 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | José Luis Marcos Camus | -- | 1935 | 2023-01-18 23:41:13 | | | |
| 2 | Jessie Wu | Meta information modification | 1935 | 2023-01-19 03:15:36 | | | | |
| 3 | Jessie Wu | + 3 word(s) | 1938 | 2023-01-19 03:25:36 | | |
Video Upload Options
Obesity-induced neuroinflammation is a chronic aseptic central nervous system inflammation that presents systemic characteristics associated with increased pro-inflammatory cytokines such as interleukin 1 beta (IL-1β) and interleukin 18 (IL-18) and the presence of microglia and reactive astrogliosis as well as the activation of the NOD-like receptor protein 3 (NLRP3) inflammasome. The obesity pandemic is associated with lifestyle changes, including an excessive intake of obesogenic foods and decreased physical activity. Brain areas such as the lateral hypothalamus (LH), lateral septum (LS), ventral tegmental area (VTA), and nucleus accumbens (NAcc) have been implicated in the homeostatic and hedonic control of feeding in experimental models of diet-induced obesity. A chronic lipid intake triggers neuroinflammation in several brain regions such as the hypothalamus, hippocampus, and amygdala.
1. Introduction
2. Inflammation
2.1. Peripheral Inflammation
2.2. Central Inflammation or Neuroinflammation
2.2.1. Neuroinflammatory Mechanisms
Blood-Brain Barrier
Fatty Acids
2.2.2. Role of Glial Cells in Obesity
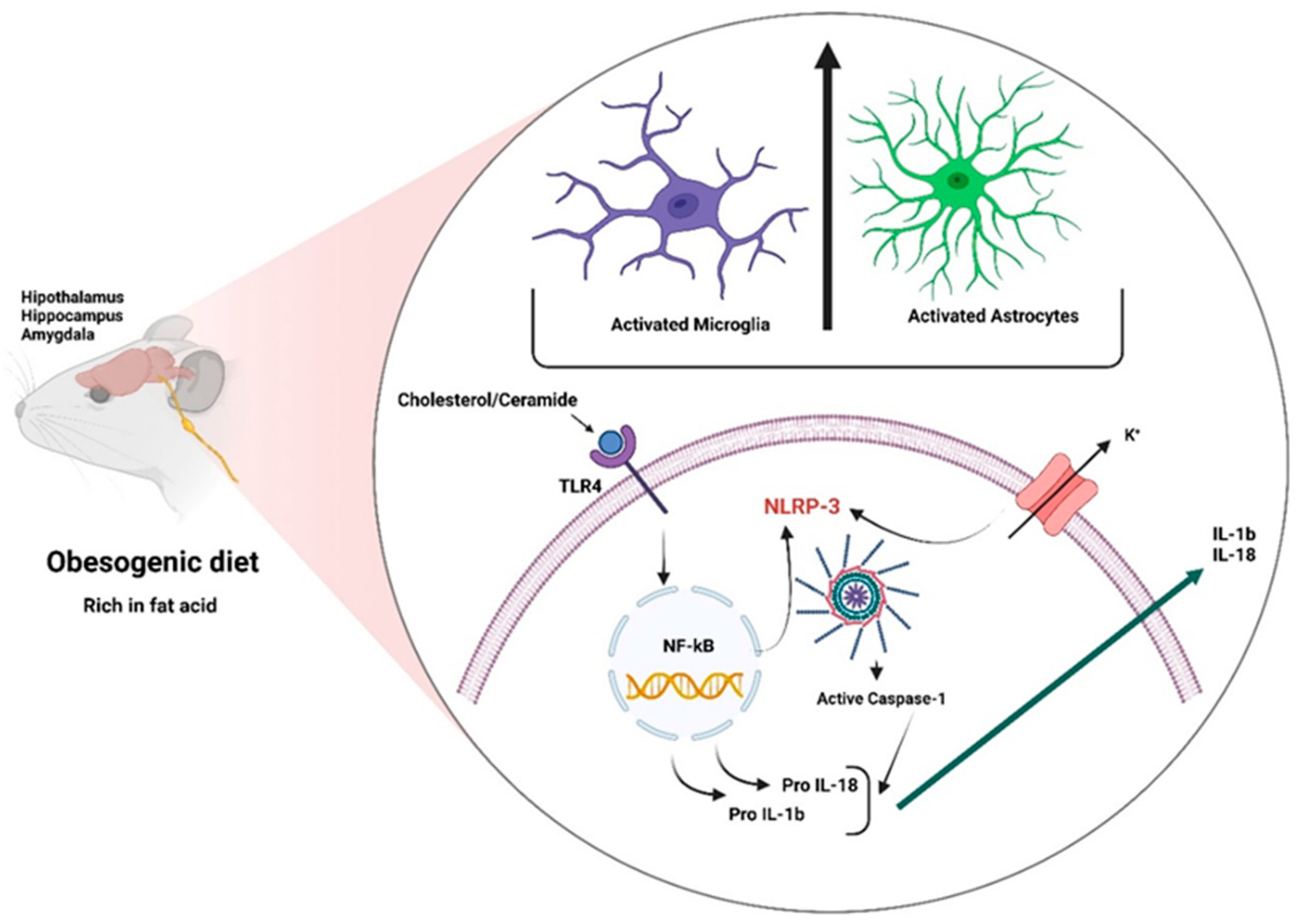
2.2.3. Inflammasomes
References
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.; Velloso, L.A. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 2005, 146, 4192–4199.
- Thaler, J.P.; Guyenet, S.J.; Dorfman, M.D.; Wisse, B.E.; Schwartz, M.W. Hypothalamic inflammation: Marker or mechanism of obesity pathogenesis? Diabetes 2013, 62, 2629–2634.
- Buckman, L.B.; Thompson, M.M.; Moreno, H.N.; Ellacott, K.L. Regional astrogliosis in the mouse hypothalamus in response to obesity. J. Comp. Neurol. 2013, 521, 1322–1333.
- Waise, T.M.Z.; Toshinai, K.; Naznin, F.; NamKoong, C.; Md Moin, A.S.; Sakoda, H.; Nakazato, M. One-day high-fat diet induces inflammation in the nodose ganglion and hypothalamus of mice. Biochem. Biophys. Res. Commun. 2015, 464, 1157–1162.
- Lee, B.C.; Lee, J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim. Biophys. Acta 2014, 1842, 446–462.
- Lee, T.J.; Hargrave, S.L.; Kinzig, K.P. Dual functions of CNS inflammation in food intake and metabolic regulation. Brain Res. 2020, 1740, 146859.
- Pan, W.; Allison, M.B.; Sabatini, P.; Rupp, A.; Adams, J.; Patterson, C.; Jones, J.C.; Olson, D.P.; Myers, M.G., Jr. Transcriptional and physiological roles for STAT proteins in leptin action. Mol. Metab. 2019, 22, 121–131.
- Dorfman, M.D.; Thaler, J.P. Hypothalamic inflammation and gliosis in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 325–330.
- Cai, M.; Wang, H.; Li, J.J.; Zhang, Y.L.; Xin, L.; Li, F.; Lou, S.J. The signaling mechanisms of hippocampal endoplasmic reticulum stress affecting neuronal plasticity-related protein levels in high fat diet-induced obese rats and the regulation of aerobic exercise. Brain Behav. Immun. 2016, 57, 347–359.
- Seong, J.; Kang, J.Y.; Sun, J.S.; Kim, K.W. Hypothalamic inflammation and obesity: A mechanistic review. Arch. Pharm. Res. 2019, 42, 383–392.
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633.
- Unamuno, X.; Gomez-Ambrosi, J.; Rodriguez, A.; Becerril, S.; Fruhbeck, G.; Catalan, V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur. J. Clin. Invest. 2018, 48, e12997.
- Trim, W.V.; Lynch, L. Immune and non-immune functions of adipose tissue leukocytes. Nat Rev Immunol 2022, 22, 371–386.
- Fritsche, K.L. The science of fatty acids and inflammation. Adv. Nutr. 2015, 6, 293S–301S.
- Wang, Z.; Liu, D.; Wang, F.; Liu, S.; Zhao, S.; Ling, E.A.; Hao, A. Saturated fatty acids activate microglia via Toll-like receptor 4/NF-kappaB signalling. Br. J. Nutr. 2012, 107, 229–241.
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668.
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997, 389, 610–614.
- Camandola, S.; Mattson, M.P. Toll-like receptor 4 mediates fat, sugar, and umami taste preference and food intake and body weight regulation. Obesity 2017, 25, 1237–1245.
- Reynolds, C.M.; McGillicuddy, F.C.; Harford, K.A.; Finucane, O.M.; Mills, K.H.; Roche, H.M. Dietary saturated fatty acids prime the NLRP3 inflammasome via TLR4 in dendritic cells-implications for diet-induced insulin resistance. Mol. Nutr. Food Res. 2012, 56, 1212–1222.
- Genser, L.; Poitou, C.; Brot-Laroche, E.; Rousset, M.; Vaillant, J.C.; Clement, K.; Thenet, S.; Leturque, A. Alteration of intestinal permeability: The missing link between gut microbiota modifications and inflammation in obesity? Med. Sci. (Paris) 2016, 32, 461–469.
- Cox, A.J.; West, N.P.; Cripps, A.W. Obesity, inflammation, and the gut microbiota. Lancet Diabetes Endocrinol 2015, 3, 207–215.
- Kang, C.; Wang, B.; Kaliannan, K.; Wang, X.; Lang, H.; Hui, S.; Huang, L.; Zhang, Y.; Zhou, M.; Chen, M.; et al. Gut Microbiota Mediates the Protective Effects of Dietary Capsaicin against Chronic Low-Grade Inflammation and Associated Obesity Induced by High-Fat Diet. MBio 2017, 8, e00470-17.
- Bauer, K.C.; Huus, K.E.; Finlay, B.B. Microbes and the mind: Emerging hallmarks of the gut microbiota-brain axis. Cell Microbiol. 2016, 18, 632–644.
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321.
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73.
- Ganong, W.F. Circumventricular organs: Definition and role in the regulation of endocrine and autonomic function. Clin. Exp. Pharm. Physiol. 2000, 27, 422–427.
- Mullier, A.; Bouret, S.G.; Prevot, V.; Dehouck, B. Differential distribution of tight junction proteins suggests a role for tanycytes in blood-hypothalamus barrier regulation in the adult mouse brain. J. Comp. Neurol. 2010, 518, 943–962.
- Rodriguez, E.M.; Blazquez, J.L.; Guerra, M. The design of barriers in the hypothalamus allows the median eminence and the arcuate nucleus to enjoy private milieus: The former opens to the portal blood and the latter to the cerebrospinal fluid. Peptides 2010, 31, 757–776.
- Buckman, L.B.; Hasty, A.H.; Flaherty, D.K.; Buckman, C.T.; Thompson, M.M.; Matlock, B.K.; Weller, K.; Ellacott, K.L. Obesity induced by a high-fat diet is associated with increased immune cell entry into the central nervous system. Brain Behav. Immun. 2014, 35, 33–42.
- Hsuchou, H.; Kastin, A.J.; Mishra, P.K.; Pan, W. C-reactive protein increases BBB permeability: Implications for obesity and neuroinflammation. Cell Physiol. Biochem. 2012, 30, 1109–1119.
- Stranahan, A.M.; Hao, S.; Dey, A.; Yu, X.; Baban, B. Blood-brain barrier breakdown promotes macrophage infiltration and cognitive impairment in leptin receptor-deficient mice. J. Cereb. Blood Flow Metab. 2016, 36, 2108–2121.
- Nerurkar, P.V.; Johns, L.M.; Buesa, L.M.; Kipyakwai, G.; Volper, E.; Sato, R.; Shah, P.; Feher, D.; Williams, P.G.; Nerurkar, V.R. Momordica charantia (bitter melon) attenuates high-fat diet-associated oxidative stress and neuroinflammation. J. Neuroinflammation 2011, 8, 64.
- Chang, H.C.; Tai, Y.T.; Cherng, Y.G.; Lin, J.W.; Liu, S.H.; Chen, T.L.; Chen, R.M. Resveratrol attenuates high-fat diet-induced disruption of the blood-brain barrier and protects brain neurons from apoptotic insults. J. Agric. Food Chem. 2014, 62, 3466–3475.
- Elahy, M.; Lam, V.; Pallebage-Gamarallage, M.M.; Giles, C.; Mamo, J.C.; Takechi, R. Nicotine Attenuates Disruption of Blood-Brain Barrier Induced by Saturated-Fat Feeding in Wild-Type Mice. Nicotine Tob. Res. 2015, 17, 1436–1441.
- Kanoski, S.E.; Zhang, Y.; Zheng, W.; Davidson, T.L. The effects of a high-energy diet on hippocampal function and blood-brain barrier integrity in the rat. J. Alzheimers Dis. 2010, 21, 207–219.
- Douglass, J.D.; Dorfman, M.D.; Fasnacht, R.; Shaffer, L.D.; Thaler, J.P. Astrocyte IKKbeta/NF-kappaB signaling is required for diet-induced obesity and hypothalamic inflammation. Mol. Metab. 2017, 6, 366–373.
- Valdearcos, M.; Robblee, M.M.; Benjamin, D.I.; Nomura, D.K.; Xu, A.W.; Koliwad, S.K. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 2014, 9, 2124–2138.
- Borg, M.L.; Omran, S.F.; Weir, J.; Meikle, P.J.; Watt, M.J. Consumption of a high-fat diet, but not regular endurance exercise training, regulates hypothalamic lipid accumulation in mice. J. Physiol. 2012, 590, 4377–4389.
- Thaler, J.P.; Choi, S.J.; Schwartz, M.W.; Wisse, B.E. Hypothalamic inflammation and energy homeostasis: Resolving the paradox. Front. Neuroendocr. 2010, 31, 79–84.
- Andre, C.; Guzman-Quevedo, O.; Rey, C.; Remus-Borel, J.; Clark, S.; Castellanos-Jankiewicz, A.; Ladeveze, E.; Leste-Lasserre, T.; Nadjar, A.; Abrous, D.N.; et al. Inhibiting Microglia Expansion Prevents Diet-Induced Hypothalamic and Peripheral Inflammation. Diabetes 2017, 66, 908–919.
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194.
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16.
- Valdearcos, M.; Xu, A.W.; Koliwad, S.K. Hypothalamic inflammation in the control of metabolic function. Annu. Rev. Physiol. 2015, 77, 131–160.
- Buttini, M.; Limonta, S.; Boddeke, H.W. Peripheral administration of lipopolysaccharide induces activation of microglial cells in rat brain. Neurochem. Int. 1996, 29, 25–35.
- Garcia-Bueno, B.; Serrats, J.; Sawchenko, P.E. Cerebrovascular cyclooxygenase-1 expression, regulation, and role in hypothalamic-pituitary-adrenal axis activation by inflammatory stimuli. J. Neurosci. 2009, 29, 12970–12981.
- Shih, R.H.; Wang, C.Y.; Yang, C.M. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci. 2015, 8, 77.
- Cintra, D.E.; Ropelle, E.R.; Moraes, J.C.; Pauli, J.R.; Morari, J.; Souza, C.T.; Grimaldi, R.; Stahl, M.; Carvalheira, J.B.; Saad, M.J.; et al. Unsaturated fatty acids revert diet-induced hypothalamic inflammation in obesity. PLoS ONE 2012, 7, e30571.
- Wang, Y.; Qian, Y.; Fang, Q.; Zhong, P.; Li, W.; Wang, L.; Fu, W.; Zhang, Y.; Xu, Z.; Li, X.; et al. Saturated palmitic acid induces myocardial inflammatory injuries through direct binding to TLR4 accessory protein MD2. Nat. Commun. 2017, 8, 13997.
- Valdearcos, M.; Douglass, J.D.; Robblee, M.M.; Dorfman, M.D.; Stifler, D.R.; Bennett, M.L.; Gerritse, I.; Fasnacht, R.; Barres, B.A.; Thaler, J.P.; et al. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab. 2017, 26, 185–197 e3.
- Cope, E.C.; LaMarca, E.A.; Monari, P.K.; Olson, L.B.; Martinez, S.; Zych, A.D.; Katchur, N.J.; Gould, E. Microglia Play an Active Role in Obesity-Associated Cognitive Decline. J. Neurosci. 2018, 38, 8889–8904.
- Butler, M.J.; Cole, R.M.; Deems, N.P.; Belury, M.A.; Barrientos, R.M. Fatty food, fatty acids, and microglial priming in the adult and aged hippocampus and amygdala. Brain Behav. Immun. 2020, 89, 145–158.
- Hahm, J.R.; Jo, M.H.; Ullah, R.; Kim, M.W.; Kim, M.O. Metabolic Stress Alters Antioxidant Systems, Suppresses the Adiponectin Receptor 1 and Induces Alzheimer’s Like Pathology in Mice Brain. Cells 2020, 9, 249.
- Jin, S.; Kim, K.K.; Park, B.S.; Kim, D.H.; Jeong, B.; Kang, D.; Lee, T.H.; Park, J.W.; Kim, J.G.; Lee, B.J. Function of astrocyte MyD88 in high-fat-diet-induced hypothalamic inflammation. J. Neuroinflammation 2020, 17, 195.
- Shi, H.; Wang, Q.; Zheng, M.; Hao, S.; Lum, J.S.; Chen, X.; Huang, X.F.; Yu, Y.; Zheng, K. Supplement of microbiota-accessible carbohydrates prevents neuroinflammation and cognitive decline by improving the gut microbiota-brain axis in diet-induced obese mice. J. Neuroinflammation 2020, 17, 77.
- Kim, J.D.; Yoon, N.A.; Jin, S.; Diano, S. Microglial UCP2 Mediates Inflammation and Obesity Induced by High-Fat Feeding. Cell Metab. 2019, 30, 952–962 e5.
- Robison, L.S.; Gannon, O.J.; Thomas, M.A.; Salinero, A.E.; Abi-Ghanem, C.; Poitelon, Y.; Belin, S.; Zuloaga, K.L. Role of sex and high-fat diet in metabolic and hypothalamic disturbances in the 3xTg-AD mouse model of Alzheimer’s disease. J. Neuroinflammation 2020, 17, 285.
- Shi, H.; Yu, Y.; Lin, D.; Zheng, P.; Zhang, P.; Hu, M.; Wang, Q.; Pan, W.; Yang, X.; Hu, T.; et al. beta-glucan attenuates cognitive impairment via the gut-brain axis in diet-induced obese mice. Microbiome 2020, 8, 143.
- Park, J.H.; Ahn, J.H.; Song, M.; Kim, H.; Park, C.W.; Park, Y.E.; Lee, T.K.; Lee, J.C.; Kim, D.W.; Lee, C.H.; et al. A 2-Min Transient Ischemia Confers Cerebral Ischemic Tolerance in Non-Obese Gerbils, but Results in Neuronal Death in Obese Gerbils by Increasing Abnormal mTOR Activation-Mediated Oxidative Stress and Neuroinflammation. Cells 2019, 8, 1126.
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328.
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298.
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 2012, 28, 137–161.
- de Rivero Vaccari, J.P.; Dietrich, W.D.; Keane, R.W. Activation and regulation of cellular inflammasomes: Gaps in our knowledge for central nervous system injury. J. Cereb. Blood Flow Metab. 2014, 34, 369–375.
- Jorquera, G.; Russell, J.; Monsalves-Alvarez, M.; Cruz, G.; Valladares-Ide, D.; Basualto-Alarcon, C.; Barrientos, G.; Estrada, M.; Llanos, P. NLRP3 Inflammasome: Potential Role in Obesity Related Low-Grade Inflammation and Insulin Resistance in Skeletal Muscle. Int. J. Mol. Sci. 2021, 22, 3254.
- Man, S.M.; Kanneganti, T.D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21.
- Rajamaki, K.; Lappalainen, J.; Oorni, K.; Valimaki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 5, e11765.
- Giordano, A.; Murano, I.; Mondini, E.; Perugini, J.; Smorlesi, A.; Severi, I.; Barazzoni, R.; Scherer, P.E.; Cinti, S. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J. Lipid Res. 2013, 54, 2423–2436.
- Rheinheimer, J.; de Souza, B.M.; Cardoso, N.S.; Bauer, A.C.; Crispim, D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: A systematic review. Metabolism 2017, 74, 1–9.
- Barra, N.G.; Henriksbo, B.D.; Anhe, F.F.; Schertzer, J.D. The NLRP3 inflammasome regulates adipose tissue metabolism. Biochem. J. 2020, 477, 1089–1107.
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 inflammasome: A sensor for metabolic danger? Science 2010, 327, 296–300.

