Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ichiro Nojima | -- | 2132 | 2023-01-23 07:55:15 | | | |
| 2 | Peter Tang | Meta information modification | 2132 | 2023-01-30 09:06:04 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nojima, I.; Wada, J. Metformin and Metabolism in Cancer Cells. Encyclopedia. Available online: https://encyclopedia.pub/entry/40480 (accessed on 12 June 2026).
Nojima I, Wada J. Metformin and Metabolism in Cancer Cells. Encyclopedia. Available at: https://encyclopedia.pub/entry/40480. Accessed June 12, 2026.
Nojima, Ichiro, Jun Wada. "Metformin and Metabolism in Cancer Cells" Encyclopedia, https://encyclopedia.pub/entry/40480 (accessed June 12, 2026).
Nojima, I., & Wada, J. (2023, January 23). Metformin and Metabolism in Cancer Cells. In Encyclopedia. https://encyclopedia.pub/entry/40480
Nojima, Ichiro and Jun Wada. "Metformin and Metabolism in Cancer Cells." Encyclopedia. Web. 23 January, 2023.
Copy Citation
Metformin has been a long-standing prescribed drug for treatment of type 2 diabetes (T2D) and its beneficial effects on virus infection, autoimmune diseases, aging and cancers are also recognized. Metformin modulates the differentiation and activation of various immune-mediated cells such as CD4+ and CD+8 T cells. The activation of adenosine 5′-monophosphate-activated protein kinase (AMPK) and mammalian target of rapamycin complex 1 (mTORC1) pathway may be involved in this process.
CD8 T cells
AMPK
mTORC
cancer
1.Introduction
The traditional herbal medicine in Europe, Galega officinalis, was found to be rich in guanidine and shown to lower blood glucose levels. Several guanidine derivatives were synthesized and used to treat diabetes in 1920s and 1930s, but they were discontinued due to severe toxicity [1]. The blood glucose lowering effects were recognized by the French physician Jean Sterne who reported the use of metformin to treat diabetes in 1957 [2]. However, metformin received the limited attention and the biguanides were generally discontinued in late 1970s due to the risk of lactic acidosis. The beneficial effects of metformin ameliorating insulin resistance without weight gain and hypoglycemia in the patients with type 2 diabetes were recognized in Europe and the long-term cardiovascular benefits [3] were identified by the UK Prospective Diabetes Study (UKPDS) in 1998, providing a new rationale to adopt metformin as an initial therapy to manage hyperglycemia in type 2 diabetes. Metformin is beneficial for aging-related morbidities such as obesity, metabolic syndrome, cardiovascular disease, and cognitive impairment [4] by its favorable action on the endothelial dysfunction [5]. In addition to risk reduction of any diabetes-related endpoint, myocardial infarction, and death from any cause in metformin [6], the reduction of cancer incidence was demonstrated in an observational study in Scotland [7] and a Chinese meta-analysis showed an overall reduction of 20% in cancer incidence in metformin users [8].
The metabolic effects of metformin with insulin sensitizing actions result in reduction of insulin and free insulin-like growth factor (IGF-1) levels may indirectly contribute the decreased incidence of cancer. In addition to the indirect effects, metformin as adenosine 5′-monophosphate-activated protein kinase (AMPK) activator and subsequent inhibition of mammalian target of rapamycin complex 1 (mTORC1) specifically retard the growth of malignant cells. Rapidly proliferating malignant cells prefer to facilitate the process of catabolic glucose metabolism. Metformin inhibits glycolysis rate limiting enzyme of glycolysis, hexokinase 2 (HK2), and mitochondrial respiratory complex 1, which result in the reduction of mitochondrial ATP production in cancer cells [9]. Recently, it has been demonstrated that metformin may also enhance the antitumor immune response by acting on the tumor infiltrating and circulating CD8 T cells. Furthermore, the advent of immune checkpoint inhibitors in cancer therapies and their prominent antitumor effects also arouse the interests to the immune mediated effects of metformin in various diseases.
2. Molecular Mechanisms of Metformin-Induced Inhibition of Gluconeogenesis and Lipogenesis in Hepatocytes
Metformin is transported by facilitated diffusion via plasma membrane monoamine transporter (PMAT) and organic transporter 3 (OCT3) in enterocytes and further transported into hepatocytes via portal vein, where metformin reaches 40-70 μM, through OCT1 and OCT3 [10]. The excretion of metformin from hepatocytes to bile or circulation occurs through multidrug and toxin extrusion 1 (MATE1) and metformin concentration is reduced to 10-40 μM. Then, metformin enters renal epithelial cells via OCT2 and is secreted by renal MATE1 and MATE2 in unchanged form and eliminated into urine [10].
In hepatocyte, metformin partially inhibits mitochondrial respiratory-chain complex 1, resulting in reduction of ATP levels and accumulation of AMP and ADP. The gluconeogenesis is an energetically costly anabolic process and required 6 ATPs per molecule and depletion of ATP limits glucose synthesis. Pyruvate carboxylase (PC), Phosphoenol pyruvate carboxykinase (PEPCK) (Figure 1), Glyceraldehyde phosphate dehydrogenase require 2 ATPs, 2 GTPs, and 2 ATPs, respectively. The increased level of AMP inhibits an important rate-limiting gluconeogenic enzyme, Fructose-1, 6-bisphsphatase (FBPase). This gluconeogenic step antagonizes the opposite reaction that forms fructose-1,6-bisphosphate from fructose-6-phosphate and ATP in Phosphofructokinase (PFK)-dependent manner, a key rate-limiting step in glycolysis. Metformin was shown to reduce the glucagon signal transduction by decreasing 3′-5′-cyclic adenosine monophosphate (cAMP) thorough inhibiting adenylate cyclase coupled with glucagon receptor [11]. Decreased cAMP content leads to reduced activity of cAMP-dependent protein kinase A (PKA), an important signal transducer of glucagon-induced gluconeogenesis [12]. Metformin selectively inhibits the mitochondrial isoform of glycerophosphate dehydrogenase (mGPD) but not cytosolic GPD (cGPD), an enzyme that catalyzes the conversion of glyceraldehyde 3-phosphate (G3P) to dihydroxyacetone phosphate (DHAP). The inhibition of mGPD induces the accumulation of G3P, NADH and reduced conversion of lactate to pyruvate. Since DHAP and pyruvate are required for gluconeogenesis, it results in reduction of use of glycerol and lactate as gluconeogenic precursors [13]. In addition, metformin-induced increase in AMP/ATP ratio also activates AMPK, which suppresses lipid and protein synthesis and enhances glycolysis, fatty acid oxidation, mitochondrial biogenesis and autophagy [14]. The activation of AMPK by metformin resulted in inhibition of Acetyl-CoA carboxylase (ACC), and reduction of Malonyl-CoA. Since Malonyl-CoA inhibits the activity of carnitine palmitonyl transferase 1 (CPT1), CPT1 is activated by the reduction of Malonyl-CoA and fatty acid oxidation is enhanced. In addition, the activation of AMPK by metformin increased the expression of Sterol regulatory element binding protein-1c (SREBP-1c) which inhibits the expression of lipogenic and fatty acid synthesis genes (Figure 2).
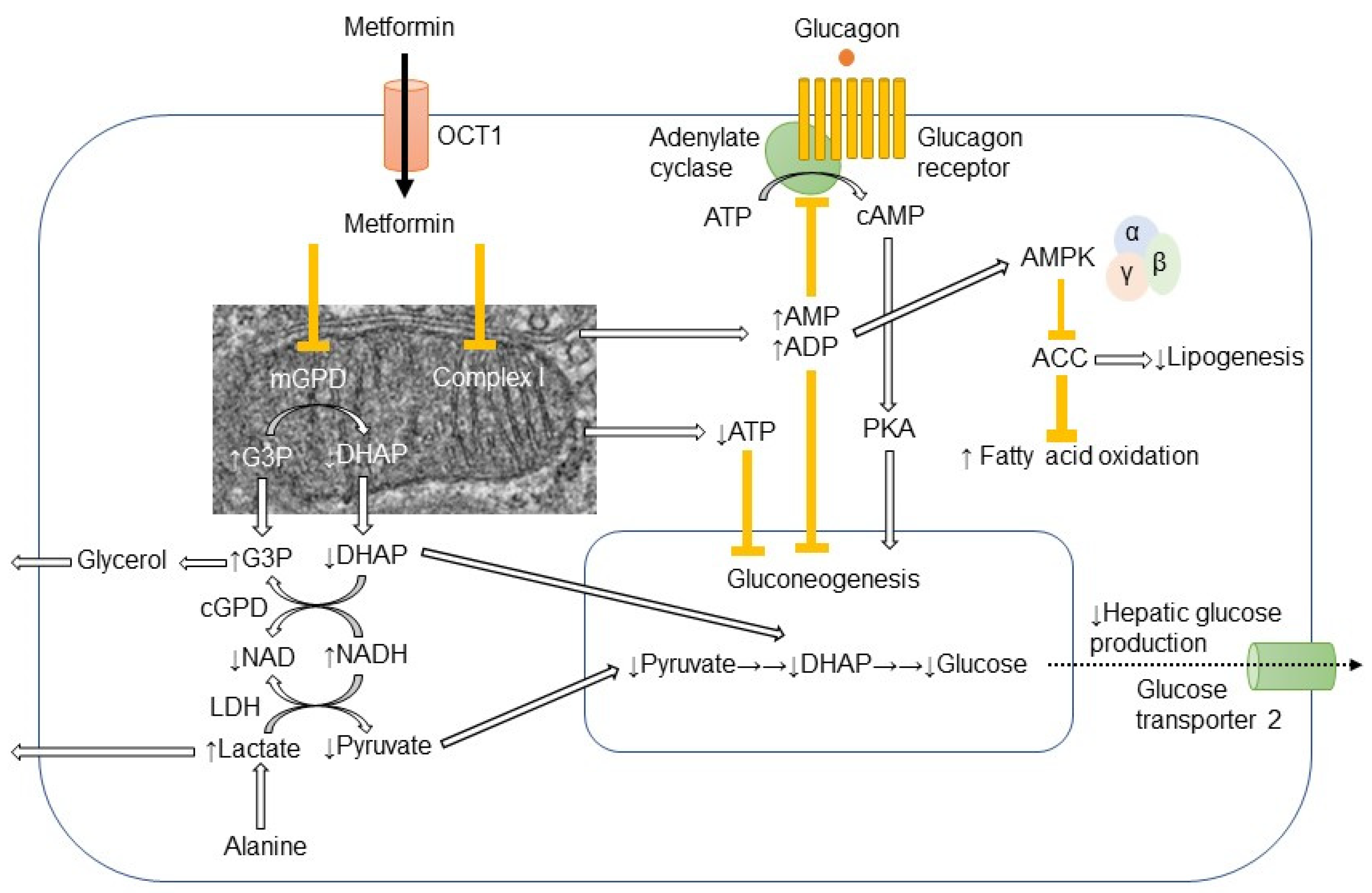
Figure 1. Action mechanism of metformin in hepatocyte.Metformin is mainly transported into hepatocyte through organic transporter 1 (OCT1) and inhibits respiratory-chain complex 1 (Complex 1). The reduction of ATP levels and increase in AMP and ADP levels result in the inhibition of gluconeogenesis. The increased AMP levels inhibit the adenylate cyclase coupled with glucagon receptor and the subsequent cAMP/protein kinase A (PKA) signaling pathway, which ultimately links to enhancement of gluconeogenesis. Metformin-induced elevation of AMP/ATP ratio also activates AMP-activated protein kinase (AMPK). Lipogenesis is inhibited and fatty acid oxidation is enhanced by the AMPK-induced inhibition of acetyl CoA carboxylase (ACC). Metformin also inhibits mitochondrial glycerophosphate dehydrogenase (mGPD) but not cytosolic GPD (cGPD). Glyceraldehyde 3-phosphate (G3P), nicotinamide adenine dinucleotide (NADH), and lactate are accumulated, while dihydroxyacetone phosphate (DHAP), NAD, and pyruvate are reduced. By the reduction of substrates for gluconeogenesis, i.e., pyruvate and DHAP, hepatic glucose production is reduced by metformin. The excess of glycerol and lactate enters into circulation.
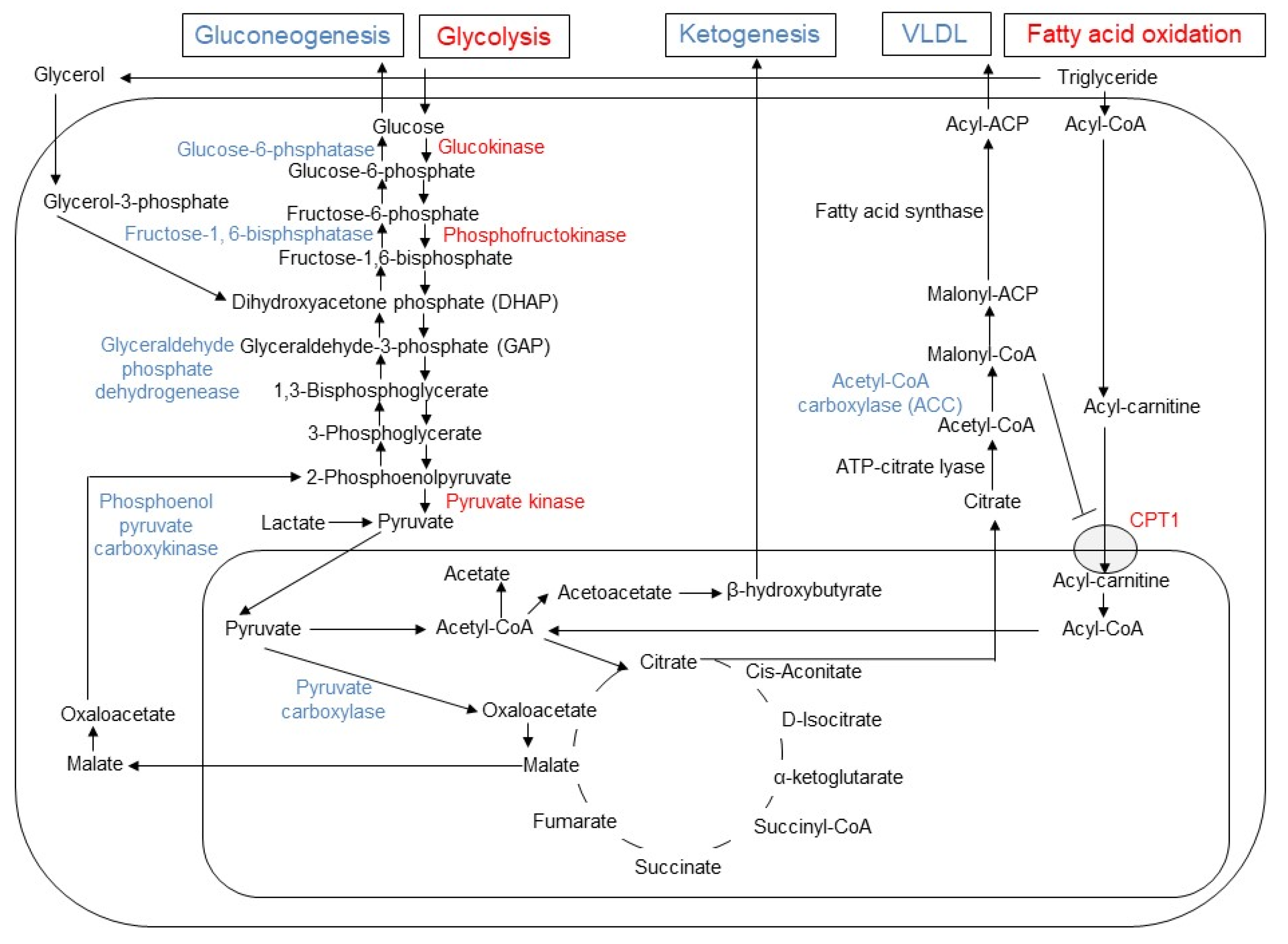
Figure 2. Changes in gluconeogenesis, glycolysis, lipogenesis, fatty acid oxidation, and ketogenesis induced by metformin in hepatocyte. In hepatocytes, metformin enhances glycolysis and fatty acid oxidation, while it inhibits gluconeogenesis, ketogenesis and release of very low-density lipoprotein (VLDL). The enzymes with enhanced activities are indicated by blue, while the reduced enzymes by red font. Since malonyl-CoA inhibits the activity of carnitine palmitoyltransferase 1 (CPT1), the reduced activity of acetyl CoA carboxylase (ACC) results in increased influx of acyl-carnitine into mitochondria and fatty acid oxidation by metformin.
Recently, the intestine has been focused as a target organ of metformin in addition to liver. Intraduodenal infusion of metformin activated the duodenal mucosal AMPK and lowered hepatic glucose production. Both glucagon-like peptide-1 receptor-PKA signaling and a neuronal-mediated gut-brain-liver pathway are required for metformin to lower hepatic glucose production [15]. The 3-day treatment with metformin in newly diagnosed patients with type 2 diabetes reduced Bacteroides fragilis and increased the bile acid glycoursodeoxycholic acid (GUDCA) in the gut, which were accompanied by inhibition of intestinal farnesoid X receptor (FXR) signaling [16]. By using Positron emission tomography-magnetic resonance imaging (PET-MRI), metformin increased the accumulation of [18F] fluorodeoxyglucose (FDG) in the intraluminal space of the intestine, suggesting that stool is one of the major disposal sites of glucose by the action of metformin targeting intestine [17].
3. Metformin and Mitochondrial Biogenesis and Dynamics
Various pharmacological activators of mitochondrial biogenesis such as AMPK activators, SIRT1 activators, nuclear receptor agonists, and cGMP modulators are possible candidate targets for the treatment of obesity, type 2 diabetes, and vascular complications [18]. Long-term administration of metformin demonstrated the increased activity of peroxisome proliferators-activated receptor γ (PPARγ) coactivator-1α (PGC-1α) and enhanced biogenesis of mitochondira [19]. in vitro models of vascular complications of diabetes demonstrated that metformin activates AMPK in human umbilical vein endothelial cells and reduces hyperglycemia-induced mitochondrial ROS production and mitochondrial biogenesis [20].
In addition to mitochondrial biogenesis, mitochondrial dynamics such as fusion and fission of mitochondria, and mitochondria-associated endoplasmic reticulum (ER) membranes (MAMs) are also involved in the process of diabetes. Once close contact between mitochondria is established, the dynamin-related outer mitochondrial membrane (OMM) proteins, such as mitofusin 1 (MFN1) and mitofusin 2 (MFN2), form homotypic (MFN1-MFN1 and MFN2-MFN2) or heterotypic (MFN1-MFN2) complexes. After tethering, inner mitochondrial membrane (IMM) fusion is mediated by optic atrophy 1 (OPA1) depending on the inner membrane potential [21]. The process of fusion retains the capacity of the mitochondria and maintains genetic and biochemical homogeneity by allowing the dilution of superoxide species and mutated DNA and the repolarization of membranes [22]. The reverse process of mitochondrial fusion, the division of mitochondria (fission) produces one or more daughter mitochondria, requires cytosolic dynamin-related protein 1 (DRP1). In the organellar interactions, MAMs function as membrane contact sites between the ER and mitochondria. The ER-mitochondria contact sites have emerged as major players in lipid metabolism, calcium signaling, autophagy and mitochondrial dynamics [23].
Disturbances in mitochondrial architecture and mitochondrial fusion-related genes are observed in situations of type 2 diabetes and obesity, leading to a highly fissioned mitochondria. Liver specific Mfn1 knockout mice (Mfn1LKO) were associated with increased complex I abundance, sensitive to hypoglycemic effects of metformin, and protected against insulin resistance induced by a high-fat diet [24]. In cybrid cells harboring mitochondrial haplogroup B4, which are more likely to develop type 2 diabetes in Chinese population, presented increased mitochondrial fission profiles, while metformin inhibited mitochondrial fission and attenuated pro-inflammation profile [25]. In IR Huh7 cells with high invasiveness ability, mitochondrial fission was increased revealed by structured illumination microscopy and metformin could inhibit mitochondrial fission, which is the feature of type 2 diabetes [26]. The disruption of MAMs by pharmacological inhibition and genetic ablation of the mitochondrial MAM protein, cyclophilin D, causes the impairment of insulin signaling and metformin improves both MAM integrity and insulin sensitivity [27].
4. Metformin and Metabolism in Cancer Cells
OCT1 is almost exclusively expressed in the liver, and it is a major target tissue for metformin. In contrast to the virtual absence of OCT1 in various tissues except liver, the tumor cells significantly express OCT1 and it may be related the antitumor effects of metformin [28]. OCT1 is also involved in the uptake of irinotecan and paclitaxel and OCT1-positive cancer cells exhibit significantly higher susceptibilities to the cytotoxic effects of these anticancer agents [29]. Liver kinase B1 (LKB1) is identified as a tumor suppressor gene and upstream activator of AMPK. The activation of LKB1-AMPK pathway by metformin results in the inhibition of Raptor-mTORC1 complex and suppression of cellular protein synthesis and cell growth. Phosphorylation of AMPK activates tuberous sclerosis complex 2 (TSC2) in a subunit of the TSC1–TSC2 complex, which further inactivates the small GTP-binding protein Ras Homolog Enriched in Brain (RHEB). The inactivated RHEB fails to promote the activity of mTORC1, suppresses the cell cycle, and reduces the proliferation of the cancer cells [9]. In addition to identification of tumor suppressor genes in cancer development, metabolic alterations induced by cancer cells was also rediscovered. The observation by Otto Warburg demonstrated that the proliferating cancer cells highly consume glucose and produce plenty of lactate and it is the reverse of Pasteur effects, where the fermentation is inhibited in the presence of O2 [30]. Recent investigations demonstrated that tumor suppressors and oncogenes converge on the prolyl hydroxylases and hypoxia-inducible factor (HIF), reverse the Pasteur effects, and thereby induce the Warburg effects. Importantly, the cancer cells carry out and enhance both aerobic glycolysis and mitochondrial respiration concurrently [30]. The tumor cells utilize the glucose for the proliferation and hypertrophy of the cells. The phosphorylation of glucose, the production of glucose-6-phosphate (G6P), is the initial step in glucose metabolism in cancer cells [9]. Hexokinase 1 (HK1) and HK2 are responsible for the production of G6P and show high affinity for glucose with Km values of μM range. They are highly sensitive to inhibition by their own product, G6P. HK1 is ubiquitously expressed, while HK2 is detected in skeletal muscle, adipose tissue and heart. HKs 1 and 2 are found in mitochondrial fraction and interact with the permeability transition pore including the voltage-dependent anion channel 1 (VDAC1) responsible for ATP flux to the cytoplasm from mitochondria. Such association between HK2 and VDAC1 facilitates the production of G6P by HK2 and protects cells from apoptosis. HK2 is highly expressed in lung and breast cancer and required for the proliferating cancer cells. Metformin directly inhibits HK2 activity by occupying the G6P binding site and induces dissociation of HK2 from mitochondria [9]. In addition to the reduction of glycolysis, metformin inhibits mTORC1 by decreasing the insulin and IGF-1 concentrations and inhibiting IGF1-induced AKT phosphorylation. Since AKT further phosphorylates HK2 at Thr473 and facilitates the association of HK2 with mitochondria, metformin decreases HK2 expression, activity and mitochondrial interaction [31]. Finally, metformin inhibits mitochondrial ATP production acting on respiratory chain complex 1 and decreasing TCA cycle intermediates, thus, metformin inhibits both glycolysis and mitochondrial respiration in cancer cells.
References
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576.
- Sterne, J. Blood sugar-lowering effect of 1,1-dimethylbiguanide. Therapie 1958, 13, 650–659.
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998, 352, 854–865.
- Salvatore, T.; Pafundi, P.C.; Morgillo, F.; Di Liello, R.; Galiero, R.; Nevola, R.; Marfella, R.; Monaco, L.; Rinaldi, L.; Adinolfi, L.E.; et al. Metformin: An old drug against old age and associated morbidities. Diabetes Res. Clin. Pract. 2020, 160, 108025.
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Rinaldi, L.; Caturano, A.; Vetrano, E.; Aprea, C.; Albanese, G.; Di Martino, A.; Ricozzi, C.; et al. Can Metformin Exert as an Active Drug on Endothelial Dysfunction in Diabetic Subjects? Biomedicines 2020, 9, 3.
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 2008, 359, 1577–1589.
- Libby, G.; Donnelly, L.A.; Donnan, P.T.; Alessi, D.R.; Morris, A.D.; Evans, J.M. New users of metformin are at low risk of incident cancer: A cohort study among people with type 2 diabetes. Diabetes Care 2009, 32, 1620–1625.
- Zhang, K.; Bai, P.; Dai, H.; Deng, Z. Metformin and risk of cancer among patients with type 2 diabetes mellitus: A systematic review and meta-analysis. Prim. Care Diabetes 2021, 15, 52–58.
- Salani, B.; Del Rio, A.; Marini, C.; Sambuceti, G.; Cordera, R.; Maggi, D. Metformin, cancer and glucose metabolism. Endocr. Relat. Cancer 2014, 21, R461–R471.
- Szymczak-Pajor, I.; Wenclewska, S.; Sliwinska, A. Metabolic Action of Metformin. Pharmaceuticals 2022, 15, 810.
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966.
- Hur, K.Y.; Lee, M.S. New mechanisms of metformin action: Focusing on mitochondria and the gut. J. Diabetes Investig. 2015, 6, 600–609.
- Ferrannini, E. The target of metformin in type 2 diabetes. N. Engl. J. Med. 2014, 371, 1547–1548.
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262.
- Duca, F.A.; Cote, C.D.; Rasmussen, B.A.; Zadeh-Tahmasebi, M.; Rutter, G.A.; Filippi, B.M.; Lam, T.K. Corrigendum: Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat. Med. 2016, 22, 217.
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 2018, 24, 1919–1929.
- Morita, Y.; Nogami, M.; Sakaguchi, K.; Okada, Y.; Hirota, Y.; Sugawara, K.; Tamori, Y.; Zeng, F.; Murakami, T.; Ogawa, W. Enhanced Release of Glucose into the Intraluminal Space of the Intestine Associated with Metformin Treatment as Revealed by Fluorodeoxyglucose PET-MRI. Diabetes Care 2020, 43, 1796–1802.
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol. Rev. 2016, 68, 20–48.
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022.
- Kukidome, D.; Nishikawa, T.; Sonoda, K.; Imoto, K.; Fujisawa, K.; Yano, M.; Motoshima, H.; Taguchi, T.; Matsumura, T.; Araki, E. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes 2006, 55, 120–127.
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532.
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287.
- Krols, M.; van Isterdael, G.; Asselbergh, B.; Kremer, A.; Lippens, S.; Timmerman, V.; Janssens, S. Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol. 2016, 131, 505–523.
- Kulkarni, S.S.; Joffraud, M.; Boutant, M.; Ratajczak, J.; Gao, A.W.; Maclachlan, C.; Hernandez-Alvarez, M.I.; Raymond, F.; Metairon, S.; Descombes, P.; et al. Mfn1 Deficiency in the Liver Protects Against Diet-Induced Insulin Resistance and Enhances the Hypoglycemic Effect of Metformin. Diabetes 2016, 65, 3552–3560.
- Chang, Y.H.; Lin, H.Y.; Shen, F.C.; Su, Y.J.; Chuang, J.H.; Lin, T.K.; Liou, C.W.; Lin, C.Y.; Weng, S.W.; Wang, P.W. The Causal Role of Mitochondrial Dynamics in Regulating Innate Immunity in Diabetes. Front. Endocrinol. 2020, 11, 445.
- Du, Y.; Zhu, Y.J.; Zeng, B.; Mu, X.L.; Liu, J.Y. Super-Resolution Quantification of T2DM-Induced Mitochondrial Morphology Changes and Their Implications in Pharmacodynamics of Metformin and Sorafenib. Front. Pharmacol. 2022, 13, 932116.
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294.
- Segal, E.D.; Yasmeen, A.; Beauchamp, M.C.; Rosenblatt, J.; Pollak, M.; Gotlieb, W.H. Relevance of the OCT1 transporter to the antineoplastic effect of biguanides. Biochem. Biophys. Res. Commun. 2011, 414, 694–699.
- Gupta, S.; Wulf, G.; Henjakovic, M.; Koepsell, H.; Burckhardt, G.; Hagos, Y. Human organic cation transporter 1 is expressed in lymphoma cells and increases susceptibility to irinotecan and paclitaxel. J. Pharmacol. Exp. Ther. 2012, 341, 16–23.
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337.
- Salani, B.; Marini, C.; Rio, A.D.; Ravera, S.; Massollo, M.; Orengo, A.M.; Amaro, A.; Passalacqua, M.; Maffioli, S.; Pfeffer, U.; et al. Metformin impairs glucose consumption and survival in Calu-1 cells by direct inhibition of hexokinase-II. Sci. Rep. 2013, 3, 2070.
More
Information
Subjects:
Medicine, General & Internal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
908
Revisions:
2 times
(View History)
Update Date:
30 Jan 2023
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No