 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jasmin Galper | -- | 2608 | 2023-01-19 00:26:36 | | | |
| 2 | Rita Xu | -5 word(s) | 2603 | 2023-01-19 03:28:18 | | |
Video Upload Options
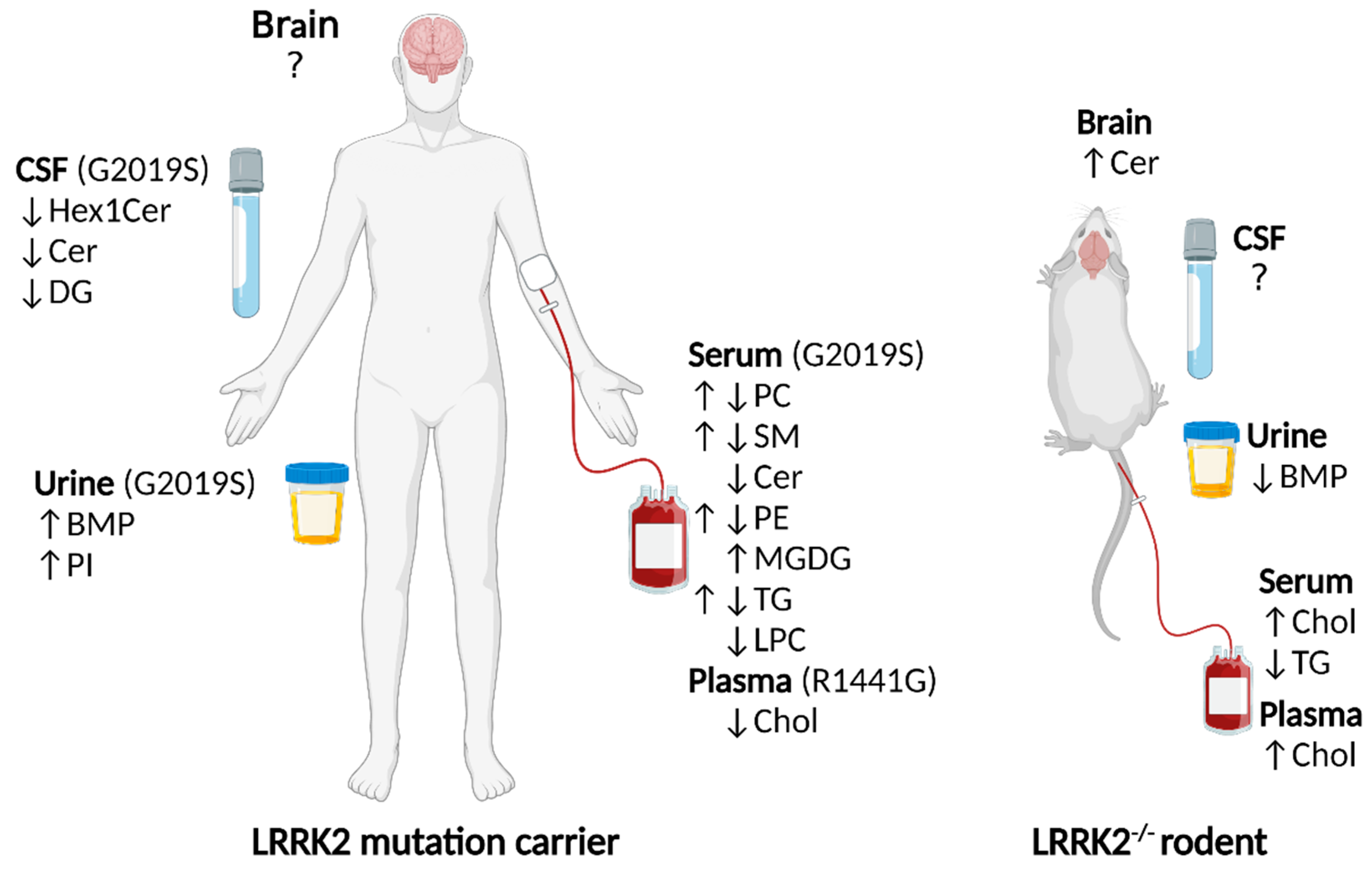
Genetic alterations in the LRRK2 gene, encoding leucine-rich repeat kinase 2, are a common risk factor for Parkinson’s disease. How LRRK2 alterations lead to cell pathology is an area of ongoing investigation, multiple lines of evidence suggest a role for LRRK2 in lipid pathways. It is increasingly recognized that in addition to being energy reservoirs and structural entities, some lipids, including neural lipids, participate in signaling cascades. Early investigations revealed that LRRK2 localized to membranous and vesicular structures, suggesting an interaction of LRRK2 and lipids or lipid-associated proteins. LRRK2 substrates from the Rab GTPase family play a critical role in vesicle trafficking, lipid metabolism and lipid storage, all processes which rely on lipid dynamics. In addition, LRRK2 is associated with the phosphorylation and activity of enzymes that catabolize plasma membrane and lysosomal lipids. Furthermore, LRRK2 knockout studies have revealed that blood, brain and urine exhibit lipid level changes, including alterations to sterols, sphingolipids and phospholipids, respectively. In human LRRK2 mutation carriers, changes to sterols, sphingolipids, phospholipids, fatty acyls and glycerolipids are reported in multiple tissues.
1. Introduction
1.1. Overview
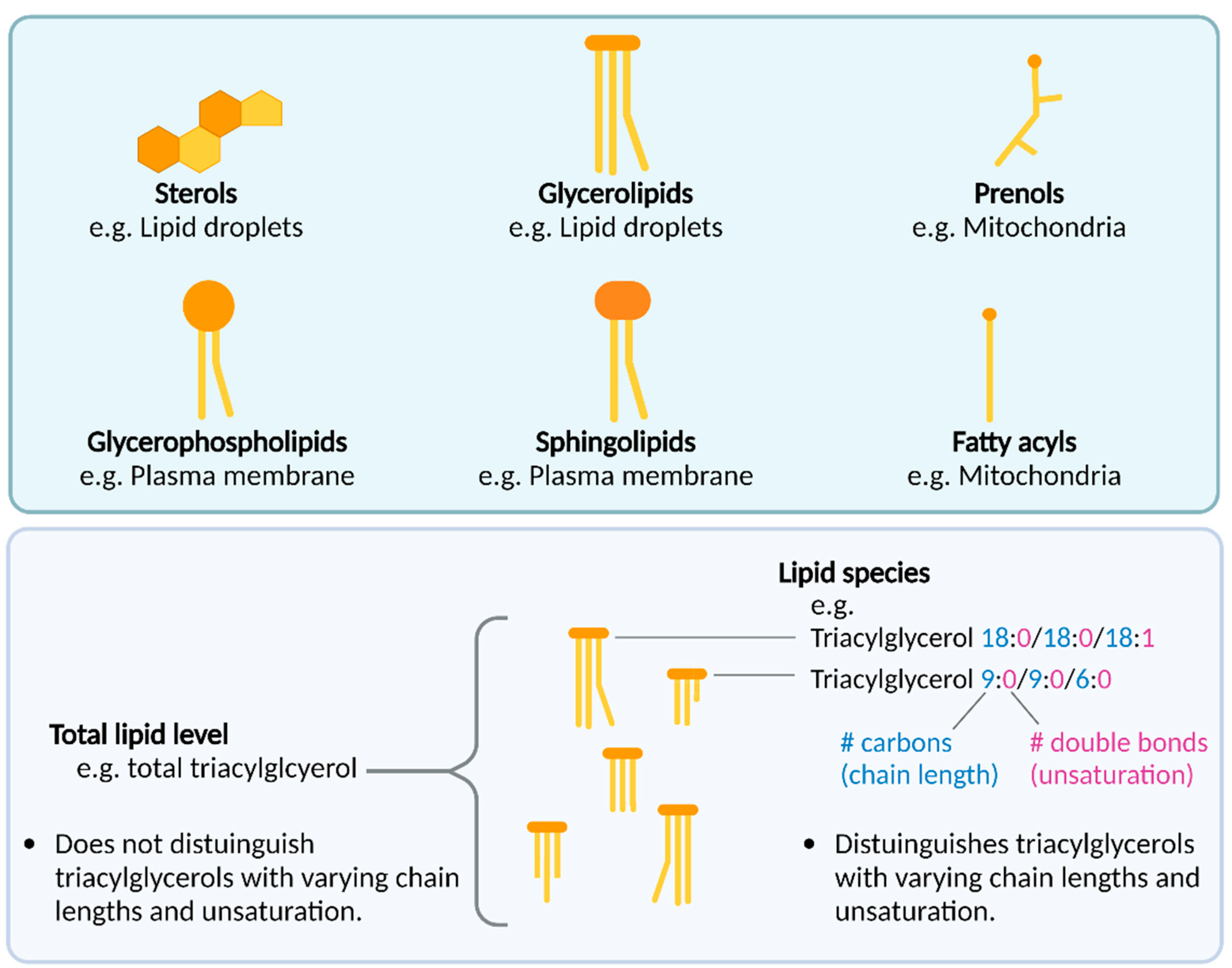
1.2. An Introduction to Lipids
1.3. Lipid Alterations in LRRK2 Knockouts
1.4. Lipid Alterations in LRRK2 Mutation Carrier Humans
2. LRRK2 and Lysosomal Lipid Metabolism
References
- Monfrini, E.; Di Fonzo, A. Leucine-Rich Repeat Kinase (LRRK2) Genetics and Parkinson’s Disease. Adv. Neurobiol. 2017, 14, 3–30.
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590.
- Gilks, W.P.; Abou-Sleiman, P.M.; Gandhi, S.; Jain, S.; Singleton, A.; Lees, A.J.; Shaw, K.; Bhatia, K.P.; Bonifati, V.; Quinn, N.P.; et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 2005, 365, 415–416.
- Biskup, S.; Moore, D.J.; Celsi, F.; Higashi, S.; West, A.B.; Andrabi, S.A.; Kurkinen, K.; Yu, S.-W.; Savitt, J.M.; Waldvogel, H.J.; et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 2006, 60, 557–569.
- Shin, N.; Jeong, H.; Kwon, J.; Heo, H.Y.; Kwon, J.J.; Yun, H.J.; Kim, C.H.; Han, B.S.; Tong, Y.; Shen, J.; et al. LRRK2 regulates synaptic vesicle endocytosis. Exp. Cell Res. 2008, 314, 2055–2065.
- Piccoli, G.; Condliffe, S.B.; Bauer, M.; Giesert, F.; Boldt, K.; De Astis, S.; Meixner, A.; Sarioglu, H.; Vogt-Weisenhorn, D.M.; Wurst, W.; et al. LRRK2 Controls Synaptic Vesicle Storage and Mobilization within the Recycling Pool. J. Neurosci. 2011, 31, 2225–2237.
- Alegre-Abarrategui, J.; Christian, H.; Lufino, M.M.; Mutihac, R.; Venda, L.L.; Ansorge, O.; Wade-Martins, R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 2009, 18, 4022–4034.
- Piomelli, D.; Astarita, G.; Rapaka, R. A neuroscientist’s guide to lipidomics. Nat. Rev. Neurosci. 2007, 8, 743–754.
- Yoon, J.H.; Seo, Y.; Jo, Y.S.; Lee, S.; Cho, E.; Cazenave-Gassiot, A.; Shin, Y.-S.; Moon, M.H.; An, H.J.; Wenk, M.R.; et al. Brain lipidomics: From functional landscape to clinical significance. Sci. Adv. 2022, 8, eadc9317.
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell. Biol. 2015, 33, 125–131.
- Holthuis, J.C.M.; Menon, A.K. Lipid landscapes and pipelines in membrane homeostasis. Nature 2014, 510, 48–57.
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14.
- Ferrazza, R.; Cogo, S.; Melrose, H.; Bubacco, L.; Greggio, E.; Guella, G.; Civiero, L.; Plotegher, N. LRRK2 deficiency impacts ceramide metabolism in brain. Biochem. Biophys. Res. Commun. 2016, 478, 1141–1146.
- Fuji, R.N.; Flagella, M.; Baca, M.; Baptista, M.A.S.; Brodbeck, J.; Chan, B.K.; Fiske, B.K.; Honigberg, L.; Jubb, A.M.; Katavolos, P.; et al. Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci. Transl. Med. 2015, 7, 273ra215.
- Baptista, M.A.S.; Dave, K.D.; Frasier, M.A.; Sherer, T.B.; Greeley, M.; Beck, M.J.; Varsho, J.S.; Parker, G.A.; Moore, C.; Churchill, M.J.; et al. Loss of Leucine-Rich Repeat Kinase 2 (LRRK2) in Rats Leads to Progressive Abnormal Phenotypes in Peripheral Organs. PLoS ONE 2013, 8, e80705.
- Ness, D.; Ren, Z.; Gardai, S.; Sharpnack, D.; Johnson, V.J.; Brennan, R.J.; Brigham, E.F.; Olaharski, A.J. Leucine-Rich Repeat Kinase 2 (LRRK2)-Deficient Rats Exhibit Renal Tubule Injury and Perturbations in Metabolic and Immunological Homeostasis. PLoS ONE 2013, 8, e66164.
- Boddu, R.; Hull, T.D.; Bolisetty, S.; Hu, X.; Moehle, M.S.; Daher, J.P.L.; Kamal, A.I.; Joseph, R.; George, J.F.; Agarwal, A.; et al. Leucine-rich repeat kinase 2 deficiency is protective in rhabdomyolysis-induced kidney injury. Hum. Mol. Genet. 2015, 24, 4078–4093.
- Abbott, S.K.; Li, H.; Muñoz, S.S.; Knoch, B.; Batterham, M.; Murphy, K.E.; Halliday, G.M.; Garner, B. Altered ceramide acyl chain length and ceramide synthase gene expression in Parkinson’s disease. Mov. Disord. 2014, 29, 518–526.
- Bardien, S.; Lesage, S.; Brice, A.; Carr, J. Genetic characteristics of leucine-rich repeat kinase 2 (LRRK2) associated Parkinson’s disease. Park. Relat. Disord. 2011, 17, 501–508.
- Yu, M.; Arshad, M.; Wang, W.; Zhao, D.; Xu, L.; Zhou, L. LRRK2 mediated Rab8a phosphorylation promotes lipid storage. Lipids Health Dis. 2018, 17, 34.
- Whiffin, N.; Armean, I.M.; Kleinman, A.; Marshall, J.L.; Minikel, E.V.; Goodrich, J.K.; Quaife, N.M.; Cole, J.B.; Wang, Q.; Karczewski, K.J.; et al. The effect of LRRK2 loss-of-function variants in humans. Nat. Med. 2020, 26, 869–877.
- Yakhine-Diop, S.M.S.; Morales-García, J.A.; Niso-Santano, M.; González-Polo, R.A.; Uribe-Carretero, E.; Martinez-Chacon, G.; Durand, S.; Maiuri, M.C.; Aiastui, A.; Zulaica, M.; et al. Metabolic alterations in plasma from patients with familial and idiopathic Parkinson’s disease. Aging (Albany NY) 2020, 12, 16690–16708.
- Taylor, M.; Alessi, D.R. Advances in elucidating the function of leucine-rich repeat protein kinase-2 in normal cells and Parkinson’s disease. Curr. Opin. Cell. Biol. 2020, 63, 102–113.
- Alcalay, R.N.; Hsieh, F.; Tengstrand, E.; Padmanabhan, S.; Baptista, M.; Kehoe, C.; Narayan, S.; Boehme, A.K.; Merchant, K. Higher Urine bis (Monoacylglycerol) Phosphate Levels in LRRK2 G2019S Mutation Carriers: Implications for Therapeutic Development. Mov. Disord. 2020, 35, 134–141.
- Gomes, S.; Garrido, A.; Tonelli, F.; Obiang, D.; Tolosa, E.; Marti, M.J.; Ruiz-Martinez, J.; Aragon, A.V.; Hernandez-Eguiazu, H.; Croitoru, I.; et al. Elevated urine BMPs—bis (Monoacylglycerol) Phosphates—in LRRK2 G2019S, R1441G/C and VPS35 D620N mutation carriers with and without Parkinson’s disease. medRxiv 2022.
- Galper, J.; Dean, N.J.; Pickford, R.; Lewis, S.J.G.; Halliday, G.M.; Kim, W.S.; Dzamko, N. Lipid pathway dysfunction is prevalent in patients with Parkinson’s disease. Brain J. Neurol. 2022.
- Stiban, J.; Perera, M. Very long chain ceramides interfere with C16-ceramide-induced channel formation: A plausible mechanism for regulating the initiation of intrinsic apoptosis. Biochim. Biophys. Acta (BBA) Biomembr. 2015, 1848, 561–567.
- Hartmann, D.; Wegner, M.-S.; Wanger, R.A.; Ferreirós, N.; Schreiber, Y.; Lucks, J.; Schiffmann, S.; Geisslinger, G.; Grösch, S. The equilibrium between long and very long chain ceramides is important for the fate of the cell and can be influenced by co-expression of CerS. Int. J. Biochem. Cell Biol. 2013, 45, 1195–1203.
- Abou-Ghali, M.; Stiban, J. Regulation of ceramide channel formation and disassembly: Insights on the initiation of apoptosis. Saudi J. Biol. Sci. 2015, 22, 760–772.
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2020.
- McGranaghan, P.; Kirwan, J.A.; Garcia-Rivera, M.A.; Pieske, B.; Edelmann, F.; Blaschke, F.; Appunni, S.; Saxena, A.; Rubens, M.; Veledar, E.; et al. Lipid Metabolite Biomarkers in Cardiovascular Disease: Discovery and Biomechanism Translation from Human Studies. Metabolites 2021, 11, 621.
- Fu, X.; Wang, Y.; He, X.; Li, H.; Liu, H.; Zhang, X. A systematic review and meta-analysis of serum cholesterol and triglyceride levels in patients with Parkinson’s disease. Lipids Health Dis. 2020, 19, 97.
- Jiang, Z.; Xu, X.; Gu, X.; Ou, R.; Luo, X.; Shang, H.; Song, W. Effects of Higher Serum Lipid Levels on the Risk of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Neurol. 2020, 11, 597.
- Thaler, A.; Shenhar-Tsarfaty, S.; Shaked, Y.; Gurevich, T.; Omer, N.; Bar-Shira, A.; Gana-Weisz, M.; Goldstein, O.; Kestenbaum, M.; Cedarbaum, J.M.; et al. Metabolic syndrome does not influence the phenotype of LRRK2 and GBA related Parkinson’s disease. Sci. Rep. 2020, 10, 9329.
- Macías-García, D.; Periñán, M.T.; Muñoz-Delgado, L.; Jimenez-Jaraba, M.V.; Labrador-Espinosa, M.Á.; Jesús, S.; Buiza-Rueda, D.; Méndez-Del Barrio, C.; Adarmes-Gómez, A.; Gómez-Garre, P.; et al. Serum lipid profile among sporadic and familial forms of Parkinson’s disease. Npj Park. Dis. 2021, 7, 59.
- Johansen, C.T.; Hegele, R.A. Genetic bases of hypertriglyceridemic phenotypes. Curr. Opin. Lipidol. 2011, 22, 247–253.
- Li, L.-H.; Dutkiewicz, E.P.; Huang, Y.-C.; Zhou, H.-B.; Hsu, C.-C. Analytical methods for cholesterol quantification. J. Food Drug Anal. 2019, 27, 375–386.
- Aviram, R.; Manella, G.; Kopelman, N.; Neufeld-Cohen, A.; Zwighaft, Z.; Elimelech, M.; Adamovich, Y.; Golik, M.; Wang, C.; Han, X.; et al. Lipidomics Analyses Reveal Temporal and Spatial Lipid Organization and Uncover Daily Oscillations in Intracellular Organelles. Mol. Cell 2016, 62, 636–648.
- Vance, J.E. Dysregulation of cholesterol balance in the brain: Contribution to neurodegenerative diseases. Dis. Model. Mech. 2012, 5, 746–755.
- Phan, K.; He, Y.; Pickford, R.; Bhatia, S.; Katzeff, J.S.; Hodges, J.R.; Piguet, O.; Halliday, G.M.; Kim, W.S. Uncovering pathophysiological changes in frontotemporal dementia using serum lipids. Sci. Rep. 2020, 10, 3640.
- Roosen, D.A.; Cookson, M.R. LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol. Neurodegener. 2016, 11, 73.
- Henry, A.G.; Aghamohammadzadeh, S.; Samaroo, H.; Chen, Y.; Mou, K.; Needle, E.; Hirst, W.D. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum. Mol. Genet. 2015, 24, 6013–6028.
- Tong, Y.; Yamaguchi, H.; Giaime, E.; Boyle, S.; Kopan, R.; Kelleher, R.J.; Shen, J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of α-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. 2010, 107, 9879–9884.
- Tong, Y.; Giaime, E.; Yamaguchi, H.; Ichimura, T.; Liu, Y.; Si, H.; Cai, H.; Bonventre, J.V.; Shen, J. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol. Neurodegener. 2012, 7, 2.
- Keller, J.N.; Dimayuga, E.; Chen, Q.; Thorpe, J.; Gee, J.; Ding, Q. Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int. J. Biochem. Cell Biol. 2004, 36, 2376–2391.
- Kobayashi, T.; Beuchat, M.-H.; Chevallier, J.; Makino, A.; Mayran, N.; Escola, J.-M.; Lebrand, C.; Cosson, P.; Kobayashi, T.; Gruenberg, J. Separation and Characterization of Late Endosomal Membrane Domains*. J. Biol. Chem. 2002, 277, 32157–32164.
- Kobayashi, T.; Stang, E.; Fang, K.S.; de Moerloose, P.; Parton, R.G.; Gruenberg, J. A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature 1998, 392, 193–197.
- Kobayashi, T.; Beuchat, M.-H.; Lindsay, M.; Frias, S.; Palmiter, R.D.; Sakuraba, H.; Parton, R.G.; Gruenberg, J. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol. 1999, 1, 113–118.
- Chevallier, J.; Chamoun, Z.; Jiang, G.; Prestwich, G.; Sakai, N.; Matile, S.; Parton, R.G.; Gruenberg, J. Lysobisphosphatidic Acid Controls Endosomal Cholesterol Levels*. J. Biol. Chem. 2008, 283, 27871–27880.
- Ilnytska, O.; Jeziorek, M.; Lai, K.; Altan-Bonnet, N.; Dobrowolski, R.; Storch, J. Lysobisphosphatidic acid (LBPA) enrichment promotes cholesterol egress via exosomes in Niemann Pick type C1 deficient cells. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2021, 1866, 158916.
- Ebner, M.; Koch, P.A.; Haucke, V. Phosphoinositides in the control of lysosome function and homeostasis. Biochem. Soc. Trans. 2019, 47, 1173–1185.
- Albanese, F.; Mercatelli, D.; Finetti, L.; Lamonaca, G.; Pizzi, S.; Shimshek, D.R.; Bernacchia, G.; Morari, M. Constitutive silencing of LRRK2 kinase activity leads to early glucocerebrosidase deregulation and late impairment of autophagy in vivo. Neurobiol. Dis. 2021, 159, 105487.
- Alcalay, R.N.; Levy, O.A.; Waters, C.H.; Fahn, S.; Ford, B.; Kuo, S.-H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain J. Neurol. 2015, 138, 2648–2658.
- Ysselstein, D.; Nguyen, M.; Young, T.J.; Severino, A.; Schwake, M.; Merchant, K.; Krainc, D. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat. Commun. 2019, 10, 5570.
- Kedariti, M.; Frattini, E.; Baden, P.; Cogo, S.; Civiero, L.; Ziviani, E.; Zilio, G.; Bertoli, F.; Aureli, M.; Kaganovich, A.; et al. LRRK2 kinase activity regulates GCase level and enzymatic activity differently depending on cell type in Parkinson’s disease. Npj Park. Dis. 2022, 8, 92.
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. New Engl. J. Med. 2009, 361, 1651–1661.
- Wang, Y.; Liu, L.; Xiong, J.; Zhang, X.; Chen, Z.; Yu, L.; Chen, C.; Huang, J.; Zhang, Z.; Mohmed, A.A.; et al. Glucocerebrosidase L444P mutation confers genetic risk for Parkinson’s disease in central China. Behav. Brain Funct. 2012, 8, 57.
- Guimarães Bde, C.; Pereira, A.C.; Rodrigues Fda, C.; dos Santos, A.V.; Campos, M., Jr.; dos Santos, J.M.; dos Santos, F.L.; de Rosso, A.L.; Nicaretta, D.H.; Pereira, J.S.; et al. Glucocerebrosidase N370S and L444P mutations as risk factors for Parkinson’s disease in Brazilian patients. Park. Relat. Disord 2012, 18, 688–689.
- Aharon-Peretz, J.; Rosenbaum, H.; Gershoni-Baruch, R. Mutations in the Glucocerebrosidase Gene and Parkinson’s Disease in Ashkenazi Jews. N. Engl. J. Med. 2004, 351, 1972–1977.
- Vaccaro, A.M.; Muscillo, M.; Suzuki, K. Characterization of human glucosylsphingosine glucosyl hydrolase and comparison with glucosylceramidase. Eur. J. Biochem. 1985, 146, 315–321.
- Smith, L.; Schapira, A.H.V. GBA Variants and Parkinson Disease: Mechanisms and Treatments. Cells 2022, 11, 1261.
- Zhao, Y.; Perera, G.; Takahashi-Fujigasaki, J.; Mash, D.C.; Vonsattel, J.P.G.; Uchino, A.; Hasegawa, K.; Jeremy Nichols, R.; Holton, J.L.; Murayama, S.; et al. Reduced LRRK2 in association with retromer dysfunction in post-mortem brain tissue from LRRK2 mutation carriers. Brain J. Neurol. 2017, 141, 486–495.
- Omer, N.; Giladi, N.; Gurevich, T.; Bar-Shira, A.; Gana-Weisz, M.; Glinka, T.; Goldstein, O.; Kestenbaum, M.; Cedarbaum, J.M.; Mabrouk, O.S.; et al. Glucocerebrosidase Activity is not Associated with Parkinson’s Disease Risk or Severity. Mov. Disord. 2022, 37, 190–195.
- Ortega, R.A.; Bodamer, O.; Peake, R.W.A.; Raymond, D.; Bressman, S.B.; Saunders-Pullman, R. Assessment of Glucocerebrosidase Enzyme Activity in Parkinson Disease Using Multiple Approaches. Mov. Disord. 2022, 37, 655–656.
- Ysselstein, D.; Young, T.J.; Nguyen, M.; Padmanabhan, S.; Hirst, W.D.; Dzamko, N.; Krainc, D. Evaluation of Strategies for Measuring Lysosomal Glucocerebrosidase Activity. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 2719–2730.
- Stoffel, W. Functional analysis of acid and neutral sphingomyelinases in vitro and in vivo. Chem. Phys. Lipids 1999, 102, 107–121.
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516.
- Gan-Or, Z.; Ozelius, L.J.; Bar-Shira, A.; Saunders-Pullman, R.; Mirelman, A.; Kornreich, R.; Gana-Weisz, M.; Raymond, D.; Rozenkrantz, L.; Deik, A.; et al. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 2013, 80, 1606–1610.
- Parveen, F.; Bender, D.; Law, S.-H.; Mishra, V.K.; Chen, C.-C.; Ke, L.-Y. Role of Ceramidases in Sphingolipid Metabolism and Human Diseases. Cells 2019, 8, 1573.
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M.; Consortium, I.P.s.D.G. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain J. Neurol. 2017, 140, 3191–3203.

