Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yoshio Nosaka | -- | 2953 | 2023-01-12 08:06:45 | | | |
| 2 | Rita Xu | Meta information modification | 2953 | 2023-01-12 08:24:25 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nosaka, Y. Water Photo-Oxidation over TiO2. Encyclopedia. Available online: https://encyclopedia.pub/entry/40097 (accessed on 14 July 2026).
Nosaka Y. Water Photo-Oxidation over TiO2. Encyclopedia. Available at: https://encyclopedia.pub/entry/40097. Accessed July 14, 2026.
Nosaka, Yoshio. "Water Photo-Oxidation over TiO2" Encyclopedia, https://encyclopedia.pub/entry/40097 (accessed July 14, 2026).
Nosaka, Y. (2023, January 12). Water Photo-Oxidation over TiO2. In Encyclopedia. https://encyclopedia.pub/entry/40097
Nosaka, Yoshio. "Water Photo-Oxidation over TiO2." Encyclopedia. Web. 12 January, 2023.
Copy Citation
Photocatalytic splitting of water is a direct and attractive approach for the utilization of solar energy by producing the most-prospective clean hydrogen fuel. In photocatalytic water splitting, oxidation of water to molecular oxygen, or oxygen evolution reaction (OER), is the most difficult process because it needs the transfer of four electrons, while the hydrogen evolution reaction (HER) is a two-electron transfer reaction.
photocatalysis
water splitting
titanium dioxide
oxygen evolution reaction
1. Introduction
The electron transfer reactions originated from the absorption of one photon for each step, the semiconducting properties of metal oxide solid materials are useful to promote the reaction at a reaction site [1]. Thus, metal complexes and metal–organic frameworks (MOF) are almost certainly unsuitable as photocatalysts for OER [2].
Photo-oxidation of water was reported a century ago with ZnO powder, and 50 years ago, TiO2 was found to be useful as the stable material for converting photoenergy to chemical energy without deterioration. By starting TiO2 as photocatalysts, this 50 years is a booming period in the research of photocatalysis. In the present report, the first section is devoted to the brief history of water photo-oxidation. In the following section, several research reports about the reaction mechanism for water photo-oxidation over TiO2 are described. The reports for the reaction mechanism were based on either an experimental approach or a theoretical calculation approach. In the experimental approach, techniques used were electron spin resonance (ESR), nuclear magnetic resonance (NMR), scanning tunneling microscopy (STM), and Fourier transfer infrared spectroscopy (FTIR). In the theoretical calculation approach, several methods of density functional theory (DFT) have been used for anatase (101) and rutile (110) surfaces.
2. History of Photocatalytic Water Photo-Oxidation
After the first report of ZnO powder as the photosensitizer in 1911 [3], Baur and Perret reported the photoinduced oxidation of water to produce O2 with ZnO powder in 1924 [4]. In this report, a new concept has been suggested, in which, after the absorption of light energy, the ZnO particle undergoes simultaneous anodic and cathodic processes. In the anodic process, OH− is oxidized to generate ¼O2 + ½H2O. On the other hand, in the cathodic process, the Ag+ ion is reduced to generate Ag by the formation of intermediate peroxide Ag2O. However, this photoinduced reaction is associated with the dissolution of ZnO to release Zn2+, and then the verified reaction scheme was
2AgNO3 + ZnO = 2Ag + Zn(NO3)2 + ½O2
Therefore, though this reaction takes place only under photoirradiation, it was not a photocatalytic reaction in researcher's time.
In the field of electrochemistry, a single crystal of ZnO was employed, and the photocurrent was observed under the high anodic polarization in water [5]. O2 evolved with the anodic photocurrent. The evolution of O2 was not attributable to the photo-oxidation of water but to the photo dissolution of ZnO, similar to the case of ZnO powder stated above. In this case, Zn2+ was dissolved in the solution. Therefore, the total reaction is simply
2ZnO + hν → 2Zn2+ + O2 + 4e−
The reaction mechanism is reported as follows. The holes photo-generated in the electrode oxidize the surface O of the electrode and the second hole attacks the neighbor surface O atom to form an (O-O)2− ion at the surface, then it becomes O2 with the other two holes [5].
Compared with ZnO, TiO2 is a relatively new material because titanium metal was identified at the end of the 18th century. It was in 1923 that TiO2 was first manufactured as a white pigment to replace basic lead carbonate which was used in the porcelain industries. Photo-induced effects on the chemical reactions over TiO2 powder have been investigated before and after World War II in the vicinity of the 1940s. However, as a photosensitizer, in other words, for photocatalytic reaction, the main substance used was ZnO since the photoeffect was first reported as described above. In 1953, Markham and Laidler [6] reported the reaction mechanism of photo-oxidation on the surface of ZnO, where they suggested that O2 is formed from two •OH radicals. In the discussion, they cited the report of an XRD study [7], which described that photo-excitation of TiO2 caused the change in the XRD pattern to α-Ti2O3 crystal. Then, Markham and Laidler described that the absorption of photon energy to TiO2 may result in the photolysis of the TiO2 crystal, as represented by [6].
TiO2 + hν → ½Ti2O3 + ½O2
At that time, this chemical equation had not been confirmed by any researchers, and TiO2 became inexperienced material in the research of solid photosensitizers.
About 50 years ago, in an electrochemical study, Fujishima and Honda used TiO2 single crystals as photoelectrodes and examined photoinduced reactions. They observed the anodic photocurrent and O2 evolution similarly to the case of ZnO. Surprisingly, in the case of the TiO2 electrode, they could not detect Ti4+ nor Ti3+ ions in solution after the evolution of oxygen [8], which was different from the case of ZnO. This observation means the oxidation of water to generate O2 with the aid of photo-energy as described by
TiO2 + hν + 2H2O → O2 + 4H+ + 4e−
They suggested that when a suitable p-type semiconductor electrode is coupled with the n-type TiO2 semiconductor electrode, efficient electrochemical photolysis of water may occur on the irradiation of both electrodes [9]. Their report was epoch-making because it was shown that the photon energy could convert to the chemical energy of hydrogen fuel by using a semiconductor electrode. Actually, Yoneyama and co-workers demonstrated the O2 evolution at the TiO2 anode and the H2 evolution at the p-GaP cathode with the open circuit voltage of 0.58 V [10]. Though the deterioration in the cell performance was observed due to the unstable p-GaP cathode, oxidation of water at the TiO2 anode was confirmed stable. After these observations, the photoelectrochemical systems with metal oxides for water splitting have been investigated widely, which are reviewed by Rajeshwar [11]. Recent development in the engineering nanostructure interface of photoanode materials toward photoelectrochemical water oxidation can be referred to with a review article [12].
For the particulate photocatalysts, the decomposition of water vapor to H2 and O2 over Fe-doped TiO2 powder has been reported by Schrauzer and Guth [13], in which a stoichiometric (2:1) evolution of H2 and O2 was confirmed. Photocatalytic water vapor decomposition was enhanced when Pt nanoparticles were photodeposited on a TiO2 particle, which constructs a small particulate electrochemical cell. Sato and White [14] reported the generation of O2 and H2 with the stoichiometric ratio of 1:2, as shown in Figure 1. When the irradiation was stopped, the recombination of O2 and H2, or reverse reaction of H2O photodecomposition, occurred at a significant rate. Photocatalytic decomposition of water vapor was recently reviewed by Suguro et al. [15].
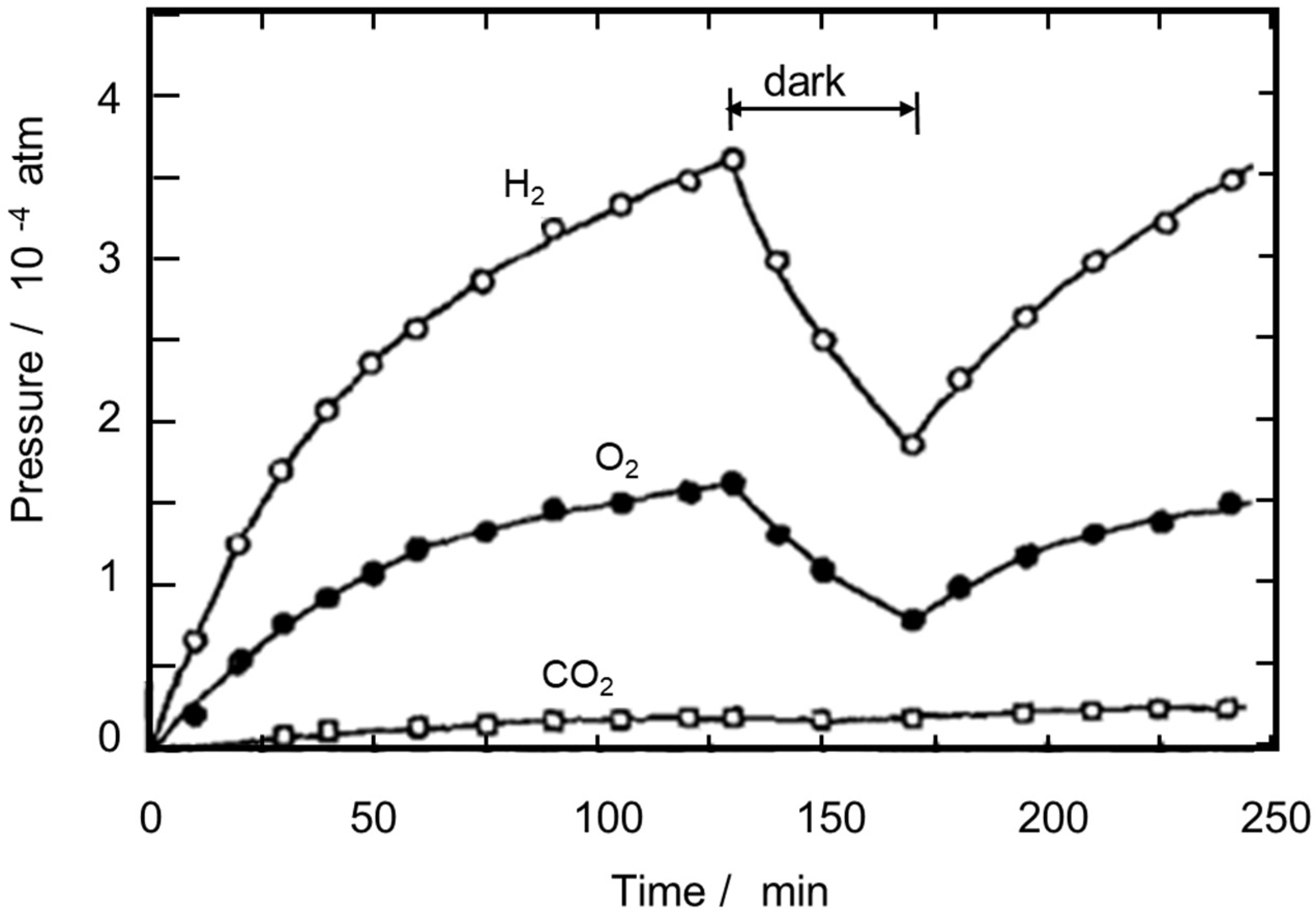
Figure 1. Photocatalytic water splitting over Pt-loaded TiO2 powder in the gas phase. Reprinted with permission from Ref [14]. Copyright 1980 Elsevier.
In the aqueous suspension system of TiO2 powder, Sayama et al. [16] found that the stoichiometric H2 and O2 production was improved by adding carbonate salts to the suspension of the photocatalyst. Recently, they confirmed the mechanism of the improvement by the DFT calculation for model molecules [17]. The adsorbed H2CO3 molecule was photo-oxidized relatively easily compared with the adsorbed H2O molecule, facilitating the formation of the peroxide intermediate and improving both the O2 evolution and the H2O2 production.
Different from the gaseous system, in the aqueous suspension system, it is difficult to inhibit the reverse reaction of the produced H2 and O2. To inhibit the reaction between H2 and O2, the surface of the co-catalyst for H2 production had to be covered with a Cr2O3 shell to protect it from the attack of the produced O2 [18]. From the point of view of a catalyst, TiO2 is not the best material for OER. The other metal oxides which show a small exchange current for OER have been explored as reviewed by Lewis and coworkers [19]. Moreover, these materials have been employed as the co-catalyst of semiconductor photocatalysts. For water oxidation, many co-catalysts, such as cobalt oxide, have been used in the newly developed photocatalysts [20]. The development of photocatalysts for water splitting of the use of such co-catalysts was compiled recently [21].
3. Mechanism for TiO2 Water Oxidation
3.1. Experimental Approach
3.1.1. ESR and NMR
In the early days of photocatalysis, a spin-trapping ESR technique was used to detect radical species in the reaction. With the UV irradiation on the suspension of Pt-deposited TiO2 powder, •OH and •O2H radicals were detected [22]. Thus, in the O2 generation by the water oxidation, •OH radical was considered as the reaction intermediate as follows.
H2O + h+ → •OH + H+
2•OH → H2O2
2H2O2 → O2 + 2H2O
The •O2H radical detected was attributed to being produced by the photocatalytic reduction of the produced O2 [22].
In the recent study, one- and two-dimensional 1H solid-state NMR techniques were employed to identify the surface hydroxyl groups and the adsorbed water molecules as well as their spatial proximity/interaction in TiO2 photocatalysts [23]. Only the bridging OH (i.e., OHbr) is in close spatial proximity to adsorbed H2O, forming hydrated OHbr. To investigate the role of hydrated OHbr in the hole transfer process, in situ ESR experiments were performed on TiO2 with variable H2O loading [23]. The ESR measurements revealed that the hydrated OHbr groups offer a channel for the transfer of photogenerated holes in the photocatalytic reaction, and the adsorbed H2O could have a synergistic effect with the neighboring OHbr groups to facilitate the formation and evolution of active paramagnetic intermediates. On the basis of these experimental observations, the detailed photocatalytic mechanism of water splitting on the surface of TiO2 was proposed, as shown in Figure 2 [23]. The surface-trapped hole Ti-O• and the adsorbed •O2− radical were identified by the ESR measurements. In this figure, as the product of the second step, they suggested the side-on coordination of O22− ions to one Ti ion. However, this structure is probably unreal conformation in an aqueous solution.
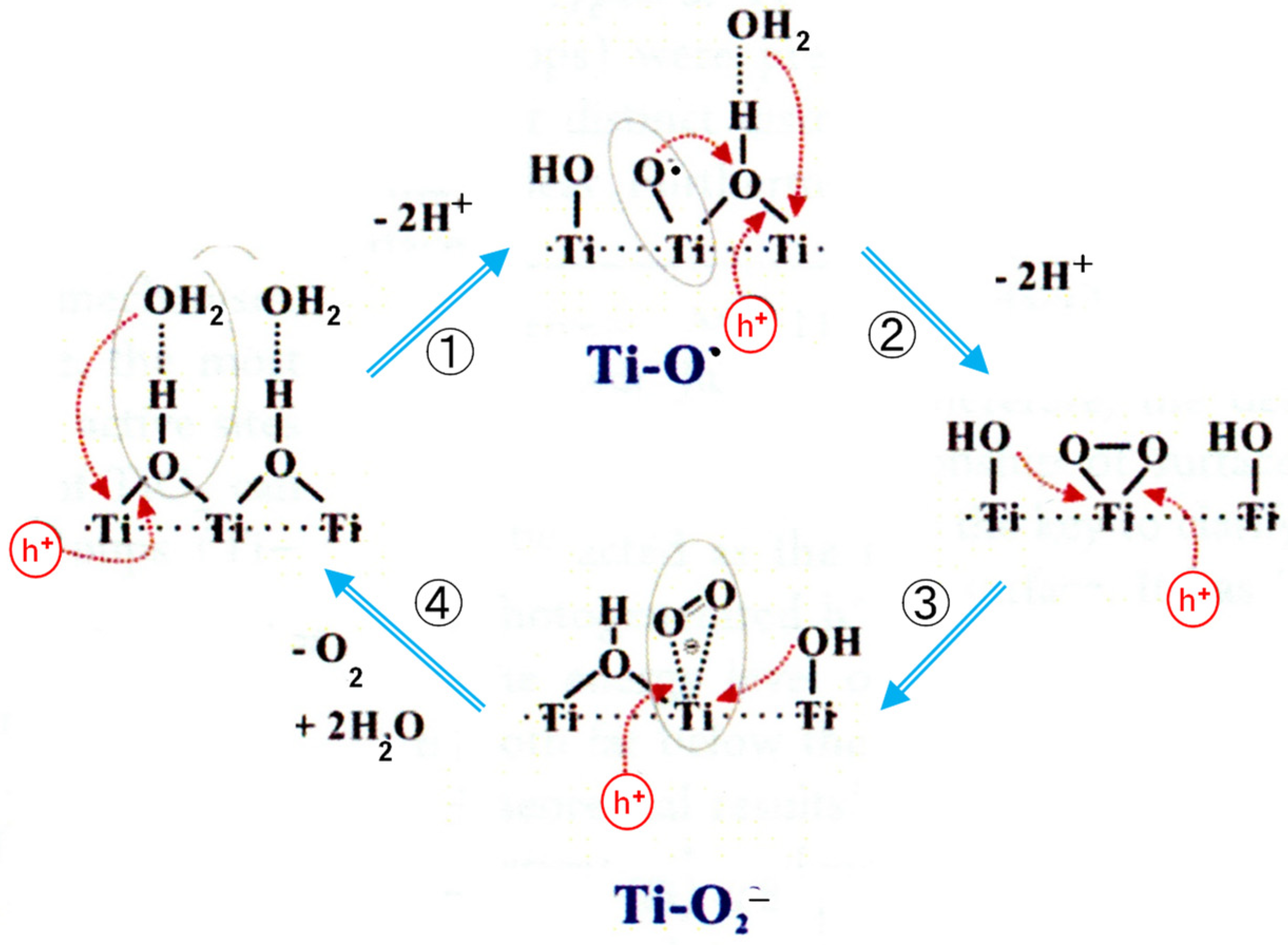
Figure 2. OER mechanism proposed from the ESR detection of radicals produced on Pt/TiO2 (P25) powder. Adapted with permission from [23]. Copyright 2017, American Chemical Society.
3.1.2. STM
By using the low-temperature STM performed at 80 K, the dissociation of individually adsorbed water molecules has been observed at the five-fold coordinated Ti (Ti5c) sites of the reduced rutile TiO2(110)-1 × 1 surface under the irradiation of UV lights with the wavelength shorter than 400 nm [24]. It was found that two kinds of hydroxyl species are involved in the process of photocatalytic water dissociation. One is always present at the adjacent bridging oxygen sites, OHbr, and the other either occurs as terminal OHt at Ti5c sites away from the original ones or even desorbs from the surface [24]. This observation is in harmony with the suggestion of the solid-state NMR described above. Thus, the initial oxidation step could be expressed by the following equation [25].
H2OTi + Obr + h+ → •OH(gas) + OHbr
Using a combination of STM and other surface science techniques, the recent progress that provides fundamental insights into TiO2 photocatalysis were reviewed through direct tracking of the evolution in single molecule photochemistry on TiO2 single crystal surfaces [25].
3.1.3. FTIR
Nakato and co-workers investigated the molecular mechanism of water photo-oxidation reaction at atomically flat n-TiO2 (rutile) single crystal photoelectrodes of (100) and (110) facet surfaces [26]. They measured the photoluminescence (PL) of the photoelectrodes in aqueous solutions of various pH values. Combining with the Fourier transfer infrared (FTIR) observations of the intermediates in the reduction of molecular oxygen as well as the oxidation of water with an electron acceptor of Fe3+ ions, the oxidation process was elucidated, as shown in Figure 3. Though the nucleophilic attack of water to Ti−O+−Ti is associated with the [Ti−O• HO−Ti] formation in Figure 3 [27], an alternative model was proposed in which the bridging oxygen radicals [Ti−OH• Ti] are photogenerated with the intrinsic band-gap surface state [28]. In both reaction models, the surface-trapped holes become bridging peroxo (Ti-OO-Ti) by combining with the hole generated secondarily in the same crystallite [29][30]. The bridging peroxo structure was regarded as the intermediate step of water photo-oxidation. Thus, the species generated by two-hole oxidation is equivalent to a chemically adsorbed H2O2.
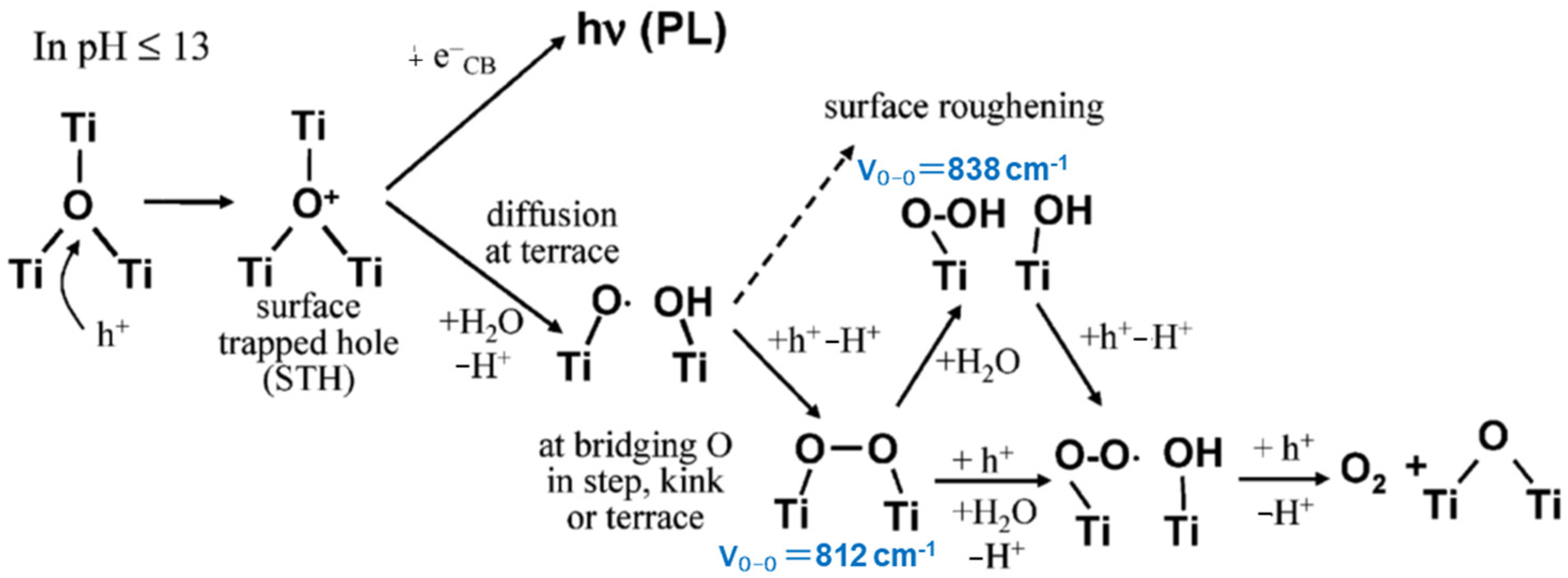
Figure 3. The proposed process of water oxidation and the peak position of the IR–spectra for the intermediates. Adapted with permission from Ref. [26]. Copyright 2007 American Chemical Society.
When H218O was used in the FTIR measurements, the observed isotope shift of the peak was small. Then, they suggested that one of the O atoms of the Ti-OOH group originated from O of the surface bridging O of the crystal [27]. Salvador [28] reported that the photoinduced holes are trapped at the bridging O of the surface, as stated above. However, in his proposed mechanism, a pair of •OH radicals are produced near the trapped hole, and they form the Ti-OO-Ti surface structure [28].
As shown in Figure 3, Nakamura et al. assigned the 838 cm−1 peak in the FTIR spectra to the OO vibration of the Ti-OOH group and the peak at 812 cm−1 to the Ti-OO-Ti group. Researchers also measured the FTIR spectra of the adsorbed H2O2 on rutile TiO2 powders and the reaction intermediates under the UV irradiation [31]. However, no absorption peaks at around 800 cm−1 could be observed, but the peaks around 1000 cm−1 and 934 cm−1, which could be assigned to Ti-OOH and Ti-OO-Ti, respectively, were observed [31]. This assignment is supported by the result of the DFT calculation described later, in which the O-O stretching mode of Ti-OOH was calculated to be 1002 cm−1 with a large intensity. Since researchers did not use an electron scavenger in the FTIR experiments, O2 in air was reduced, and the signals of superoxo Ti-OO− were increased. On the other hand, Nakamura et al. used Fe3+ ions as the sacrificial electron scavenger. Since the FTIR signal of Fe(IV)=O appears at 830–840 cm−1 as reported in the literature [32], the signal at 838 cm−1 probably originates from an Fe ion compound and should not be attributable to Ti-OOH. Due to the uncertainty of the assignments of FTIR spectra, it is not clearly concluded that the bridging O atom is involved in the Ti-OO-Ti structure.
3.1.4. Experimentally Suggested Mechanism for Rutile and Anatase
Researchers have experimentally investigated the reactive oxygen species, such as •OH, H2O2, and •O2−, which are generated from H2O by TiO2 photocatalysis and photoelectrodes [33]. Nakabayashi and Nosaka examined the facet dependence of water oxidation at anodically polarized TiO2 single-crystal photoelectrodes [34]. Though the Faraday efficiencies of the oxygen evolution were almost 100%, the intrinsic photocurrent was increased in the order of (100) < (110) < (001). On the other hand, the formation of •OH radicals simultaneously measured was reverse order; that is, the Faraday efficiencies were 0.59, 0.23, and 0.13%. Only for the crystal of (100) facet does the photocurrent decrease with the irradiation time, but it could be recovered by depolarization of the electrode. This observation showed that the surface structure changed to inhibit the O2 production at the surface of (100) single crystals. Furthermore, by the addition of H2O2, the formation of •OH radical was increased for the (100) and (110) crystals [35], indicating that H2O2 was not formed from •OH radical but H2O2 produced •OH radical at the TiO2 photo-anodes. Namely, the Ti-OO-Ti structure promotes the •OH radical generation. Thus, •OH radical could be formed in the process of the oxidation of Ti-OO-Ti species, but •OH radical is not the precursor of the Ti-OO-Ti formation. This conclusion contradicts the reaction mechanism suggested by Salvador [28] in which a pair of •OH radicals form the Ti-OO-Ti structure.
When the rutile surface is the ideal crystal structure without reorganization, the Ti-OO-Ti structure of each crystal surface can be illustrated as in Figure 4. The difference in the O2 production efficiency could be explained by the Ti-OO-Ti configurations for three kinds of crystal surfaces [34]. When the two Ti ions forming bridged O could not alter their position, the formation of these structures seems difficult. Furthermore, since the dihedral angle of H2O2 is 93.4°, the Ti-OO-Ti structure cannot lay on a plane, but the Ti-O-Ti can. Thus, the formation of Ti-OO-Ti at the rutile surfaces of (100), (110), and (001) seems to have some difficulty in configuring the structure.
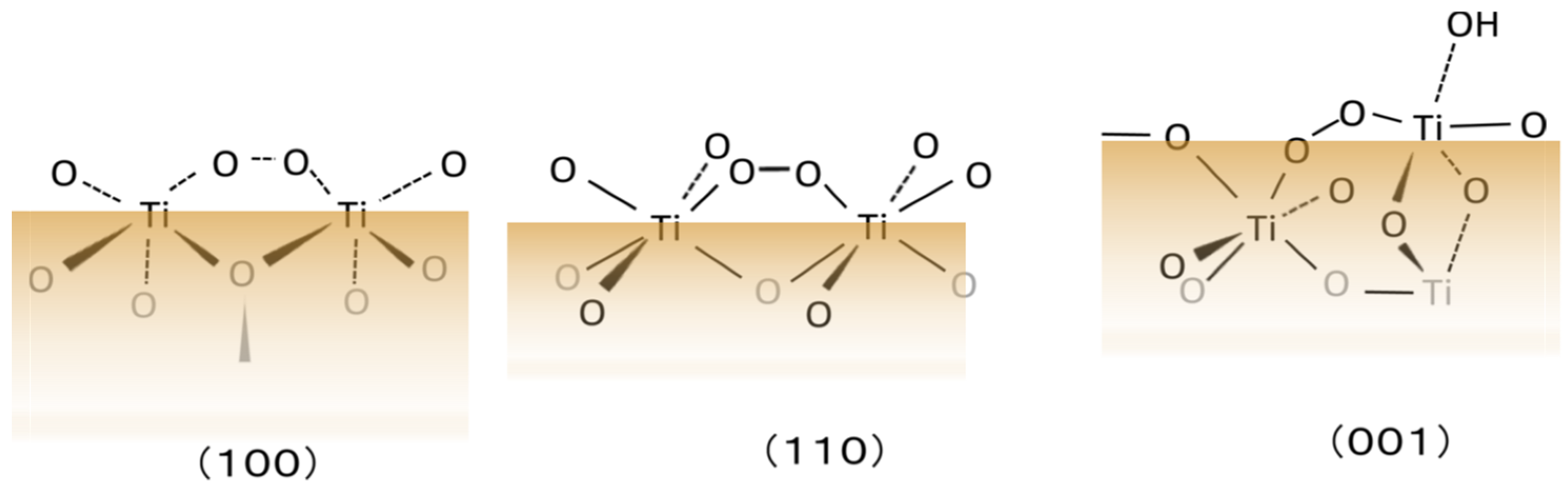
Figure 4. Local configuration of peroxo, Ti-OO-Ti, at distinct rutile TiO2 surfaces. Adapted with permission from Ref. [34]. Copyright 2013 American Chemical Society.
In order to discuss a rational reaction model for the oxidation of water at the surface of TiO2 crystal, researchers simply suggested that the oxidation and reduction sites are assigned to bridging OH and terminal Ti-OH, respectively [33], based on the fact that the surface Ti is positively charged while the surface Obr is negatively charged, as expected from the following equations of ionization equilibrium in aqueous solution.
≡Ti-OH ⇄ ≡Ti+ + OH−
[Ti-O−-Ti] + H+ ⇄ [Ti-OH-Ti]
Figure 5 shows the plausible water oxidation processes at the bridging O site of the anatase and rutile TiO2 surfaces [31]. A photoinduced hole attacks the bridging O atom at first, followed by the attack of water resulting in the formation of a pair of Ti-O• and Ti-OH [29]. As shown in Figure 5, at the anatase surface, since the Ti-Ti distance is too large to form peroxo Ti-OO-Ti structure, a surface-trapped hole should be isolated, and it is useful to oxidize organic molecules RH. A certain fraction of the trapped holes desorbs as •OH radicals into the solution [36][37]. On the other hand, at the surface of rutile, when a second hole is generated in the particle, it migrates to combine with the existing hole to form bridging peroxo species at the surface [29][38]. The distance between Ti atoms at the rutile surface (2.96 Å) could be shorter than that of anatase (3.79 Å) and be favorable to forming the Ti-OO-Ti structure [31][39]. Thus, rutile crystal is relatively more active for the water oxidation to O2 evolution against anatase crystal [33][36]. This explanation by the surface Ti-Ti distance for the difference in the water oxidation between rutile and anatase was supported by a theoretical calculation in a recent report [40].
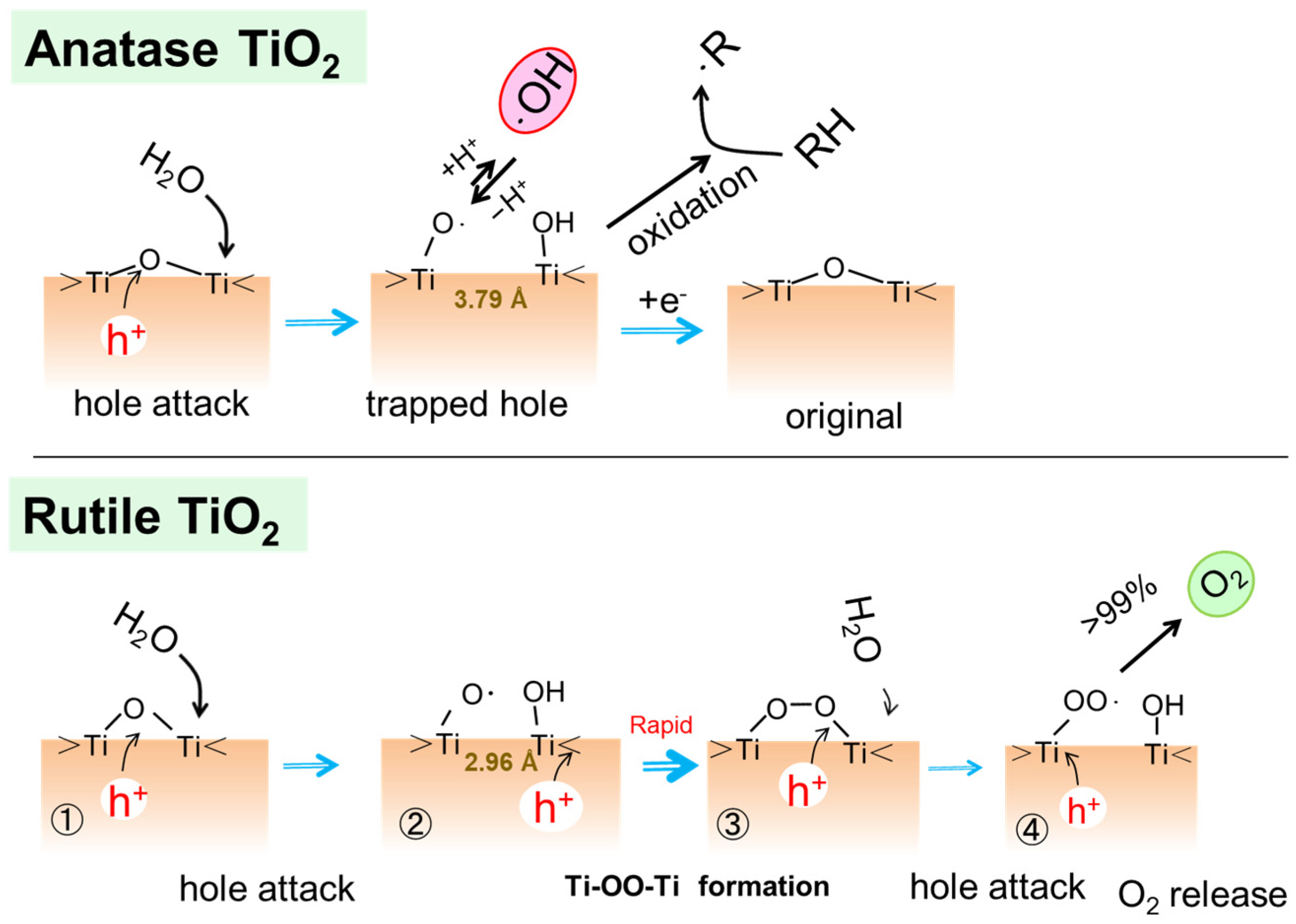
Figure 5. Plausible photocatalytic oxidation paths at the surface of TiO2 of anatase and rutile polymorphs. Reproduced from Ref. [31] with permission from the PCCP Owner Societies.
References
- Nosaka, Y.; Nosaka, A. Introduction to Photocatalysis—From Basic Science to Applications; Royal Society of Chemistry: Cambridge, UK, 2016; 272p.
- Chen, J.; Abazari, R.; Adegoke, K.A.; Maxakato, N.W.; Bello, O.S.; Tahir, M.; Tasleem, S.; Sanati, S.; Kirillovg, A.M.; Zhou, Y. Metal–organic frameworks and derived materials as photocatalysts for water splitting and carbon dioxide reduction, Coord. Chem. Rev. 2022, 469, 214664.
- Eibner, A. Action of light on pigments. Chem. Ztg. 1911, 35, 753–774.
- Baur, E.; Perret, A. The action of light on dissolved silver salts in the presence of zinc oxide. Helv. Chim. Acta 1924, 7, 910–915.
- Gerischer, H. Electrochemical behavior of semiconductors under illumination. J. Electrochem. Soc. 1966, 113, 1174–1182.
- Markham, M.C.; Laidler, K.J. A kinetic study of photo-oxidations on the surface of zinc oxide in aqueous suspensions. J. Phys. Chem. 1953, 57, 363–369.
- Jacobsen, A.E. Titanium dioxide pigments, correlation between photochemical reactivity and chalking. Ind. Eng. Chem. 1949, 41, 523–526.
- Fujishima, A.; Honda, K.; Kikuchi, S. Photosensitized electrolytic oxidation at TiO2 semiconductor electrodes. Kogyo Kagaku Zasshi 1969, 72, 108–113.
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38.
- Yoneyama, H.; Sakamoto, H.; Tamura, H. A Photo-electochemical cell with production of hydrogen and oxygen by a cell reaction. Electrochim. Acta 1975, 20, 341–345.
- Rajeshwar, K. Hydrogen generation at irradiated oxide semiconductor–solution interfaces. J. Appl. Electrochem. 2007, 37, 765–787.
- Tang, R.; Zhou, S.; Zhang, Z.; Zheng, R.; Huang, J. Engineering nanostructure–interface of photoanode materials toward photoelectrochemical water oxidation. Adv. Mater. 2021, 33, 2005389.
- Schrauzer, G.N.; Guth, T.D. Photolysis of water and photoreduction of nitrogen on titanium dioxide. J. Am. Chem. Soc. 1977, 99, 7189–7193.
- Sato, S.; White, J.M. Photodecomposition of water over Pt/TiO2 catalysts. Chem. Phys. Lett. 1980, 72, 83–86.
- Suguro, T.; Kishimoto, F. Photocatalytic hydrogen production under water vapor feeding—A minireview. Energy Fuels 2022, 36, 8978–8994.
- Sayama, K.; Arakawa, H. Significant effect of carbonate addition on stoichiometric photodecomposition of liquid water into hydrogen and oxygen from platinum–titanium(IV) oxide suspension. J. Chem. Soc. Chem. Commun. 1992, 150–152.
- Kusama, H.; Kodera, M.; Yamashita, K.; Sayama, K. Insights into the carbonate effect on water oxidation over metal oxide photocatalysts/photoanodes. Phys. Chem. Chem. Phys. 2022, 24, 5894–5902.
- Maeda, K.; Domen, K. Photocatalytic water splitting: Recent progress and future challenges. J. Phys. Chem. Lett. 2010, 18, 2655–2661.
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Solar water splitting cells. Chem. Rev. 2010, 110, 6446–6473.
- Tsuneda, T.; Ten-no, S.L. Water–oxidation mechanism of cobalt phosphate co-catalyst in artificial photosynthesis: A theoretical study. Phys. Chem. Chem. Phys. 2022, 24, 4674–4682.
- Wang, Q.; Domen, K. Particulate photocatalysts for light-driven water splitting: Mechanisms, challenges, and design strategies. Chem. Rev. 2020, 120, 919–985.
- Jaeger, C.D.; Bard, A.J. Spin trapping and electron spin resonance detection of radical intermediates in the photodecomposition of water at TiO2 particulate systems. J. Phys. Chem. 1979, 83, 3146–3152.
- Liu, F.; Feng, N.; Wang, Q.; Xu, J.; Qi, G.; Wang, C.; Deng, F. Transfer channel of photoinduced holes on a TiO2 surface as revealed by solid-state nuclear magnetic resonance and electron spin resonance spectroscopy. J. Am. Chem. Soc. 2017, 139, 10020–10028.
- Tan, S.; Feng, H.; Ji, Y.; Wang, Y.; Zhao, J.; Zhao, A.; Wang, B.; Luo, Y.; Yang, J.; Hou, J.G. Observation of photocatalytic dissociation of water on terminal Ti sites of TiO2(110)-1×1 surface. J. Am. Chem. Soc. 2012, 134, 9978–9985.
- Guo, Q.; Ma, Z.; Zhou, C.; Ren, Z.; Yang, X. Single molecule photocatalysis on TiO2 surfaces. Chem. Rev. 2019, 119, 11020–11041.
- Imanishi, A.; Okamura, T.; Ohashi, N.; Nakamura, R.; Nakato, Y. Mechanism of water photooxidation reaction at atomically flat TiO2 (rutile) (110) and (100) surfaces: Dependence on solution pH. J. Am. Chem. Soc. 2007, 129, 11569–11578.
- Nakamura, R.; Nakato, Y. Primary intermediates of oxygen photoevolution reaction on TiO2 (Rutile) particles, revealed by in situ FTIR absorption and photoluminescence measurements. J. Am. Chem. Soc. 2004, 126, 1290–1298.
- Salvador, P. Mechanisms of water photooxidation at n-TiO2 rutile single crystal oriented electrodes under UV illumination in competition with photocorrosion. Prog. Surf. Sci. 2011, 86, 41–58.
- Nakamura, R.; Nakato, Y. Molecular mechanism of water oxidation reaction at photo-irradiated TiO2 and related metal oxide surfaces. Solid State Phenom. 2010, 162, 1–27.
- Nakamura, R.; Okamura, T.; Ohashi, N.; Imanishi, A.; Nakato, Y. Molecular mechanisms of photoinduced oxygen evolution, PL emission, and surface roughening at atomically smooth (110) and (100) n-TiO2 (rutile) surfaces in aqueous acidic solutions. J. Am. Chem. Soc. 2005, 127, 12975–12983.
- Kakuma, Y.; Nosaka, A.Y.; Nosaka, Y. Difference in TiO2 photocatalytic mechanism between rutile and anatase studied by the detection of active oxygen and surface species in water. Phys. Chem. Chem. Phys. 2015, 17, 18691–18698.
- Zhang, M.; De Respinis, M.; Frei, H. Time-resolved observations of water oxidation intermediates on a cobalt oxide nanoparticles catalyst. Nat. Chem. 2014, 6, 362–367.
- Nosaka, Y.; Nosaka, A.Y. Generation and detection of reactive oxygen species in photocatalysis. Chem. Rev. 2017, 117, 11302–11336.
- Nakabayashi, Y.; Nosaka, Y. OH radical formation at distinct faces of rutile TiO2 crystal in the procedure of photoelectrochemical water oxidation. J. Phys. Chem. C 2013, 117, 23832–23839.
- Nakabayashi, Y.; Nosaka, Y. The pH dependence of OH radical formation in photo-electrochemical water oxidation with rutile TiO2 single crystals. Phys. Chem. Chem. Phys. 2015, 17, 30570–30576.
- Kim, W.; Tachikawa, T.; Moon, G.; Majima, T.; Choi, W. Molecular-level understanding of the photocatalytic activity difference between anatase and rutile nanoparticles. Angew. Chem. Int. Ed. 2014, 53, 14036–14041.
- Zhang, J.; Nosaka, Y. Photocatalytic oxidation mechanism of methanol and the other reactants in irradiated TiO2 aqueous suspension investigated by OH radical detection. Appl. Catal. B Environ. 2015, 166–167, 32–36.
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 photocatalysis: Mechanisms and materials. Chem. Rev. 2014, 114, 9919–9986.
- Sahel, K.; Elsellami, L.; Mirali, I.; Dappozze, F.; Bouhent, M.; Guillard, C. Hydrogen peroxide and photocatalysis. Appl. Catal. B Environ. 2016, 188, 106–112.
- Li, F.; Chen, J.-F.; Gong, X.-Q.; Hu, P.; Wang, D.; Li, F. Subtle structure matters: The vicinity of surface Ti5c cations alters the photooxidation behaviors of anatase and rutile TiO2 under aqueous environments. ACS Catal. 2022, 12, 8242–8251.
More
Information
Subjects:
Chemistry, Physical
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Revisions:
2 times
(View History)
Update Date:
16 Jan 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No