Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Moncef Zouali | -- | 2043 | 2023-01-12 14:57:12 | | | |
| 2 | Beatrix Zheng | Meta information modification | 2043 | 2023-01-13 02:02:30 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zouali, M. B Cells in Autoinflammatory Disorders. Encyclopedia. Available online: https://encyclopedia.pub/entry/40121 (accessed on 09 August 2026).
Zouali M. B Cells in Autoinflammatory Disorders. Encyclopedia. Available at: https://encyclopedia.pub/entry/40121. Accessed August 09, 2026.
Zouali, Moncef. "B Cells in Autoinflammatory Disorders" Encyclopedia, https://encyclopedia.pub/entry/40121 (accessed August 09, 2026).
Zouali, M. (2023, January 12). B Cells in Autoinflammatory Disorders. In Encyclopedia. https://encyclopedia.pub/entry/40121
Zouali, Moncef. "B Cells in Autoinflammatory Disorders." Encyclopedia. Web. 12 January, 2023.
Copy Citation
Whereas autoimmune diseases are mediated primarily by T and B cells, auto-inflammatory syndromes (AIFS) involve natural killer cells, macrophages, mast cells, dendritic cells, different granulocyte subsets and complement components. In contrast to autoimmune diseases, the immune response of patients with AIFS is not associated with a breakdown of immune tolerance to self-antigens.
B cell
autoinflammation
autoimmunity
mevalonate kinase deficiency syndrome
1. Introduction
In response to exposure to invading pathogens or to endogenous stimuli derived from damaged cells, the immune system mounts inflammatory processes that can lead to tissue repair or pathology. Deciphering the mechanisms and roles of inflammation during physiological and pathological immune responses recently has undergone important advances, driven, in part, by single-cell analysis and high-throughput profiling. In this research arena, studies of a particular group of diseases, called auto-inflammatory syndromes (AIFS), are providing important insights into the links between inflammatory processes and human pathology.
Two decades ago, studies of a subset of human diseases characterized by systemic inflammation and involving cells of the innate immune system, but lacking attributes of autoimmune diseases, led to the concept of autoinflammation. Specifically, investigation of autosomal dominant periodic fever syndromes allowed identification of mis-sense mutations of the gene encoding tumor necrosis factor receptor (TNFR1) associated with low levels of soluble plasma TNFR1 [1]. It was proposed that deregulation of innate immunity pathways can give rise to an auto-inflammatory phenotype. This view implied a clear distinction between autoimmune disease and AIFS, the latter being characterized by deregulation of the innate immune system that causes a hyper-inflammatory state. Neutrophils and monocytes are thought to be the central effector cells, and the adaptive branch of immunity, including B cells and autoantibody production, is considered to play a marginal role, if any [2][3]. On the other hand, loss of immune tolerance to self-antigens is a hallmark of autoimmune diseases, and activation of T cells and B cells can give rise to the production of autoantibodies and autoreactive T cells, resulting in tissue damage of multiple organs [4].
AIFS can be categorized in two subgroups. First, monogenic autoinflammatory disorders result from inborn monogenetic mutations that affect the innate immune system and culminate in undesirable inflammation. They include autosomal dominant TNF receptor-associated periodic syndrome (TRAPS) [5], Familial Mediterranean Fever (FMF), Pyrin-Associated Autoinflammation with Neutrophilic Dermatosis (PAAND), Cryopyrin-associated periodic syndromes (CAPS), NLRP12-associated disorder (NLRP12AD), and mevalonate kinase deficiency (MKD). These conditions are characterized by recurrent febrile episodes, but no infectious agent can be identified [2][3]. Second, polygenic, complex AIFS comprise disorders of unknown etiology, including hereditary and multifactorial disorders. Clinical manifestations are nonspecific, and include arthritis, cutaneous lesions, fever, abdominal pain, arthritis, cutaneous lesions, central nervous system (CNS) involvement and hearing loss. Complex AIFS comprise idiopathic recurrent pericarditis (IRP), adult-onset Still disease (AOSD), Schnitzler syndrome, crystal-induced arthropathies, systemic juvenile idiopathic arthritis (sJIA), Kawasaki disease, idiopathic recurrent pericarditis (IRP), Behçet’ disease and gout, a disorder in which the central event of inflammation is the activation of white blood cells by monosodium urate crystals [6].
2. Multiple Roles of B Cells in Autoinflammatory Disorders
Restriction of inflammatory responses depends, at least in part, on cholesterol metabolism [7]. As a result, patients exhibiting mutations in genes implicated in cholesterol metabolism pathways develop severe and recurring auto-inflammation, together with abnormal B cell responses [8]. By modulating TLR9 signaling, cholesterol metabolism is essential for the induction of human IL-10-secreting regulatory B cells known to play a role in different contexts, including cancer, infection, and autoimmune disease [9]. Downstream of TLR9 engagement, BLIMP1 acts as a transcriptional regulator of IL-10 expression. In humans, loss-of-function mutations in the gene encoding mevalonate kinase underly development of mevalonate kinase deficiency (MKD) syndrome, also called Hyper IgD syndrome. MKD patients are typically diagnosed in early childhood with high serum circulating levels of mevalonic acid, IgA, IgD, and IL-1β [8]. Consequently, the ability of MKD patients to convert mevalonate to mevalonate-5-phosphate is severely impaired, and the diseased subjects exhibit poor regulatory B cell responses [10], significant B cell cytopenia, hypogammaglobulinemia, and an autoinflammatory symptomatology [11]. A combination of prednisone, azathioprine, and intravenous immunoglobulins reduces the incidence and severity of the febrile attacks. In addition to a reduced ability to produce IL-10, a functional impairment in restricting T cell responses has been observed in these patients [10]. It is possible that this B cell defect contributes to the relapsing and remitting episodes of the disease.
Kawasaki syndrome (KS) is responsible for an acquired cardiac disorder in children [12]. It is characterized by fever and coronary artery inflammation and aneurysm. The diagnosis is based on four out of five clinical criteria, and several pathogenic mechanisms have been proposed, including post-infectious autoimmune inflammation [13]. Several animal models of coronary arteritis have been used to gain insight into the pathogenesis of KD [14]. In one of them, a single intraperitoneal injection of a cell wall extract from Lactobacillus casei triggered development of proximal coronary arteritis that exhibits striking similarities in the histopathology and kinetics with human KS. In this model, both innate and adaptive immunity were found to be essential for development of coronary lesions [15].
The implications of B cells in KS pathogenesis comes from human genetic and histopathogy studies. First, genome-wide association studies suggest that genes involved in B cell activation are implicated [16][17]. In both Japanese and Taiwanese KS patients, significant associations in the FAM167A-BLK region at 8p22–23 and in the CD40 region at 20q13 were identified [16][17]. BLK codes for the Src family kinase Blk, which is responsible for downstream signaling of the B cell receptor and affects the proliferation/differentiation and tolerance of B cells [18][19]. Blk is also required for T-cell-mediated proinflammatory cytokine production [20]. The cell surface receptor CD40 is expressed on antigen-presenting cells, such as B cells, and plays a key role in proliferation, differentiation, and activation of B cells, and in T cell-dependent immune responses [21]. Thus, the kinase Blk and the receptor CD40, which play key roles in the B cell compartment, seem to be prominent in determining the risk for KS.
Second, progress has been made in understanding the pathogenesis of KS through examination of tissue samples from fatal cases. Studies of human autopsy specimens revealed early infiltration by neutrophils, followed by mixed lymphocytes, plasma cells and macrophages [22][23]. The B cell and plasma cell infiltration in specimens from KS patients suggest that B lymphocytes could play a role in tissue injury. They could be the result of the activity of a B cell superantigen [24][25] or of an infectious agent. They also could represent the product of B cell hyperactivation by ligands that can lead to polyclonal activation.
Initially, several groups suggested that KS is caused by multiple infectious agents, and several microorganisms have been isolated from patients with KS. Consistently, sequence analysis of the IgM transcripts expressed by peripheral B cells of KS patients in the acute phase of the disease revealed oligoclonal expansions of Ig variable region genes, suggesting that KS is caused by stimulations in response to an antigenic stimulus [26]. In further investigations, oligoclonal IgA plasma cells were documented to infiltrate inflamed tissues, including the coronary arteries of patients with acute KD [27]. Remarkably, this infiltration contrasts with the decrease of absolute numbers of IgA B lymphocytes in peripheral blood of patients with acute KS [28]. Further characterization of the Ig heavy-chain-genes present in the arterial wall of children who had died of acute KS exhibited a restricted pattern of CDR3 usage and somatic mutation, a marker of an antigen-driven response [29]. Synthetic antibody versions of these oligoclonal KS antibodies are bound to an antigen present in inflamed acute KS ciliated bronchial epithelium [30].
These observations are consistent with the view that, following infection of ciliated bronchial epithelium of the respiratory tract by a pathogen, intra-cytoplasmic inclusion bodies are formed. In order to neutralize the presence of the pathogen, antigen-specific plasma cells infiltrate the infected tissue. However, a collateral effect of this infiltration may lead to damage of the coronary arteries by products of activated lymphocytes and, possibly, other inflammatory cells. A better identification of the B cell subsets present at the site of inflammation could provide further insight into the physiopathology of KS.
Relevant to innate immunity and auto-inflammation are the inflammasomes, multiprotein cytoplasmic complexes that act as regulators of immune recognition trajectories, including viral, bacterial, and fungal pathogens [31]. Inflammasome activation leads to IL-1β and IL-18 maturation, a key process in the host defense against potential threats. Not surprisingly, deregulation of NLRP3 has been linked to the physio-pathology of AIFS, as well as several degenerative and metabolic disorders. For example, mutations in genes coding for components of the NLRP3 inflammasome lead to an AIFS called Cryopyrin-Associated Periodic Syndromes (CAPS) [32]. In human B cells, NLRP3 has been reported to be essential for pro-inflammatory cytokine secretion, and IL-1β secretion was modulated by NLRP3 and involved potassium efflux and Caspase-1 [33]. Consistently, ablation of the NLRP3 inflammasome in mice leads to altered B cell development in the bone marrow, and distorted expression of B cell subsets that play innate-like functions, i.e., marginal zone B cells in the spleen and B-1a cells in the peritoneal cavity, indicating that the inflammasome plays a role in B cell development, homing, and retention in lymphoid organs [34]. Since the NLRP3 inflammasome acts as a modulator of B lymphocyte functions, further work is required to understand this role in auto-inflammatory disorders.
Patients affected with Schnitzler syndrome manifest cardinal features of AIFS, but also have a monoclonal IgM gammopathy associated with a kappa light-chain in over 98% of cases [35], suggesting that B cells could play a role. To determine whether B cells exhibit shared clonality, deep sequencing of the immunoglobulin variable region genes from Schnitzler syndrome patients has been performed, allowing characterization of the immunoglobulin variable region gene usage, and the length and amino acid composition of the third hypervriable regions [36]. The investigators found evidence of various degrees of B cell clonality in each individual case. However, they could not document shared B cell clonality among all the patients studied. This research suggests that the adaptive branch of the immune system plays a role in this autoinflammatory condition. However, since the investigators undertook sequencing of immunoglobulin genes of peripheral blood B cells, it is likely that resident B cell clones present in the bone marrow or secondary lymphoid organs have been missed. Further longitudinal studies of resident B cell clones in these patients could provide insight into the role of B cells in Schnitzler syndrome.
Inflammatory bone disorders are characterized by production of pro-inflammatory cytokines and a variety of danger-associated molecular patterns (DAMPs), such as purine metabolites and fatty acids [37]. It is possible that autoinflammation plays a role in systemic bone loss associated with various rheumatic and musculoskeletal diseases, potentially leading to osteoporosis and fragility fractures. In support of this view, B lymphocytes have the potential to engage in multi-directional cross-talk with cells of the skeletal system, including osteocytes, osteoclasts and osteoblasts [38]. For example, osteoprotegerin produced by B cells is key in regulating bone remodeling mediated by the RANK-RANK-L pair (Figure 1). Given the tight relationships between B lymphocytes and the skeletal system, the role of B cells in autoinflammation in bone disorders deserves further consideration.
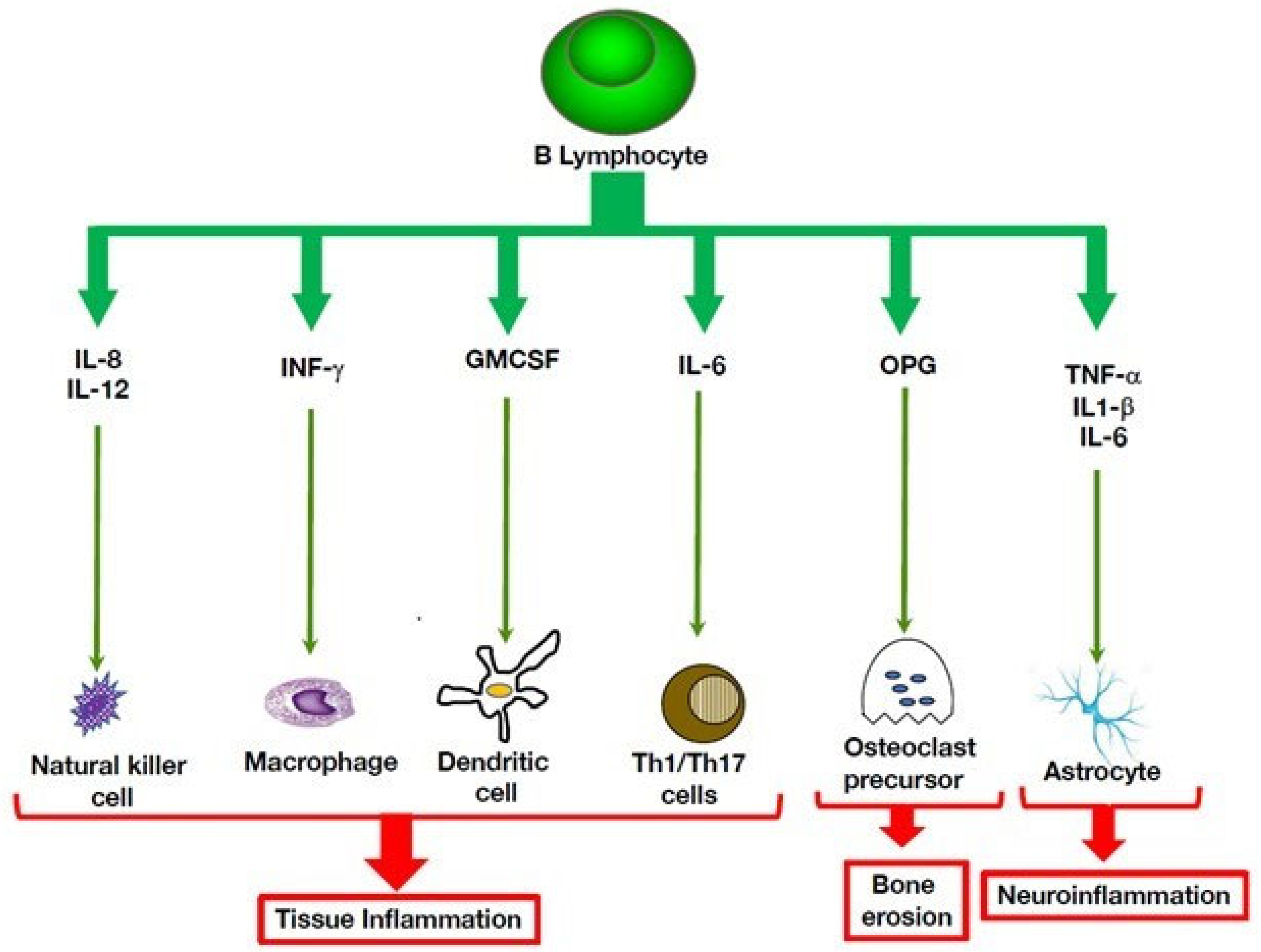
Figure 1. B cell cytokine-mediated pro-inflammatory pathways that act on innate immunity. B cells can exert multiple immune activities that promote inflammatory processes by producing cytokines that act on immune cells that mediate inflammation, such as macrophages and natural killer cells. Through their interactions with the skeletal system, they can promote bone erosion 46. In the central nervous system, their potential to secrete inflammatory cytokines can aggravate neuroinflammation.
Behçet’s disease (BD) is an auto-inflammatory vasculitis that affects predominantly the mucosa, skin and eyes [39]. In addition to elevated serum cytokine levels, such as IL-1β, TNF-α, and IL-18, there is a distorted expression of toll-like receptors (TLR). Cells that express these receptors can recognize conserved motifs present on pathogens, termed pathogen-associated molecular patterns (PAMPs) and DAMPs, which lead to activation of a signaling cascade that culminates in production of pro-inflammatory cytokines, including TNF-α [37]. A recent study of TLR expression in Behçet’s disease revealed deregulated expression of different cell subsets, including monocytes, granulocytes and T cells [40]. In addition, TLR1 and TLR2, TLR4 and TLR5 were abnormally expressed on B cells, suggesting that B cells contribute to the prominent inflammatory response in this disease. Further mechanistic studies are required to determine the B cell subsets overexpressing TLRs in Behçet’s disease.
References
- McDermott, M.F.; Aksentijevich, I.; Galon, J.; McDermott, E.M.; Ogunkolade, B.W.; Centola, M.; Mansfield, E.; Gadina, M.; Karenko, L.; Pettersson, T.; et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999, 97, 133–144.
- Betrains, A.; Staels, F.; Schrijvers, R.; Meyts, I.; Humblet-Baron, S.; De Langhe, E.; Wouters, C.; Blockmans, D.; Vanderschueren, S. Systemic autoinflammatory disease in adults. Autoimmun. Rev. 2021, 20, 102774.
- Szekanecz, Z.; McInnes, I.B.; Schett, G.; Szamosi, S.; Benko, S.; Szucs, G. Autoinflammation and autoimmunity across rheumatic and musculoskeletal diseases. Nat. Rev. Rheumatol. 2021, 17, 585–595.
- Hedrich, C.M. Shaping the spectrum—From autoinflammation to autoimmunity. Clin. Immunol. 2016, 165, 21–28.
- Melek, M.; Gellert, M. RAG1/2-mediated resolution of transposition intermediates: Two pathways and possible consequences. Cell 2000, 101, 625–633.
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241.
- Perucha, E.; Melchiotti, R.; Bibby, J.A.; Wu, W.; Frederiksen, K.S.; Roberts, C.A.; Hall, Z.; LeFriec, G.; Robertson, K.A.; Lavender, P.; et al. The cholesterol biosynthesis pathway regulates IL-10 expression in human Th1 cells. Nat. Commun. 2019, 10, 498.
- Ter Haar, N.M.; Jeyaratnam, J.; Lachmann, H.J.; Simon, A.; Brogan, P.A.; Doglio, M.; Cattalini, M.; Anton, J.; Modesto, C.; Quartier, P.; et al. The Phenotype and Genotype of Mevalonate Kinase Deficiency: A Series of 114 Cases from the Eurofever Registry. Arthritis Rheumatol. 2016, 68, 2795–2805.
- Shen, P.; Roch, T.; Lampropoulou, V.; O’Connor, R.A.; Stervbo, U.; Hilgenberg, E.; Ries, S.; Dang, V.D.; Jaimes, Y.; Daridon, C.; et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 2014, 507, 366–370.
- Bibby, J.A.; Purvis, H.A.; Hayday, T.; Chandra, A.; Okkenhaug, K.; Rosenzweig, S.; Aksentijevich, I.; Wood, M.; Lachmann, H.J.; Kemper, C.; et al. Cholesterol metabolism drives regulatory B cell IL-10 through provision of geranylgeranyl pyrophosphate. Nat. Commun. 2020, 11, 3412.
- Sornsakrin, M.; Wenner, K.; Ganschow, R. B cell cytopenia in two brothers with hyper-IgD and periodic fever syndrome. Eur. J. Pediatr. 2009, 168, 825–831.
- Kawasaki, T. Kawasaki disease. Acta Paediatr. 1995, 84, 713–715.
- Lindquist, M.E.; Hicar, M.D. B Cells and Antibodies in Kawasaki Disease. Int. J. Mol. Sci. 2019, 20, 1834.
- Orenstein, J.M.; Rowley, A.H. An evaluation of the validity of the animal models of Kawasaki disease vasculopathy. Ultrastruct. Pathol. 2014, 38, 245–247.
- Schulte, D.J.; Yilmaz, A.; Shimada, K.; Fishbein, M.C.; Lowe, E.L.; Chen, S.; Wong, M.; Doherty, T.M.; Lehman, T.; Crother, T.R.; et al. Involvement of innate and adaptive immunity in a murine model of coronary arteritis mimicking Kawasaki disease. J. Immunol. 2009, 183, 5311–5318.
- Onouchi, Y.; Ozaki, K.; Burns, J.C.; Shimizu, C.; Terai, M.; Hamada, H.; Honda, T.; Suzuki, H.; Suenaga, T.; Takeuchi, T.; et al. A genome-wide association study identifies three new risk loci for Kawasaki disease. Nat. Genet. 2012, 44, 517–521.
- Chen, M.R.; Chang, T.Y.; Chiu, N.C.; Chi, H.; Yang, K.D.; Chang, L.; Huang, D.T.; Huang, F.Y.; Lien, Y.P.; Lin, W.S.; et al. Validation of genome-wide associated variants for Kawasaki disease in a Taiwanese case-control sample. Sci. Rep. 2020, 10, 11756.
- Reth, M.; Wienands, J. Initiation and processing of signals from the B cell antigen receptor. Annu. Rev. Immunol. 1997, 15, 453–479.
- Hasler, P.; Zouali, M. B cell receptor signaling and autoimmunity. FASEB J. 2001, 15, 2085–2098.
- Samuelson, E.M.; Laird, R.M.; Papillion, A.M.; Tatum, A.H.; Princiotta, M.F.; Hayes, S.M. Reduced B lymphoid kinase (Blk) expression enhances proinflammatory cytokine production and induces nephrosis in C57BL/6-lpr/lpr mice. PLoS ONE 2014, 9, e92054.
- van Kooten, C.; Banchereau, J. CD40-CD40 ligand. J. Leukoc. Biol. 2000, 67, 2–17.
- Takahashi, K.; Oharaseki, T.; Naoe, S.; Wakayama, M.; Yokouchi, Y. Neutrophilic involvement in the damage to coronary arteries in acute stage of Kawasaki disease. Pediatr. Int. 2005, 47, 305–310.
- Fujiwara, T.; Fujiwara, H.; Nakano, H. Pathological features of coronary arteries in children with Kawasaki disease in which coronary arterial aneurysm was absent at autopsy. Quantitative analysis. Circulation 1988, 78, 345–350.
- Aksentijevich, I.; Zhou, Q. NF-kappaB Pathway in Autoinflammatory Diseases: Dysregulation of Protein Modifications by Ubiquitin Defines a New Category of Autoinflammatory Diseases. Front. Immunol. 2017, 8, 399.
- Zouali, M. B-cell superantigens: Implications for selection of the human antibody repertoire. Immunol. Today 1995, 16, 399–405.
- Lee, H.H.; Park, I.H.; Shin, J.S.; Kim, D.S. Immunoglobulin V(H) chain gene analysis of peripheral blood IgM-producing B cells in patients with Kawasaki disease. Yonsei Med. J. 2009, 50, 493–504.
- Rowley, A.H.; Shulman, S.T.; Mask, C.A.; Finn, L.S.; Terai, M.; Baker, S.C.; Galliani, C.A.; Takahashi, K.; Naoe, S.; Kalelkar, M.B.; et al. IgA plasma cell infiltration of proximal respiratory tract, pancreas, kidney, and coronary artery in acute Kawasaki disease. J. Infect. Dis. 2000, 182, 1183–1191.
- Shingadia, D.; O’Gorman, M.; Rowley, A.H.; Shulman, S.T. Surface and cytoplasmic immunoglobulin expression in circulating B-lymphocytes in acute Kawasaki disease. Pediatr. Res. 2001, 50, 538–543.
- Rowley, A.H.; Shulman, S.T.; Spike, B.T.; Mask, C.A.; Baker, S.C. Oligoclonal IgA response in the vascular wall in acute Kawasaki disease. J. Immunol. 2001, 166, 1334–1343.
- Rowley, A.H.; Shulman, S.T.; Garcia, F.L.; Guzman-Cottrill, J.A.; Miura, M.; Lee, H.L.; Baker, S.C. Cloning the arterial IgA antibody response during acute Kawasaki disease. J. Immunol. 2005, 175, 8386–8391.
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489.
- Mortimer, L.; Moreau, F.; MacDonald, J.A.; Chadee, K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat. Immunol. 2016, 17, 1176–1186.
- Ali, M.F.; Dasari, H.; Van Keulen, V.P.; Carmona, E.M. Canonical Stimulation of the NLRP3 Inflammasome by Fungal Antigens Links Innate and Adaptive B-Lymphocyte Responses by Modulating IL-1beta and IgM Production. Front. Immunol. 2017, 8, 1504.
- Hsu, M.; Zouali, M. Inflammasome is a central player in B cell development and homing. Life Sci. Alliance 2022, 6, e202201700.
- de Koning, H.D. Schnitzler’s syndrome: Lessons from 281 cases. Clin. Transl. Allergy 2014, 4, 41.
- Pathak, S.; Rowczenio, D.; Lara-Reyna, S.; Kacar, M.; Owen, R.; Doody, G.; Krause, K.; Lachmann, H.; Doffinger, R.; Newton, D.; et al. Evidence of B Cell Clonality and Investigation Into Properties of the IgM in Patients with Schnitzler Syndrome. Front. Immunol. 2020, 11, 569006.
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216.
- Manilay, J.O.; Zouali, M. Tight relationships between B lymphocytes and the skeletal system. Trends. Mol. Med. 2014, 20, 405–412.
- Sakane, T.; Takeno, M.; Suzuki, N.; Inaba, G. Behcet’s disease. N. Engl. J. Med. 1999, 341, 1284–1291.
- van der Houwen, T.B.; Dik, W.A.; Goeijenbier, M.; Hayat, M.; Nagtzaam, N.M.A.; van Hagen, M.; van Laar, J.A.M. Leukocyte toll-like receptor expression in pathergy positive and negative Behcet’s disease patients. Rheumatology 2020, 59, 3971–3979.
More
Information
Subjects:
Medicine, Research & Experimental
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
858
Revisions:
2 times
(View History)
Update Date:
13 Jan 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No