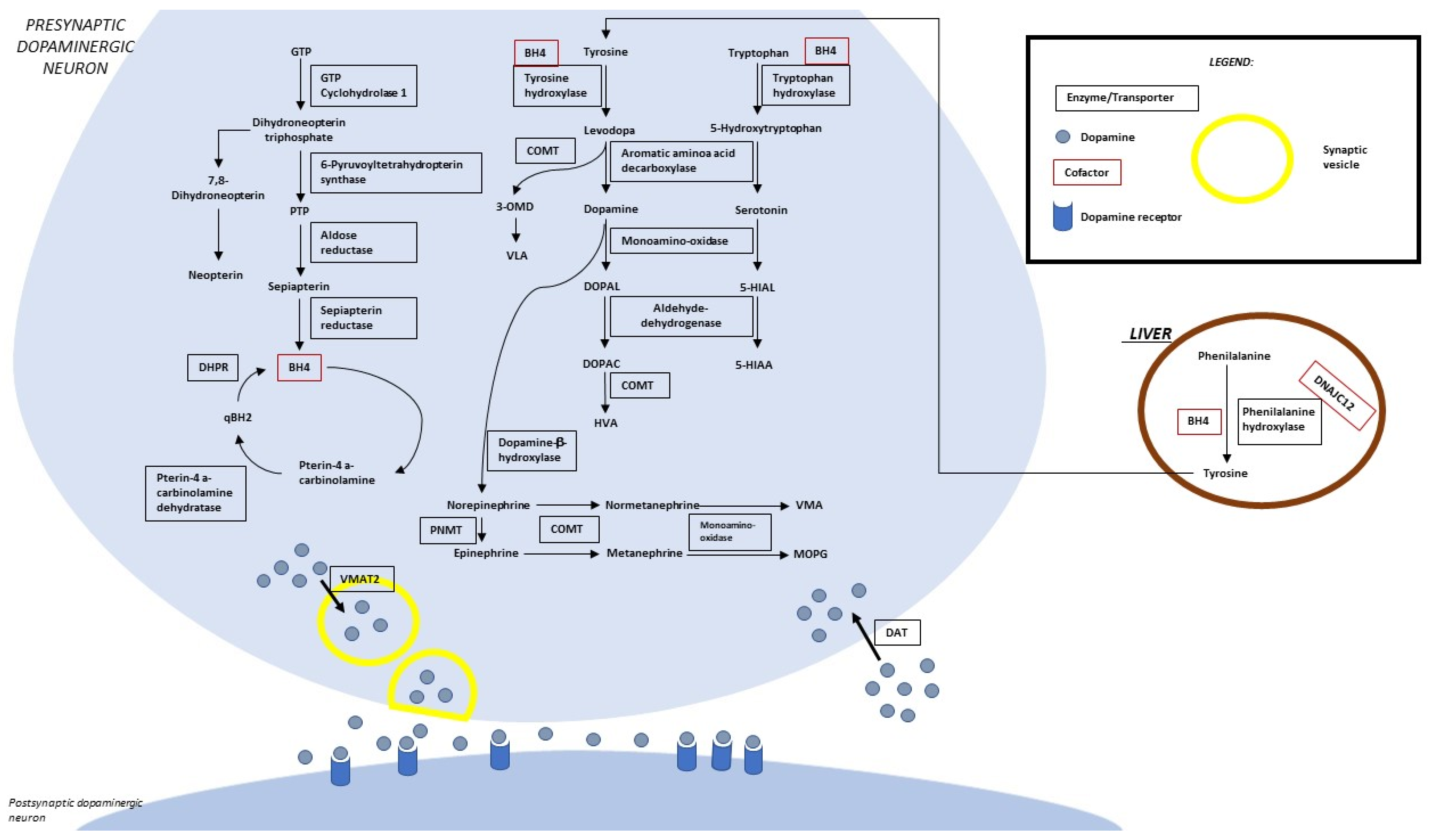
Figure 1. Biogenic amine and pterin metabolism. LEGEND: 5-HIAA, 5-hydroxyindoleacetic acid; 5-HIAL, 5-hydroxyindoleacetaldehyde; BH4, tetrahydrobiopterin; DHPR, dihydropterine reductase; DOPAC, 3,4-dihydroxyphenylacetic acid; DOPAL, 3,4-dihydroxyphenylacetaldehyde; DAT, dopamine transporter; GTP, guanosine-5′-triphosphate; HVA, homovanillic acid; MOPG, methoxylhydroxyphenylglycol; PLP, pyridoxal 5′-phosphate; PNMT, phenylethanolamine N-methyltransferase; PTP, 6-pyruvoyltetrahydropterin; qBH2, quinonoid dihydrobiopterin; VLA, vanillyllactic acid; VMA, vanillylmandelic acid; VMAT 2, vesicular monoamine transporter.
Several peculiarities justify the interest in these conditions: (a) as a group, they are among the most frequent genetic causes of movement disorders in children [1][2][3][4]; (b) many of them are treatable conditions, i.e., early therapy can bias the natural history of the disease, preventing severe neurological disabilities characterizing untreated patients [1][2][3][4]; (c) they have revealed the crucial function of serotonin and dopamine for the normal development of the central nervous system [1][2][3][4]. On clinical grounds, despite their relative rarity, each disease shares a unique pattern of recurrent clinical signs caused by dopamine and serotonin deficiency occurring in the immature brain [1][2][3][4]. From a biochemical viewpoint, they are considered a very early onset form of parkinsonism and parkinsonism-dystonia [1][2][3][4].
2. Epidemiology
A reliable estimate of the frequency of this set of conditions is available for hyperphenylalaninemia-associated disorders, which are intercepted by neonatal screening programs for phenylketonuria (autosomal recessive DYT/PARK-GCH1, DYT/PARK-PTS, DYT/PARK-QDPR, and DNAJC12-related disorders), all including 2–3% of the overall number of patients with neonatal hyperphenylalaninemia (http://www.biopku.org/; last access 7 October 2022).
The first reported prevalence of autosomal dominant DYT/PARK-GCH1 was approximately 0.5–1.0 per million, with a remarkably higher penetrance of DYT/PARK-GCH1 variant carriers among females (87% vs. 38% in males) [5][6]. A more recent Serbian study estimated higher figures up to a prevalence of 2.96 per million [5][7]. A much lower prevalence of 1.4 per 100,000 patients under 18 was estimated in Estonia [8]. Autosomal recessive DYT/PARK-GCH1 probably affects less than 1 per 1,000,000 patients [3].
No adequate epidemiological data are available for other primary biogenic amine disorders. An indirect estimate of their frequency can be drawn on the basis of cases entered in the International Working Group on Neurotransmitter Related Disorders (iNTD patient registry (http://intd-online.org, last access 7 October 2022), which collected 350 patients with disorders of biogenic amine metabolism: 161 patients with BH4 deficiency, 56 patients with DYT/PARK-PTS, 37 patients with DYT/PARK-QDPR, 36 patients with autosomal dominant DYT/PARK-GCH1, 18 patients with autosomal recessive DYT/PARK-GCH1, 14 patients with DYT/PARK-SR, 131 patients with DYT-DDC, 44 patients with DYT/PARK-TH, 5 with DYT/PARK-SLC6A3, 5 with DNAJC12 deficiency, and 4 patients with MAO A deficiency [2].
The prevalence of DYT-DDC was also studied through other indirect evaluations [9][10]. The frequency analysis of 216 known pathogenic DDC variants (19 homozygous and 39 compound heterozygous) suggested a global prevalence of 1800 living patients with DYT-DDC [9]. An estimated prevalence of 1/42,000 live births per year was calculated for the same disease by analyzing biological samples from 19,684 American patients with neurological disorders of unknown origin [10].
3. Clinical Presentation
Table 1 summarizes the main clinical features of inherited biogenic amine neurotransmitter metabolism disorders. The onset of symptoms is in the first months or year of life for almost all these conditions, with two extremes represented by DYT/PARK-PTS, including the first possible manifestations also in the fetal life, and by autosomal dominant DYT/PARK-GCH1, with a prominent later presentation during school age or later [1][2][4]. An increased rate of prematurity or very low birth weight and congenital microcephaly was observed in DYT/PARK-PTS, DYT/PARK-TH, and DYT-DDC [2][11]. Exceptional late-onset presentations in adulthood have been reported for DYT/PARK-GCH1, DYT/PARK-SR, DYT/PARK-TH, DNAJC12, and DYT/PARK-SLC6A3 [2].
The pattern of neurological impairment, the age at the onset of symptoms, and their severity are usually correlated with the severity of biogenic amine depletion. The earlier the onset of the disease, the more severe and pervasive the neurological impairment is, resulting in a neurodevelopmental delay leading to intellectual disability, behavioral problems, and movement disorders [2][11][12][13]. Speech and language impairment are typical clinical traits of late or untreated conditions and, similar to other higher cortical functions, they respond less than movement disorders to pharmacological treatments [2]. A more severe cognitive impairment has been reported in patients with DYT/PARK-SR, DYT/PARK-TH, and DYT-DDC [11][12].
The onset of symptoms after three is usually associated with the selective impairment of motor functions, as usually happens for autosomal dominant DYT/PARK-GCH1 [13]. An actual neurological regression was reported for DYT/PARK-SLC6A3 only, miming levodopa unresponsive degenerative parkinsonism [1].
The individual response to dopaminergic and serotoninergic treatment [1][13] may represent an additional source of clinical variability.
Movement disorders are a significant part of the clinical spectrum of inherited disorders of biogenic amine metabolism, both as an isolated disorder or in the context of infantile encephalopathy [13].
A remarkable source of complication for their management includes the lack of consensus for the classification of movement disorders in children under 3 years of life and the relevant differences between their clinical presentations and those typical of adult-onset parkinsonism [13].
Initial presentations may include rigidity of limbs and trunk hypotonia in patients with a global delay in developmental milestones [13]. Hypotonia is often associated with a dorsal trunk extension that might be considered a dystonic opisthotonus or an abnormal persistence of the fetal righting reflex [13].
In older children or adolescents, lead-pipe rigidity may often be combined with bradykinesia or focal arrhythmic jerks [13]. Akinesia and amimia may be difficult to assess in children because of higher interindividual variability of spontaneous motor activity and mimic expressions during the first months and years of life [13]. Classic akinetic-rigid syndrome may be preceded by a delay in antigravity motor development or pathological postural patterns [13]. Rest tremors are sporadic in early infancy. The whole semiology of rhythmic or pseudo-rhythmic oscillatory involuntary movements in infants cannot be categorized according to the tremor classification of tremors in adults [13].
References
- Ng, J.; Papandreou, A.; Heales, S.J.; Kurian, M.A. Monoamine neurotransmitter disorders—Clinical advances and future perspectives. Nat. Rev. Neurol. 2015, 11, 567–584.
- Kuseyri Hübschmann, O.; Horvath, G.; Cortès-Saladelafont, E.; Yıldız, Y.; Mastrangelo, M.; Pons, R.; Friedman, J.; Mercimek-Andrews, S.; Wong, S.N.; Pearson, T.S.; et al. Insights into the expanding phenotypic spectrum of inherited disorders of biogenic amines. Nat. Commun. 2021, 12, 5529.
- Brennenstuhl, H.; Jung-Klawitter, S.; Assmann, B.; Opladen, T. Inherited Disorders of Neurotransmitters: Classification and Practical Approaches for Diagnosis and Treatment. Neuropediatrics 2019, 50, 2–14.
- Kurian, M.A.; Gissen, P.; Smith, M.; Heales, S., Jr.; Clayton, P.T. The monoamine neurotransmitter disorders: An expanding range of neurological syndromes. Lancet Neurol. 2011, 10, 721–733.
- Kikuchi, A.; Takeda, A.; Fujihara k Kimpara, T.; Shiga, Y.; Tanji, H.; Nagai, M.; Ichinose, H.; Urano, F.; Okamura, N.; Arai, H.; et al. Arg(184)His mutant GTP cyclohydrolase I, causing recessive hyperphenylalaninemia, is responsible for dopa-responsive dystonia with parkinsonism: A case report. Mov. Disord. 2004, 19, 590–593.
- Himmelreich, N.; Blau, N.; Thöny, B. Molecular and metabolic bases of tetrahydrobiopterin (BH4) deficiencies. Mol. Genet. Metab. 2021, 133, 123–136.
- Dobricic, V.; Tomic, A.; Brankovic, V.; Kresojevic, N.; Jankovic, M.; Westenberger, A.; Rasic, V.M.; Klein, C.; Novakovic, I.; Svetel, M.; et al. GCH1 mutations are common in Serbian patients with dystonia-parkinsonism: Challenging previously reported prevalence rates of DOPA-responsive dystonia. Park. Relat. Disord. 2017, 45, 81–84.
- Talvik, I.; Segawa, M.; Veri, K.; Gross-Paju, K.; Talvik, T. Cases of dopa-responsive dystonia (Segawa disease) in Estonia. Brain Dev. 2010, 32, 428–431.
- Hyland, K.; Peters, M.; Schu, M.; Erickson, S.; Croxford, J.; Whitehead, N. Estimated prevalence of aromatic l-amino acid decarboxylase (AADC) deficiency in the United States, European Union, and Japan. Hum. Gene Ther. 2018, 29, A131.
- Hyland, K.; Reott, M. Prevalence of Aromatic l-Amino Acid Decarboxylase Deficiency in At-Risk Populations. Pediatr. Neurol. 2020, 106, 38–42.
- Pearson, T.S.; Gilbert, L.; Opladen, T.; Garcia-Cazorla, A.; Mastrangelo, M.; Leuzzi, V.; Tay, S.K.H.; Sykut-Cegielska, J.; Pons, R.; Mercimek-Andrews, S.; et al. AADC deficiency from infancy to adulthood: Symptoms and developmental outcome in an international cohort of 63 patients. J. Inherit. Metab. Dis. 2020, 43, 1121–1130.
- Keller, M.; Brennenstuhl, H.; Kuseyri Hübschmann, O.; Manti, F.; Julia Palacios, N.A.; Friedman, J.; Yıldız, Y.; Koht, J.A.; Wong, S.N.; Zafeiriou, D.I.; et al. Assessment of intellectual impairment, health-related quality of life, and behavioral phenotype in patients with neurotransmitter related disorders: Data from the iNTD registry. J. Inherit. Metab. Dis. 2021, 44, 1489–1502.
- Mastrangelo, M.; Carducci, C.; Carducci, C.; Leuzzi, V. The Spectrum of Early Movement Disorders in Congenital Defects of Biogenic Amine Metabolism. J. Pediatr. Neurol. 2015, 13, 213–224.
More
Information
Subjects:
Others
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
719
Revisions:
2 times
(View History)
Update Date:
30 Jan 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No