Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Archittapon Nokkeaw | -- | 2806 | 2023-01-29 11:58:38 | | | |
| 2 | Dean Liu | -1 word(s) | 2805 | 2023-01-30 01:45:09 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nokkeaw, A.; Thamjamrassri, P.; Tangkijvanich, P.; Ariyachet, C. CircRNAs and Hepatic Stellate Cell Activation. Encyclopedia. Available online: https://encyclopedia.pub/entry/40521 (accessed on 26 July 2026).
Nokkeaw A, Thamjamrassri P, Tangkijvanich P, Ariyachet C. CircRNAs and Hepatic Stellate Cell Activation. Encyclopedia. Available at: https://encyclopedia.pub/entry/40521. Accessed July 26, 2026.
Nokkeaw, Archittapon, Pannathon Thamjamrassri, Pisit Tangkijvanich, Chaiyaboot Ariyachet. "CircRNAs and Hepatic Stellate Cell Activation" Encyclopedia, https://encyclopedia.pub/entry/40521 (accessed July 26, 2026).
Nokkeaw, A., Thamjamrassri, P., Tangkijvanich, P., & Ariyachet, C. (2023, January 29). CircRNAs and Hepatic Stellate Cell Activation. In Encyclopedia. https://encyclopedia.pub/entry/40521
Nokkeaw, Archittapon, et al. "CircRNAs and Hepatic Stellate Cell Activation." Encyclopedia. Web. 29 January, 2023.
Copy Citation
Chronic liver injury induces the activation of hepatic stellate cells (HSCs) into myofibroblasts, which produce excessive amounts of extracellular matrix (ECM), resulting in tissue fibrosis.
non-coding RNA
circular RNA
microRNA
1. Introduction
HSC activation is associated with several cytokines and regulatory networks and promotes expression of HSC activation markers, including α-smooth muscle actin (α-SMA) and type I collagen [1]. Many signaling pathways have been linked to increasing expression levels of α-SMA and/or type I collagen, including TGF-β [2], JAK/STAT [3], PDGF [4], PI3K/Akt [5], Wnt/β-catenin [6], Notch [7], Hedgehog [8][9], Hippo [10], and inflammasome signaling pathways [11]. Thus, circRNAs targeting these signaling pathways can regulate HSC activation and ultimately affect liver fibrosis (Figure 1 and Figure 2).
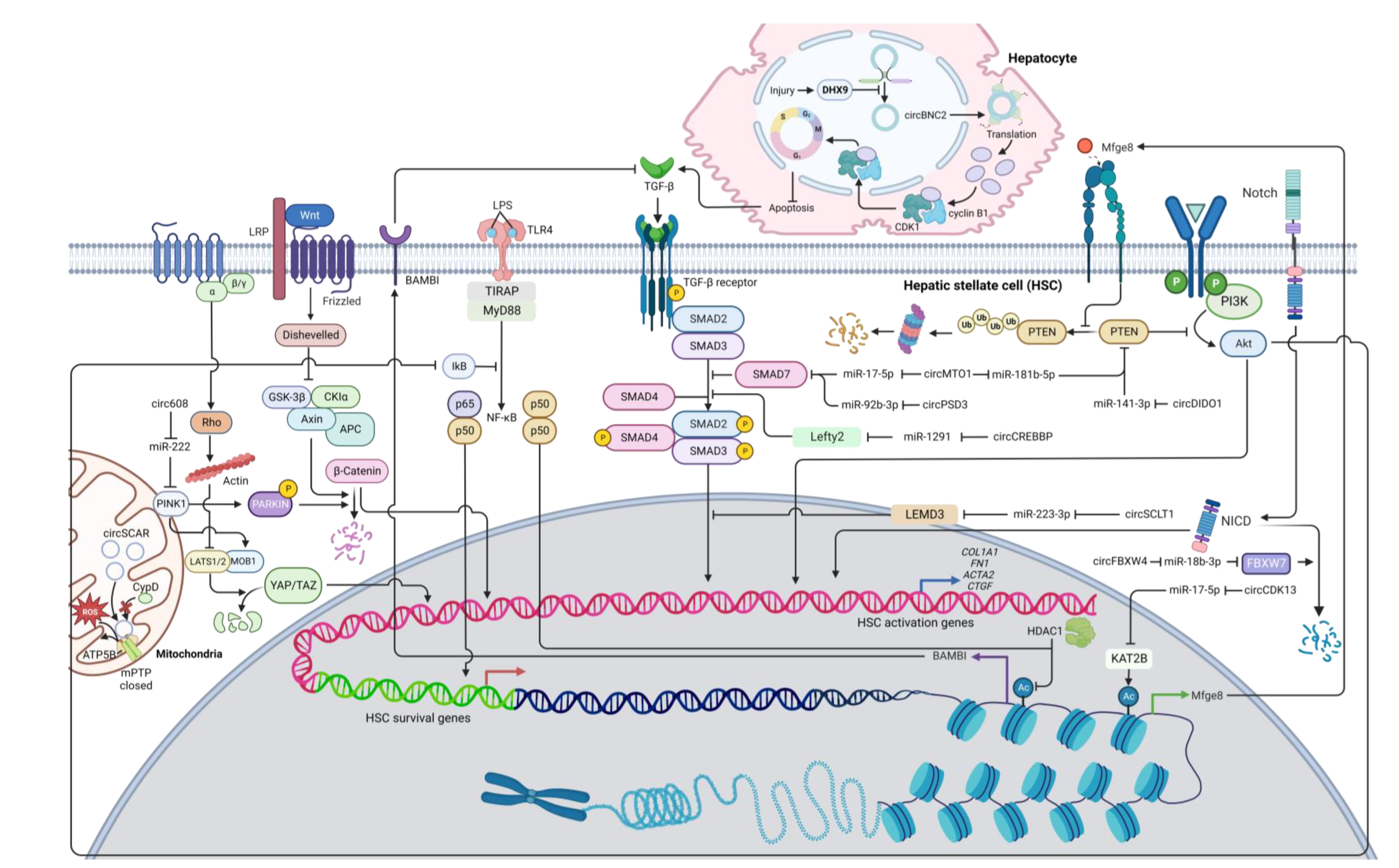
Figure 1. A diagram illustrating the regulatory networks of HSC activation modulated by anti-fibrotic circRNAs. Arrows represent activation, whereas bars symbolize inhibition. Abbreviation: COL1A1: collagen type I alpha 1 chain; CypD: Cyclophilin D; FBXW7: F-box/WD repeat-containing protein 7; KAT2B: lysine acetyltransferase 2B; LEMD3: LEM domain containing 3; Lefty2: left-right determination factor 2; mPTP: mitochondrial permeability transition pore; PINK1: PTEN-induced kinase 1; PTEN: phosphatase and tensin homolog; TGF-β: transforming growth factor beta; TLR4: toll-like receptor 4.
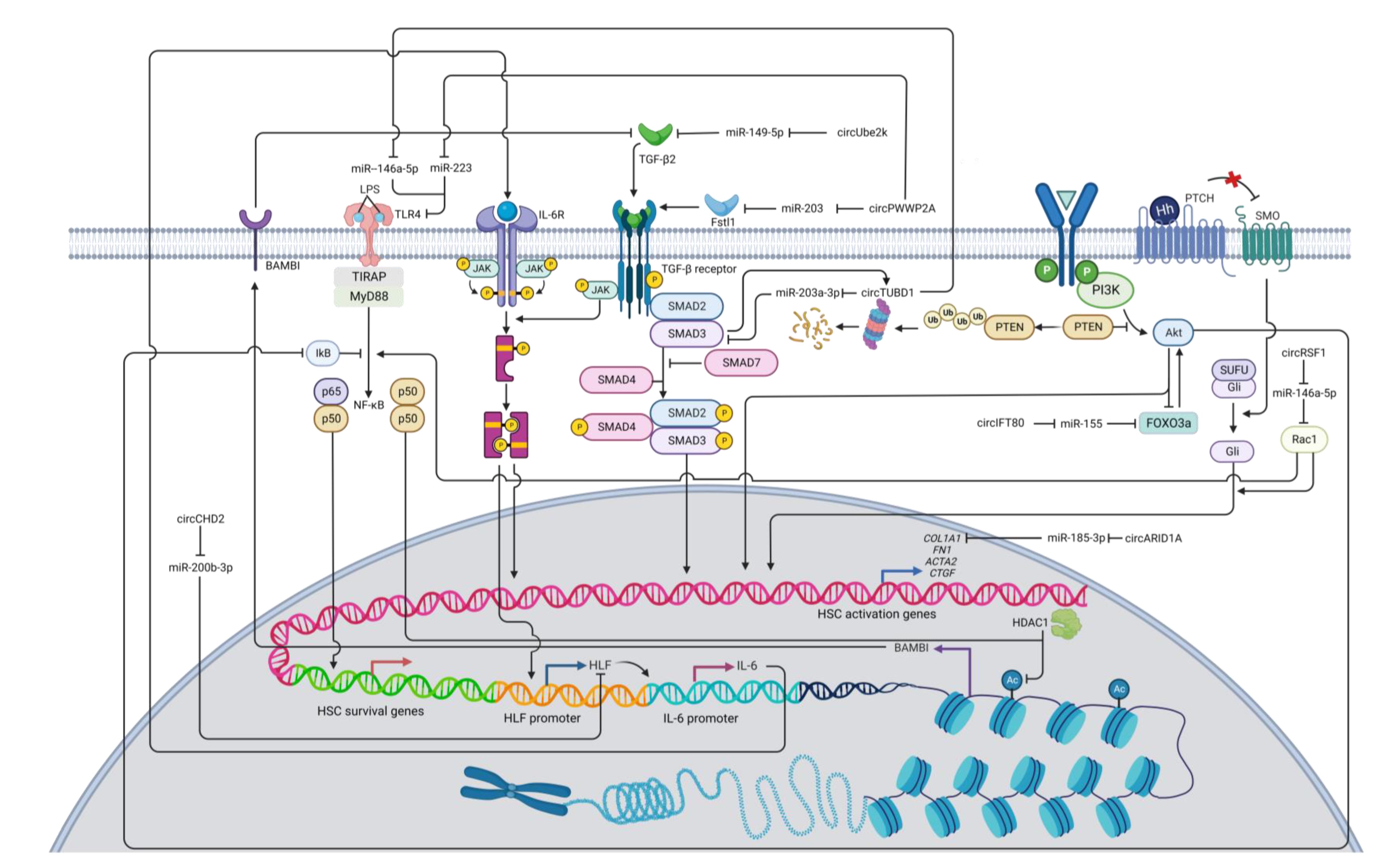
Figure 2. A diagram illustrating the regulatory networks of HSC activation modulated by pro-fibrotic circRNAs. Arrows represent activation, whereas bars symbolize inhibition. Abbreviation: COL1A1: collagen type I alpha 1 chain; HLF: hepatic leukemia factor; HDAC1: histone deacetylase 1; IL-6: interleukin 6; PTEN: phosphatase and tensin homolog; RAC1: Ras-related C3 botulinum toxin substrate 1; TGF-β: transforming growth factor beta; TLR4: toll-like receptor 4.
2. Anti-Fibrotic circRNAs
TGF-β signaling is regarded as the primary fibrogenic pathway that stimulates HSC activation and ECM synthesis [2]. TGF-β is found in trace amounts in healthy livers [12]. Following liver damage, macrophages start producing TGF-β and PDGF, which can activate excessive ECM production from HSCs and lead to the development of liver fibrosis [13]. Most reported circRNAs involved in HSC activation target miRNAs and proteins in the TGF-β pathway. One of these circRNAs is circPSD3 (mmu_circ_0001682), which is downregulated in primary HSCs and liver tissues from mice with carbon tetrachloride (CCl4)-induced liver fibrosis [14]. CircPSD3 can function as a miR-92b-3p sponge, consequently promoting Smad7 expression [14]. Smad7 can block the activation of receptor-regulated Smads (R-Smads) and thus inhibits the TGF-β signaling pathway [15][16]. Furthermore, circPSD3 can preclude HSC proliferation and forestall fibrosis progression in vivo [14].
CircCREBBP acts as a miR-1291 sponge, consequently increasing the expression of left-right determination factor 2 (LEFTY2) [17], which can inhibit the phosphorylation of Smad2/3 [18][19], a key signaling molecule in the TGF-β pathway [20]. Moreover, circCREBBP can also inhibit cell proliferation and arrest the cell cycle in HSCs [17]. With a similar mode of action, hsa_circ_0070963, also known as circSCLT1, has the ability to sponge miR-223-3p, which can target LEM domain containing 3 (LEMD3) [21], an inhibitory molecule that can antagonize Smad2/3 signaling and perturb the TGF-β signaling pathway [22]. In addition to suppressing HSC activation, the overexpression of circSCLT1 can induce cell cycle arrest and suppress cell proliferation in HSCs [21]. However, the circSCLT1/miR-223 axis is subject to further investigation as most studies suggest the anti-fibrotic roles of miR-223 in various fibrosis models [23][24][25][26]. Another circRNA is mmu_circ_34116, which is shown to suppress HSC activation upon overexpression [27]. Using bioinformatic techniques, mmu_circ_34116 is predicted to target the miR-22-3p/BMP7 axis [27]. Bone morphogenetic protein 7 (BMP7) can activate Smad1/5/8 but inhibits Smad3 activation, thus antagonizing TGF-β signaling [28][29].
CircMTO1 has been extensively studied for their anti-fibrotic roles. Its expression levels are observed to downregulate in liver tissues of fibrosis patients [30]. Patients with higher liver fibrosis stages have significantly less circMTO1 expression [30]. By interacting with miR-17-5p, circMTO1 can positively regulate the expression of Smad7 and thereby negatively regulate the TGF-β signaling pathway [30]. Moreover, circMTO1 is found to act as a miR-181b-5p sponge [31]. miR-181b-5p can target phosphatase and tensin homolog (PTEN), a negative regulator in the phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) signaling pathway [31], which promotes HSC activation [5][32]. Therefore, circMTO1 plays an anti-fibrotic role by downregulating miR-181b-5p and enhancing PTEN activity [31]. Additionally, Akt signaling can also lead to the activation of NF-κB [33], protecting activated HSCs against apoptosis and sustaining cell survival [34]. As a consequence, by suppressing PI3K/Akt signaling via PTEN, circMTO1 overexpression potentially enhances HSC apoptosis and diminishes ECM production.
Some circRNAs play a significant role in other liver cell types but can indirectly have an impact on HSC activation. For example, in normal hepatocytes, circBNC2 expression is high but drastically decreased upon liver injury [35]. Interestingly, expression of activation markers (α-SMA and type I collagen) is significantly increased in HSCs incubated with conditional medium from circBNC2 knockout hepatocytes, which appear to contain high levels of the pro-fibrotic cytokines including TGF-β [35]. In contrast, the overexpression of circBNC2 in hepatocytes can reduce expression levels of these cytokines upon liver injury, and conditional medium from these hepatocytes can suppress expression of α-SMA and type I collagen in cultured HSCs, indicating the anti-fibrotic roles of circBNC2 [35]. Mechanistically, circBNC2 contains an open reading frame (ORF) and an IRES, suggesting their function as a protein template [35]. The circBNC2-translated protein (ctBNC2) is a protein product derived from circBNC2 translation [35]. This 681-amino acid protein can bind to CDK1 and cyclin B1 and promote CDK/cyclin complex translocation into the nucleus, a critical step to initiate mitosis and prevent apoptosis [35]. As ctBNC2 levels decrease in a damaged liver, the translocation of the CDK/cyclin complex could be impaired and induce apoptosis, which triggers the secretion of TGF-β and DAMPs from hepatocytes [36][37]. As previously mentioned, these molecules can activate ECM production from HSCs and promote fibrosis progression. Therefore, by protecting hepatocytes from apoptosis upon injury, circBNC2 could play an anti-fibrotic role by suppressing production of inflammatory cytokines [35].
Another key signaling pathway that involves HSC activation is the network of PTEN/PI3K/Akt [5]. PI3K/Akt signaling can activate the serine/threonine kinase p70 ribosomal protein S6 kinase (p70S6K) via the mammalian target of rapamycin complex 1 (mTORC1), whereas PTEN inhibits activation of PI3K/Akt signaling [5][38]. Evidently, p70S6K facilitates HSC proliferation and collagen expression [38], and thus, the PI3K/Akt signaling pathway is considered pro-fibrotic [5]. A recent report shows that circDIDO1 inhibits HSC activation through the miR-141-3p/PTEN axis [39]. The overexpression of circDIDO1 sponges miR-141-3p and increases PTEN expression, which impairs PI3K/Akt signaling and subsequently suppresses HSC activation [39]. Similarly, circCDK13 forestalls liver fibrosis by repressing the activation and proliferation of HSCs via the miR-17-5p/KAT2B/MFGE8/PTEN axis [40]. By sponging miR-17-5p, circCDK13 can promote the expression of lysine acetyltransferase 2B (KAT2B), a protein that can regulate histone protein acetylation [40]. The acetylation of histones can enhance the accessibility of transcriptional factors to DNA templates, thereby increasing transcriptional activity [41]. Specifically, KAT2B facilitates milk fat globulin-epidermal growth factor 8 (MFGE8) transcription by promoting histone H3 acetylation [40]. Because MFGE8 can inhibit PTEN ubiquitination and degradation [42], KAT2B promotes PTEN stability, which in turn suppresses the PI3K/Akt signaling pathway [40]. Inactivation of Akt can also lead to the reduction of NF-κB activity, thus impairing the survival of fibrogenic HSCs [40][43]. Collectively, circDIDO1 and circCDK13 can exert the anti-fibrotic mechanism via suppression of PI3K/Akt signaling.
In addition to TGF-β and PI3K/Akt signaling pathways, Wnt/β-catenin and hippo signaling could be regulated by circRNAs in HSCs. For example, circ608 increases PTEN-induced kinase 1 (PINK1) expression through sponging miR-222 [44]. PINK1 can then phosphorylate Parkin, an E3 ubiquitin-protein ligase, which in turn triggers ubiquitination and degradation of β-catenin, thereby inhibiting Wnt/β-catenin transduction [45][46]. PINK1 can also promote expression of Mps one binder kinase activator 1B (MOB1B) [47], a regulator of large tumor suppressor kinases 1/2 (LATS1/2) [48]. The MOB1/LATS complex can inactivate YAP and TAZ [48], the effectors in the Hippo signaling pathway [49], therefore inhibiting the Hippo signaling cascade and subsequently HSC activation. PINK1/Parkin can promote mitophagy [47], which is suppressed in fibrogenic HSCs [50]. Taken together, circ608 is an anti-fibrotic mediator via PINK1-mediated suppression of Wnt/β-catenin and hippo signaling.
For the Notch signaling pathway, circFBXW4 positively regulates F-box/WD repeat-containing protein 7 (FBXW7) [51], which induces Notch intracellular domain (NICD) degradation [52]. By interacting with miR-18b-3p, circFBXW4 can upregulate FBXW7 expression and inhibit Notch signaling, thereby inhibiting HSC activation and proliferation as well as promoting apoptosis [51].
A mitochondrial circRNA called circSCAR is found to be downregulated in NASH patients [53]. Lipid accumulation can induce endoplasmic reticulum (ER) stress, thus increasing the expression of the ER stress mediator CCAAT-enhancer-binding protein homologous protein (CHOP) [53]. CHOP can inhibit the expression of PGC-1α, which positively regulates circSCAR transcription and thus decreases circSCAR levels [53]. CircSCAR can interact with ATP5B, a regulator of the mitochondrial permeability transition pore (mPTP) [53]. Binding between circSCAR and ATP5B hinders the opening of mPTP and consequently the efflux of reactive oxygen species (ROS) from mitochondria into the cytosol [53]. Previous studies suggest that ROS promotes NF-κB signaling pathway and HSC activation [54][55]. Therefore, circSCAR may play an anti-fibrotic role by preventing leakage of ROS from mitochondria [53].
Some circRNAs are found to suppress HSC activation, but their underlying mechanisms are not fully understood. One example is hsa_circ_0004018 or circSMYD4, which is proposed to suppress HSC activation and proliferation via miR-660-3p [56]. By using bioinformatics tools, telomerase-associated protein 1 (TEP1) is predicted to be a target of miR-660-3p and experimentally confirmed by a luciferase assay [56]. While functions of TEP1 in HSCs have not been reported, higher levels of TEP1 in hepatocytes can indirectly suppress the proliferation and activation of HSCs, potentially because TEP1 prevents formation of critically short telomeres and reduces DNA-damage response, which normally triggers hepatocyte senescence and sends apoptotic signals to activate HSCs [56][57]. Intriguingly, although TEP1 play a role in telomere length regulation in many cell types [58][59], a study shows that TEP1 is not essential for telomere length regulation in murine liver [60]. This finding suggests the roles of TEP1 in other cellular functions that could modulate HSC activation [60]. In addition to being a component of the telomerase complex, TEP1 has been found in vault ribonucleoprotein complexes (VRCs) [61]. Although the function of VRCs in HSC activation is not yet known, major vault protein knock-out mice can intensify hepatic steatosis and induce fibrotic responses [62]. Further investigations are needed to expand the knowledge of how the circSMYD4/miR-660-3p/TEP1 axis inhibits HSC activation. Other examples are hsa_circ_0089761 and hsa_circ_0089763, which are two mitochondrial-encoded circRNAs downregulated in HSCs during LPS stimulation [63]. Their downregulation indicates their potential roles as anti-fibrotic circRNAs, but their mechanisms and targets remain to be further studied. Lastly, in a hepatitis B virus (HBV)-induced activation model of LX-2 cells, circMTM1 is upregulated along with interleukin 7 receptor (IL7R), potentially via absorbing miR-122-5p [64]. It is shown that circMTM1 knockdown expression and miR-122-5p overexpression reduce the expression levels of activated HSC markers, while the upregulation of IL7R attenuates the anti-fibrotic function of miR-122-5p [64]. Normally, IL7R is expressed in T cells and plays a pivotal role in T cell homeostasis [65]. Despite interesting results, the exact roles of circMTM1 and IL7R in HSC functions need a further investigation.
3. Pro-Fibrotic circRNAs
While most reported circRNAs impedes the activation of TGF-β signaling pathway in HSCs, some circRNAs play the opposite roles. These circRNAs include circPWWP2A [66], circUBE2k [67], and circTUBD1 [68][69]. CircPWWP2A can enhance TGF-β signaling by decreasing miR-203 and miR-223 expression and thus increasing expression levels of follistatin-like 1 (Fstl1) and Toll-like receptor 4 (TLR4), respectively [66]. Fstl1 is a glycoprotein ligand and can promote TGF-β signaling by binding to TGF-β1 type II receptors (TβRII) [70][71]. TLR4 can recognize lipopolysaccharide (LPS) and sequentially trigger signaling cascades that produce the NF-κBp50 homodimer to interact with histone deacetylase 1 (HDAC1). This protein complex can transcriptionally suppress BMP and activin membrane-bound inhibitor (BAMBI), a pseudoreceptor of TGF-β [66][72][73]. Since BAMBI can inhibit TGF-β/Smad signaling [72], TLR4-mediated downregulation of BAMBI can induce fibrotic HSCs via amplification of TGF-β signaling [66]. In addition, TLR4 can activate NF-κB to support the survival of the activated HSCs [74][75]. Together, circPWWP2A can promote HSC activation via sponging miRNAs that suppress the expression of Fstl1 and TLR4.
The overexpression of circUBE2K induces liver fibrosis by sponging miR-149-5p, which negatively regulates expression of TGF-β2 [67]. Conversely, inhibiting circUBE2K expression can impair TGF-β signaling and induce cell cycle arrest in HSCs, confirming the pro-fibrotic roles of circUBE2K in liver fibrosis progression [67]. Finally, circTUBD1 can interact with both miR-203a-3p and miR-146a-5p [68][69]. By sponging miR-203a-3p, circTUBD1 can positively regulate TGF-β signaling by increasing Smad3 expression [68]. Moreover, Smad3 is found to positively regulate circTUBD1 via a feedback loop, thus further enhancing Smad3 expression [68]. TLR4 is targeted by miR-146a-5p, and by downregulating miR-146a-5p via the circTUBD1 sponge, expression of TLR4 increases, thus promoting TGF-β and NF-κB signaling pathways, which can help activate and support cell survival of HSCs, respectively [69][72][74][75][76]. Furthermore, circTUBD1 can also promote HSC proliferation and inhibit the apoptosis of HSCs by increasing the expression of the anti-apoptotic protein B-cell lymphoma-2 (BCL-2), thereby amplifying the population of fibrogenic cells [69].
Interleukin-6 (IL-6) can activate HSC through the MAPK and JAK/STAT signaling pathways [3]. CircCHD2 can enhance hepatic leukemia factor (HLF) expression by interacting with miR-200b-3p [77]. HLF can promote the expression of IL-6 that can bind to the IL-6 receptor and activate the JAK/STAT3 pathway [78]. The JAK/STAT pathway is also a part of a non-SMAD pathway of TGF-β signal transduction that is essential for HSC activation [79]. The JAK/STAT3 pathway also promotes HLF expression, thereby regulating signal transduction in a feed-forward circuit manner [78].
Additionally, hsa_circ_0071410 (circPALLD) and hsa-circ-0067835 (circIFT80) are pro-fibrotic circRNAs that promote the PI3K/Akt signaling pathway [80][81][82]. CircPALLD can induce HSC activation and increase cell viability by interacting with miR-9-5p [81], which is reported to target annexin A2 (ANXA2) [82]. ANXA2 has been considered to play a role in HSC activation, proliferation, and apoptosis [82], possibly by activating the FAK/PI3K/Akt signaling pathway [83]. CircIFT80 can activate HSC by regulating the miR-155/Forkhead box O3 (FOXO3)/Akt axis [80][84]. As CircIFT80 alleviates FOXO3 expression by sponging miR-155, this transcription factor can transactivate PI3K/Akt through a positive feedback loop and promote PI3K/Akt signal transduction in fibrogenic HSCs [85][86].
The Hedgehog signaling pathway plays a significant role in HSC activation [8][9]. A recent study shows that circRSF1 sponges miR-146a-5p. Subsequently, this sequestration promotes Ras-related C3 botulinum toxin substrate 1 (RAC1) expression and Hedgehog signal transduction, resulting in HSC activation and proliferation [87]. Rac1 has been shown to be involved in the Hedgehog signaling pathway [88]. Activated Rac1 induces glioma-associated oncogene (Gli) nuclear translocation, which is necessary for the Hedgehog signaling pathway [89]. Rac1 can also influence NF-κB and JNK signaling by promoting their transduction [90][91]. JNK signal transduction can phosphorylate Smad2 at the C-terminal and linker regions [92]. Initial findings reveal that phosphorylation of Smad2 in the linker region hinders its nuclear translocation and cellular signaling, but recent evidence shows that phosphorylation of the Smad linker can stimulate expression of fibrotic genes [92][93]. One study discovers that the phosphorylated Smad2 linker region is associated with increased expression of glycosaminoglycans (GAG) [94], and hyaluronan (HA), a class of GAG, is found to be able to activate HSCs via Notch1 [95], so the phosphorylation of the Smad2 linker region may be associated with HSC activation by increasing the expression of HA and promoting the Notch1 fibrogenic signaling pathway. However, this proposed mechanism of circRSF1/miR-146a-5p/Rac1 with subsequent signal transductions remains to be further explored.
Moreover, the direct regulation of circRNAs on fibrotic genes have been reported. For instance, the circARID1A/miR-185-3p axis post-transcriptionally regulates expression of COL1A1, a key marker of HSC activation [96]. The pro-fibrotic circARID1A can also promote the proliferation and migration of HSCs as well as inhibit their apoptosis [96]. Mechanistically, increased type I collagen expression can lead to a positive feedback loop of HSC activation in which accumulation of collagen further activates HSCs by increasing ECM stiffness [97]. This mechanical tension in turn leads to YAP activation in the Hippo signaling pathway and Akt activation in the PI3K/Akt signaling pathway [97][98].
The above discussion mainly focuses on the intrinsic expression of circRNAs in HSCs as most in vitro studies rely on the single cell type for their analysis. However, apart from HSCs, liver is a complex organ comprising of several cell types including hepatocytes, Kupffer cells, liver sinusoidal endothelial cells, and cholangiocytes, which are known to communicate with one another to maintain liver functions [99]. Some of these cells were reported to modulate liver fibrosis by transferring circRNAs to HSCs such as hepatocyte-derived circBNC2 [35]. Alternatively, circRNAs that regulate the production of inflammatory cytokines in Kupffer cells can have an indirect impact on the state of HSC activation. These circRNAs include circMcph1 [100][101] and circ1639 [102][103][104][105]. Nevertheless, a study of cell-cell interaction in mediating liver fibrosis through circRNAs is still lacking and need to be further investigated.
References
- Bennett, R.G.; Kharbanda, K.K.; Tuma, D.J. Inhibition of markers of hepatic stellate cell activation by the hormone relaxin. Biochem. Pharmacol. 2003, 66, 867–874.
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419.
- Kagan, P.; Sultan, M.; Tachlytski, I.; Safran, M.; Ben-Ari, Z. Both MAPK and STAT3 signal transduction pathways are necessary for IL-6-dependent hepatic stellate cells activation. PLoS One 2017, 12, e0176173.
- Park, H.-J.; Kim, H.-G.; Wang, J.-H.; Choi, M.-K.; Han, J.-M.; Lee, J.-S.; Son, C.-G. Comparison of TGF-β, PDGF, and CTGF in hepatic fibrosis models using DMN, CCl4, and TAA. Drug Chem. Toxicol. 2016, 39, 111–118.
- Cai, C.X.; Buddha, H.; Castelino-Prabhu, S.; Zhang, Z.; Britton, R.S.; Bacon, B.R.; Neuschwander-Tetri, B.A. Activation of Insulin-PI3K/Akt-p70S6K Pathway in Hepatic Stellate Cells Contributes to Fibrosis in Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2017, 62, 968–978.
- Ge, W.-S.; Wang, Y.-J.; Wu, J.-X.; Fan, J.-G.; Chen, Y.-W.; Zhu, L. β-catenin is overexpressed in hepatic fibrosis and blockage of Wnt/β-catenin signaling inhibits hepatic stellate cell activation. Mol. Med. Rep. 2014, 9, 2145–2151.
- Bansal, R.; van Baarlen, J.; Storm, G.; Prakash, J. The interplay of the Notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis. Sci. Rep. 2015, 5, 18272.
- Sicklick, J.K.; Li, Y.-X.; Choi, S.S.; Qi, Y.; Chen, W.; Bustamante, M.; Huang, J.; Zdanowicz, M.; Camp, T.; Torbenson, M.S.; et al. Role for Hedgehog signaling in hepatic stellate cell activation and viability. Lab. Investig. 2005, 85, 1368–1380.
- Xie, G.; Karaca, G.; Swiderska-Syn, M.; Michelotti, G.A.; Krüger, L.; Chen, Y.; Premont, R.T.; Choi, S.S.; Diehl, A.M. Cross-talk between Notch and Hedgehog regulates hepatic stellate cell fate in mice. Hepatology 2013, 58, 1801–1813.
- Mannaerts, I.; Leite, S.B.; Verhulst, S.; Claerhout, S.; Eysackers, N.; Thoen, L.F.; Hoorens, A.; Reynaert, H.; Halder, G.; van Grunsven, L.A. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 2015, 63, 679–688.
- Tao, Y.; Wang, N.; Qiu, T.; Sun, X. The Role of Autophagy and NLRP3 Inflammasome in Liver Fibrosis. Biomed. Res. Int. 2020, 2020, 7269150.
- Milani, S.; Herbst, H.; Schuppan, D.; Stein, H.; Surrenti, C. Transforming growth factors beta 1 and beta 2 are differentially expressed in fibrotic liver disease. Am. J. Pathol. 1991, 139, 1221–1229.
- Sun, Y.-Y.; Li, X.-F.; Meng, X.-M.; Huang, C.; Zhang, L.; Li, J. Macrophage Phenotype in Liver Injury and Repair. Scand. J. Immunol. 2017, 85, 166–174.
- Bu, F.-T.; Zhu, Y.; Chen, X.; Wang, A.; Zhang, Y.-F.; You, H.-M.; Yang, Y.; Yang, Y.-R.; Huang, C.; Li, J. Circular RNA circPSD3 alleviates hepatic fibrogenesis by regulating the miR-92b-3p/Smad7 axis. Mol. Ther.—Nucleic Acids 2021, 23, 847–862.
- Yan, X.; Liu, Z.; Chen, Y. Regulation of TGF-beta signaling by Smad7. Acta Biochim. Biophys. Sin. 2009, 41, 263–272.
- Dooley, S.; Hamzavi, J.; Breitkopf, K.; Wiercinska, E.; Said, H.M.; Lorenzen, J.; Dijke, P.T.; Gressner, A.M. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology 2003, 125, 178–191.
- Yang, Y.R.; Hu, S.; Bu, F.T.; Li, H.; Huang, C.; Meng, X.M.; Zhang, L.; Lv, X.W.; Li, J. Circular RNA CREBBP Suppresses Hepatic Fibrosis Via Targeting the hsa-miR-1291/LEFTY2 Axis. Front. Pharmacol. 2021, 12, 741151.
- Yang, Y.-r.; Bu, F.-t.; Yang, Y.; Li, H.; Huang, C.; Meng, X.-m.; Zhang, L.; Lv, X.-w.; Li, J. LEFTY2 alleviates hepatic stellate cell activation and liver fibrosis by regulating the TGF-1/Smad3 pathway. Mol. Immunol. 2020, 126, 31–39.
- Ulloa, L.; Tabibzadeh, S. Lefty inhibits receptor-regulated Smad phosphorylation induced by the activated transforming growth factor-beta receptor. J. Biol. Chem. 2001, 276, 21397–21404.
- Lin, A.H.; Luo, J.; Mondshein, L.H.; ten Dijke, P.; Vivien, D.; Contag, C.H.; Wyss-Coray, T. Global analysis of Smad2/3-dependent TGF-beta signaling in living mice reveals prominent tissue-specific responses to injury. J. Immunol. 2005, 175, 547–554.
- Ji, D.; Chen, G.-F.; Wang, J.-C.; Ji, S.-H.; Wu, X.-W.; Lu, X.-J.; Chen, J.-L.; Li, J.-T. Hsa_circ_0070963 inhibits liver fibrosis via regulation of miR-223-3p and LEMD3. Aging 2020, 12, 1643–1655.
- Chambers, D.M.; Moretti, L.; Zhang, J.J.; Cooper, S.W.; Chambers, D.M.; Santangelo, P.J.; Barker, T.H. LEM domain-containing protein 3 antagonizes TGF-SMAD2/3 signaling in a stiffness-dependent manner in both the nucleus and cytosol. J. Biol. Chem. 2018, 293, 15867–15886.
- Ariyachet, C.; Chuaypen, N.; Kaewsapsak, P.; Chantaravisoot, N.; Jindatip, D.; Potikanond, S.; Tangkijvanich, P. MicroRNA-223 Suppresses Human Hepatic Stellate Cell Activation Partly via Regulating the Actin Cytoskeleton and Alleviates Fibrosis in Organoid Models of Liver Injury. Int. J. Mol. Sci. 2022, 23, 9380.
- Wang, X.; Seo, W.; Park, S.H.; Fu, Y.; Hwang, S.; Rodrigues, R.M.; Feng, D.; Gao, B.; He, Y. MicroRNA-223 restricts liver fibrosis by inhibiting the TAZ-IHH-GLI2 and PDGF signaling pathways via the crosstalk of multiple liver cell types. Int. J. Biol. Sci. 2021, 17, 1153–1167.
- Wang, L.; Wang, Y.; Quan, J. Exosomal miR-223 derived from natural killer cells inhibits hepatic stellate cell activation by suppressing autophagy. Mol. Med. 2020, 26, 1–9.
- Calvente, C.J.; Del Pilar, H.; Tameda, M.; Johnson, C.D.; Feldstein, A.E. MicroRNA 223 3p Negatively Regulates the NLRP3 Inflammasome in Acute and Chronic Liver Injury. Mol. Ther. 2019, 28, 653–663.
- Zhou, Y.; Lv, X.; Qu, H.; Zhao, K.; Fu, L.; Zhu, L.; Ye, G.; Guo, J. Preliminary screening and functional analysis of circular RNAs associated with hepatic stellate cell activation. Gene 2018, 677, 317–323.
- Zou, G.L.; Zuo, S.; Lu, S.; Hu, R.H.; Lu, Y.Y.; Yang, J.; Deng, K.S.; Wu, Y.T.; Mu, M.; Zhu, J.J.; et al. Bone morphogenetic protein-7 represses hepatic stellate cell activation and liver fibrosis via regulation of TGF-/Smad signaling pathway. World J. Gastroenterol. 2019, 25, 4222–4234.
- Meng, X.M.; Chung, A.C.; Lan, H.Y. Role of the TGF-/BMP-7/Smad pathways in renal diseases. Clin. Sci. 2013, 124, 243–254.
- Wang, W.; Dong, R.; Guo, Y.; He, J.; Shao, C.; Yi, P.; Yu, F.; Gu, D.; Zheng, J. CircMTO1 inhibits liver fibrosis via regulation of miR-17-5p and Smad7. J. Cell Mol. Med. 2019, 23, 5486–5496.
- Jin, H.; Li, C.; Dong, P.; Huang, J.; Yu, J.; Zheng, J. Circular RNA cMTO1 Promotes PTEN Expression Through Sponging miR-181b-5p in Liver Fibrosis. Front. Cell Dev. Biol. 2020, 8, 714.
- Takashima, M.; Parsons, C.J.; Ikejima, K.; Watanabe, S.; White, E.S.; Rippe, R.A. The tumor suppressor protein PTEN inhibits rat hepatic stellate cell activation. J. Gastroenterol. 2009, 44, 847–855.
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.; Baldwin, A.S. Akt-dependent regulation of NF-B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500.
- Gao, R.; Brigstock, D.R. Activation of nuclear factor kappa B (NF-kappaB) by connective tissue growth factor (CCN2) is involved in sustaining the survival of primary rat hepatic stellate cells. Cell Commun. Signal. 2005, 3, 14.
- Wang, P.; Huang, Z.; Peng, Y.; Li, H.; Lin, T.; Zhao, Y.; Hu, Z.; Zhou, Z.; Zhou, W.; Liu, Y.; et al. Circular RNA circBNC2 inhibits epithelial cell G2-M arrest to prevent fibrotic maladaptive repair. Nat. Commun. 2022, 13, 1–19.
- Paik, Y.H.; Kim, J.; Aoyama, T.; De Minicis, S.; Bataller, R.; Brenner, D.A. Role of NADPH Oxidases in Liver Fibrosis. Antioxid. Redox Signal. 2014, 20, 2854–2872.
- Ignat, S.-R.; Dinescu, S.; Hermenean, A.; Costache, M. Cellular Interplay as a Consequence of Inflammatory Signals Leading to Liver Fibrosis Development. Cells 2020, 9, 461.
- Reiter, F.P.; Ye, L.; Ofner, A.; Schiergens, T.S.; Ziesch, A.; Brandl, L.; Ben Khaled, N.; Hohenester, S.; Wimmer, R.; Artmann, R.; et al. p70 Ribosomal Protein S6 Kinase Is a Checkpoint of Human Hepatic Stellate Cell Activation and Liver Fibrosis in Mice. Cell. Mol. Gastroenterol. Hepatol. 2021, 13, 95–112.
- Ma, L.; Wei, J.; Zeng, Y.; Liu, J.; Xiao, E.; Kang, Y.; Kang, Y. Mesenchymal stem cell-originated exosomal circDIDO1 suppresses hepatic stellate cell activation by miR-141-3p/PTEN/AKT pathway in human liver fibrosis. Drug Deliv. 2022, 29, 440–453.
- Ma, J.; Li, Y.; Chen, M.; Wang, W.; Zhao, Q.; He, B.; Zhang, M.; Jiang, Y. hMSCs-derived exosome circCDK13 inhibits liver fibrosis by regulating the expression of MFGE8 through miR-17-5p/KAT2B. Cell Biol. Toxicol. 2022, 1–22.
- Struhl, K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998, 12, 599–606.
- Khalifeh-Soltani, A.; Gupta, D.; Ha, A.; Podolsky, M.J.; Datta, R.; Atabai, K. The Mfge8-α8β1;1-PTEN pathway regulates airway smooth muscle contraction in allergic inflammation. FASEB J. 2018, 32, 5927–5936.
- Kane, L.P.; Shapiro, V.S.; Stokoe, D.; Weiss, A. Induction of NF-κB by the Akt/PKB kinase. Curr. Biol. 1999, 9, 601–604.
- Xu, Z.-X.; Li, J.-Z.; Li, Q.; Xu, M.-Y.; Li, H.-Y. CircRNA608-microRNA222-PINK1 axis regulates the mitophagy of hepatic stellate cells in NASH related fibrosis. Biochem. Biophys. Res. Commun. 2022, 610, 35–42.
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166.
- Berwick, D.; Harvey, K. The regulation and deregulation of Wnt signaling by PARK genes in health and disease. J. Mol. Cell Biol. 2013, 6, 3–12.
- Fan, S.; Price, T.; Huang, W.; Plue, M.; Warren, J.; Sundaramoorthy, P.; Paul, B.; Feinberg, D.; Maciver, N.; Chao, N.; et al. PINK1-Dependent Mitophagy Regulates the Migration and Homing of Multiple Myeloma Cells via the MOB1B-Mediated Hippo-YAP/TAZ Pathway. Adv. Sci. 2020, 7, 1900860.
- Kulaberoglu, Y.; Lin, K.; Holder, M.; Gai, Z.; Gomez, M.; Shifa, B.A.; Mavis, M.; Hoa, L.; Sharif, A.A.D.; Lujan, C.; et al. Stable MOB1 interaction with Hippo/MST is not essential for development and tissue growth control. Nat. Commun. 2017, 8, 1–16.
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The Biology of YAP/TAZ: Hippo Signaling and Beyond. Physiol. Rev. 2014, 94, 1287–1312.
- Dou, S.D.; Zhang, J.N.; Xie, X.L.; Liu, T.; Hu, J.L.; Jiang, X.Y.; Wang, M.M.; Jiang, H.D. MitoQ inhibits hepatic stellate cell activation and liver fibrosis by enhancing PINK1/parkin-mediated mitophagy. Open Med. 2021, 16, 1718–1727.
- Chen, X.; Li, H.-D.; Bu, F.-T.; Li, X.-F.; Chen, Y.; Zhu, S.; Wang, J.-N.; Chen, S.-Y.; Sun, Y.-Y.; Pan, X.-Y.; et al. Circular RNA circFBXW4 suppresses hepatic fibrosis via targeting the miR-18b-3p/FBXW7 axis. Theranostics 2020, 10, 4851–4870.
- Kar, R.; Jha, S.K.; Ojha, S.; Sharma, A.; Dholpuria, S.; Raju, V.S.R.; Prasher, P.; Chellappan, D.K.; Gupta, G.; Singh, S.K.; et al. The FBXW7-NOTCH interactome: A ubiquitin proteasomal system-induced crosstalk modulating oncogenic transformation in human tissues. Cancer Rep. 2021, 4, e1369.
- Zhao, Q.; Liu, J.; Deng, H.; Ma, R.; Liao, J.-Y.; Liang, H.; Hu, J.; Li, J.; Guo, Z.; Cai, J.; et al. Targeting Mitochondria-Located circRNA SCAR Alleviates NASH via Reducing mROS Output. Cell 2020, 183, 76–93.e22.
- Xie, H.; Xie, D.; Zhang, J.; Jin, W.; Li, Y.; Yao, J.; Pan, Z.; Xie, D. ROS/NF-kB Signaling Pathway-Mediated Transcriptional Activation of TRIM37 Promotes HBV-Associated Hepatic Fibrosis. Mol. Ther.-Nucleic Acids 2020, 22, 114–123.
- Ramos-Tovar, E.; Muriel, P. Molecular Mechanisms that Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants 2020, 9, 1279.
- Li, S.; Song, F.; Lei, X.; Li, J.; Li, F.; Tan, H. hsa_circ_0004018 suppresses the progression of liver fibrosis through regulating the hsa-miR-660-3p/TEP1 axis. Aging 2020, 12, 11517–11529.
- Chaiteerakij, R.; Roberts, L.R. Telomerase mutation: A genetic risk factor for cirrhosis. Hepatology 2011, 53, 1430–1432.
- Duan, X.; Wang, H.; Yang, Y.; Wang, P.; Zhang, H.; Liu, B.; Wei, W.; Yao, W.; Zhou, X.; Zhao, J.; et al. Genetic variants in telomerase-associated protein 1 are associated with telomere damage in PAH-exposed workers. Ecotoxicol. Environ. Saf. 2021, 223, 112558.
- Chang, J.T.-C.; Chen, Y.-L.; Yang, H.-T.; Chen, C.-Y.; Cheng, A.-J. Differential regulation of telomerase activity by six telomerase subunits. JBIC J. Biol. Inorg. Chem. 2002, 269, 3442–3450.
- Liu, Y.; Snow, B.; Hande, P.; Baerlocher, G.; Kickhoefer, V.; Yeung, D.; Wakeham, A.; Itie, A.; Siderovski, D.; Lansdorp, P.; et al. Telomerase-Associated Protein TEP1 Is Not Essential for Telomerase Activity or Telomere Length Maintenance In Vivo. Mol. Cell. Biol. 2000, 20, 8178–8184.
- Kickhoefer, V.A.; Stephen, A.G.; Harrington, L.; Robinson, M.; Rome, L. Vaults and Telomerase Share a Common Subunit, TEP1. J. Biol. Chem. 1999, 274, 32712–32717.
- Hahne, J.C.; Lampis, A.; Valeri, N. Vault RNAs: Hidden gems in RNA and protein regulation. Cell. Mol. Life Sci. 2020, 78, 1487–1499.
- Liu, B.; Tian, Y.; He, J.; Gu, Q.; Jin, B.; Shen, H.; Li, W.; Shi, L.; Yu, H.; Shan, G.; et al. The potential of mecciRNA in hepatic stellate cell to regulate progression of nonalcoholic hepatitis. J. Transl. Med. 2022, 20, 1–17.
- Li, B.; Li, Y.; Li, S.; Li, H.; Liu, L.; Yu, H. Circ_MTM1 knockdown inhibits the progression of HBV-related liver fibrosis via regulating IL7R expression through targeting miR-122-5p. Am. J. Transl. Res. 2022, 14, 2199–2211.
- Jacobs, S.R.; Michalek, R.D.; Rathmell, J.C. IL-7 Is Essential for Homeostatic Control of T Cell Metabolism In Vivo. J. Immunol. 2010, 184, 3461–3469.
- Liu, W.; Feng, R.; Li, X.; Li, D.; Zhai, W. TGF-β- and lipopolysaccharide-induced upregulation of circular RNA PWWP2A promotes hepatic fibrosis via sponging miR-203 and miR-223. Aging 2019, 11, 9569–9580.
- Zhu, S.; Chen, X.; Wang, J.N.; Xu, J.J.; Wang, A.; Li, J.J.; Wu, S.; Wu, Y.Y.; Li, X.F.; Huang, C.; et al. Circular RNA circUbe2k promotes hepatic fibrosis via sponging miR-149-5p/TGF-2 axis. Faseb J. 2021, 35, e21622.
- Niu, H.; Zhang, L.; Wang, B.; Zhang, G.C.; Liu, J.; Wu, Z.F.; Du, S.S.; Zeng, Z.C. CircTUBD1 Regulates Radiation-induced Liver Fibrosis Response via a circTUBD1/micro-203a-3p/Smad3 Positive Feedback Loop. J. Clin. Transl. Hepatol. 2022, 10, 680–691.
- Niu, H.; Zhang, L.; Chen, Y.-H.; Yuan, B.-Y.; Wu, Z.-F.; Cheng, J.C.-H.; Lin, Q.; Zeng, Z.-C. Circular RNA TUBD1 Acts as the miR-146a-5p Sponge to Affect the Viability and Pro-Inflammatory Cytokine Production of LX-2 Cells through the TLR4 Pathway. Radiat. Res. 2020, 193, 383–393.
- Li, X.; Fang, Y.; Jiang, D.; Dong, Y.; Liu, Y.; Zhang, S.; Guo, J.; Qi, C.; Zhao, C.; Jiang, F.; et al. Targeting FSTL1 for Multiple Fibrotic and Systemic Autoimmune Diseases. Mol. Ther. 2020, 29, 347–364.
- Shang, H.; Liu, X.; Guo, H. Knockdown of Fstl1 attenuates hepatic stellate cell activation through the TGF-1/Smad3 signaling pathway. Mol. Med. Rep. 2017, 16, 7119–7123.
- Liu, C.; Chen, X.; Yang, L.; Kisseleva, T.; Brenner, D.A.; Seki, E. Transcriptional Repression of the Transforming Growth Factor (TGF-) Pseudoreceptor BMP and Activin Membrane-bound Inhibitor (BAMBI) by Nuclear Factor Signaling in Hepatic Stellate Cells*. J. Biol. Chem. 2014, 289, 7082–7091.
- Seki, E.; De Minicis, S.; Österreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.; Schwabe, R.F. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332.
- Lang, A.; Schoonhoven, R.; Tuvia, S.; A Brenner, D.; A Rippe, R. Nuclear factor κB in proliferation, activation, and apoptosis in rat hepatic stellate cells. J. Hepatol. 2000, 33, 49–58.
- Luedde, T.; Schwabe, R.F. NF-B in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118.
- Liu, W.; Wu, Y.-H.; Zhang, L.; Xue, B.; Wang, Y.; Liu, B.; Liu, X.-Y.; Zuo, F.; Yang, X.-Y.; Chen, F.-Y.; et al. MicroRNA-146a suppresses rheumatoid arthritis fibroblastlike synoviocytes proliferation and inflammatory responses by inhibiting the TLR4/NFkB signaling. Oncotarget 2018, 9, 24050.
- Hu, P.; Guo, J.; Zhao, B.; Zhang, Z.; Zhu, J.; Liu, F. CircCHD2/miR-200b-3p/HLF Axis Promotes Liver Cirrhosis. J. Environ. Pathol. Toxicol. Oncol. 2022, 41, 1–10.
- Xiang, D.-M.; Sun, W.; Ning, B.-F.; Zhou, T.-F.; Li, X.-F.; Zhong, W.; Cheng, Z.; Xia, M.-Y.; Wang, X.; Deng, X.; et al. The HLF/IL-6/STAT3 feedforward circuit drives hepatic stellate cell activation to promote liver fibrosis. Gut 2018, 67, 1704–1715.
- Tang, L.-Y.; Heller, M.; Meng, Z.; Yu, L.-R.; Tang, Y.; Zhou, M.; Zhang, Y.E. Transforming Growth Factor-β (TGF-β) Directly Activates the JAK1-STAT3 Axis to Induce Hepatic Fibrosis in Coordination with the SMAD Pathway. J. Biol. Chem. 2017, 292, 4302–4312.
- Zhu, L.; Ren, T.; Zhu, Z.; Cheng, M.; Mou, Q.; Mu, M.; Liu, Y.; Yao, Y.; Cheng, Y.; Zhang, B.; et al. Thymosin-4 Mediates Hepatic Stellate Cell Activation by Interfering with CircRNA-0067835/miR-155/FoxO3 Signaling Pathway. Cell Physiol. Biochem. 2018, 51, 1389–1398.
- Chen, Y.; Yuan, B.; Wu, Z.; Dong, Y.; Zhang, L.; Zeng, Z. Microarray profiling of circular RNAs and the potential regulatory role of hsa_circ_0071410 in the activated human hepatic stellate cell induced by irradiation. Gene 2017, 629, 35–42.
- Liao, J.; Zhang, Z.; Yuan, Q.; Luo, L.; Hu, X. The mouse Anxa6/miR-9-5p/Anxa2 axis modulates TGF-1-induced mouse hepatic stellate cell (mHSC) activation and CCl4-caused liver fibrosis. Toxicol. Lett. 2022, 362, 38–49.
- Wang, J.; He, Z.; Liu, X.; Xu, J.; Jiang, X.; Quan, G.; Jiang, J. LINC00941 promotes pancreatic cancer malignancy by interacting with ANXA2 and suppressing NEDD4L-mediated degradation of ANXA2. Cell Death Dis. 2022, 13, 1–15.
- Liu, Y.; Ao, X.; Ding, W.; Ponnusamy, M.; Wu, W.; Hao, X.; Yu, W.; Wang, Y.; Li, P.; Wang, J. Critical role of FOXO3a in carcinogenesis. Mol. Cancer 2018, 17, 1–12.
- Hui, R.C.-Y.; Gomes, A.R.; Constantinidou, D.; Costa, J.R.; Karadedou, C.T.; de Mattos, S.F.; Wymann, M.P.; Brosens, J.J.; Schulze, A.; Lam, E.W.-F. The Forkhead Transcription Factor FOXO3a Increases Phosphoinositide-3 Kinase/Akt Activity in Drug-Resistant Leukemic Cells through Induction of PIK3CA Expression. Mol. Cell. Biol. 2008, 28, 5886–5898.
- Lu, M.; Hartmann, D.; Braren, R.; Gupta, A.; Wang, B.; Wang, Y.; Mogler, C.; Cheng, Z.; Wirth, T.; Friess, H.; et al. Oncogenic Akt-FOXO3 loop favors tumor-promoting modes and enhances oxidative damage-associated hepatocellular carcinogenesis. BMC Cancer 2019, 19, 1–13.
- Chen, Y.; Yuan, B.; Chen, G.; Zhang, L.; Zhuang, Y.; Niu, H.; Zeng, Z. Circular RNA RSF1 promotes inflammatory and fibrotic phenotypes of irradiated hepatic stellate cell by modulating miR-146a-5p. J. Cell. Physiol. 2020, 235, 8270–8282.
- Choi, S.S.; Witek, R.P.; Yang, L.; Omenetti, A.; Syn, W.-K.; Moylan, C.A.; Jung, Y.; Karaca, G.F.; Teaberry, V.S.; Pereira, T.A.; et al. Activation of Rac1 promotes hedgehog-mediated acquisition of the myofibroblastic phenotype in rat and human hepatic stellate cells. Hepatology 2010, 52, 278–290.
- Tang, C.; Wu, X.; Ren, Q.; Yao, M.; Xu, S.; Yan, Z. Hedgehog signaling is controlled by Rac1 activity. Theranostics 2022, 12, 1303–1320.
- Arbibe, L.; Mira, J.P.; Teusch, N.; Kline, L.; Guha, M.; Mackman, N.; Godowski, P.J.; Ulevitch, R.J.; Knaus, U.G. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat. Immunol. 2000, 1, 533–540.
- Coso, O.A.; Chiariello, M.; Yu, J.-C.; Teramoto, H.; Crespo, P.; Xu, N.; Miki, T.; Gutkind, J.S. The small GTP-binding proteins Rac1 and Cdc42regulate the activity of the JNK/SAPK signaling pathway. Cell 1995, 81, 1137–1146.
- Wang, J.; Liu, G.; Li, Q.; Wang, F.; Xie, F.; Zhai, R.; Guo, Y.; Chen, T.; Zhang, N.; Ni, W.; et al. Mucin1 promotes the migration and invasion of hepatocellular carcinoma cells via JNK-mediated phosphorylation of Smad2 at the C-terminal and linker regions. Oncotarget 2015, 6, 19264–19278.
- Kamato, D.; Little, P.J. Smad2 linker region phosphorylation is an autonomous cell signalling pathway: Implications for multiple disease pathologies. Biomed. Pharmacother. 2020, 124, 109854.
- Afroz, R.; Zhou, Y.; Little, P.J.; Xu, S.; Mohamed, R.; Stow, J.; Kamato, D. Toll-like Receptor 4 Stimulates Gene Expression via Smad2 Linker Region Phosphorylation in Vascular Smooth Muscle Cells. ACS Pharmacol. Transl. Sci. 2020, 3, 524–534.
- Yang, Y.M.; Noureddin, M.; Liu, C.; Ohashi, K.; Kim, S.Y.; Ramnath, D.; Powell, E.E.; Sweet, M.J.; Roh, Y.S.; Hsin, I.-F.; et al. Hyaluronan synthase 2–mediated hyaluronan production mediates Notch1 activation and liver fibrosis. Sci. Transl. Med. 2019, 11, aat9824.
- Li, B.; Zhou, J.; Luo, Y.; Tao, K.; Zhang, L.; Zhao, Y.; Lin, Y.; Zeng, X.; Yu, H. Suppressing circ_0008494 inhibits HSCs activation by regulating the miR-185-3p/Col1a1 axis. Front. Pharmacol. 2022, 13, 1050093.
- Zhubanchaliyev, A.; Temirbekuly, A.; Kongrtay, K.; Wanshura, L.C.; Kunz, J. Targeting Mechanotransduction at the Transcriptional Level: YAP and BRD4 Are Novel Therapeutic Targets for the Reversal of Liver Fibrosis. Front. Pharmacol. 2016, 7, 462.
- Liu, Z.; Mo, H.; Liu, R.; Niu, Y.; Chen, T.; Xu, Q.; Tu, K.; Yang, N. Matrix stiffness modulates hepatic stellate cell activation into tumor-promoting myofibroblasts via E2F3-dependent signaling and regulates malignant progression. Cell Death Dis. 2021, 12, 1134.
- Marrone, G.; Shah, V.H.; Gracia-Sancho, J. Sinusoidal communication in liver fibrosis and regeneration. J. Hepatol. 2016, 65, 608–617.
- Wesche, H.; Gao, X.; Li, X.; Kirschning, C.J.; Stark, G.R.; Cao, Z. IRAK-M is a novel member of the Pelle/interleukin-1 receptor-associated kinase (IRAK) family. J. Biol. Chem. 1999, 274, 19403–19410.
- Xu, J.J.; Chen, X.; Zhu, S.; Jiang, L.F.; Ma, W.X.; Chen, S.Y.; Meng, X.M.; Huang, C.; Li, J. Myc-mediated circular RNA circMcph1/miR-370-3p/Irak2 axis is a progressive regulator in hepatic fibrosis. Life Sci. 2023, 312, 121182.
- Csak, T.; Bala, S.; Lippai, D.; Satishchandran, A.; Catalano, D.; Kodys, K.; Szabo, G. microRNA-122 regulates hypoxia-inducible factor-1 and vimentin in hepatocytes and correlates with fibrosis in diet-induced steatohepatitis. Liver Int. 2015, 35, 532–541.
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002, 12, 735–739.
- Halász, T.; Horváth, G.; Pár, G.; Werling, K.; Kiss, A.; Schaff, Z.; Lendvai, G. miR-122 negatively correlates with liver fibrosis as detected by histology and FibroScan. World J. Gastroenterol. 2015, 21, 7814–7823.
- Lu, X.; Liu, Y.; Xuan, W.; Ye, J.; Yao, H.; Huang, C.; Li, J. Circ_1639 induces cells inflammation responses by sponging miR-122 and regulating TNFRSF13C expression in alcoholic liver disease. Toxicol. Lett. 2019, 314, 89–97.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
30 Jan 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No