 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Arlene Gonçalves Corrêa | -- | 1266 | 2023-01-09 13:55:46 | | | |
| 2 | Amina Yu | -2 word(s) | 1264 | 2023-01-10 02:28:19 | | |
Video Upload Options
The chirality resulting from restricted rotation around a single bond is called atropisomerism (axial chirality). This phenomenon was first described by Christie and Kenner in 1922 when investigating the biaryl 6,6’-dinitro-2,2’-diphenic acid, and the term “atropisomer”, derived from the Greek where “a” means “not” and “tropes” means “turn”, was created by Kuhn. Atropisomers belong to the class of axially chiral compounds; however, here, the enantiomers exist due to restricted rotation around a single bond. Axial chirality has also been considered as an important structural element of many natural products and bioactive compounds, whose enantiomers generally exhibit different pharmacological activities and metabolic processes in vivo and in vitro.
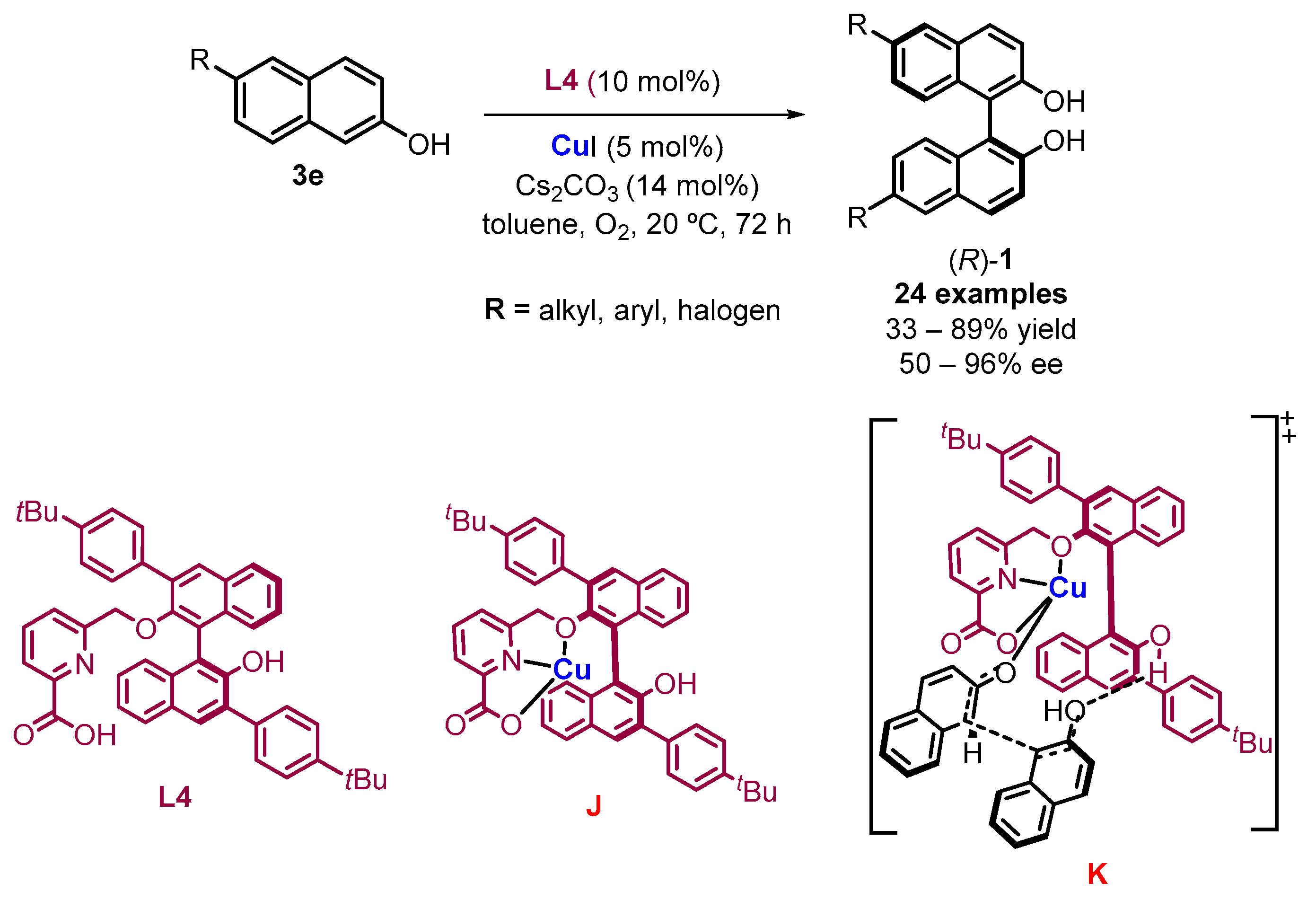
1. Metal-Mediated Oxidative Enantioselective Coupling
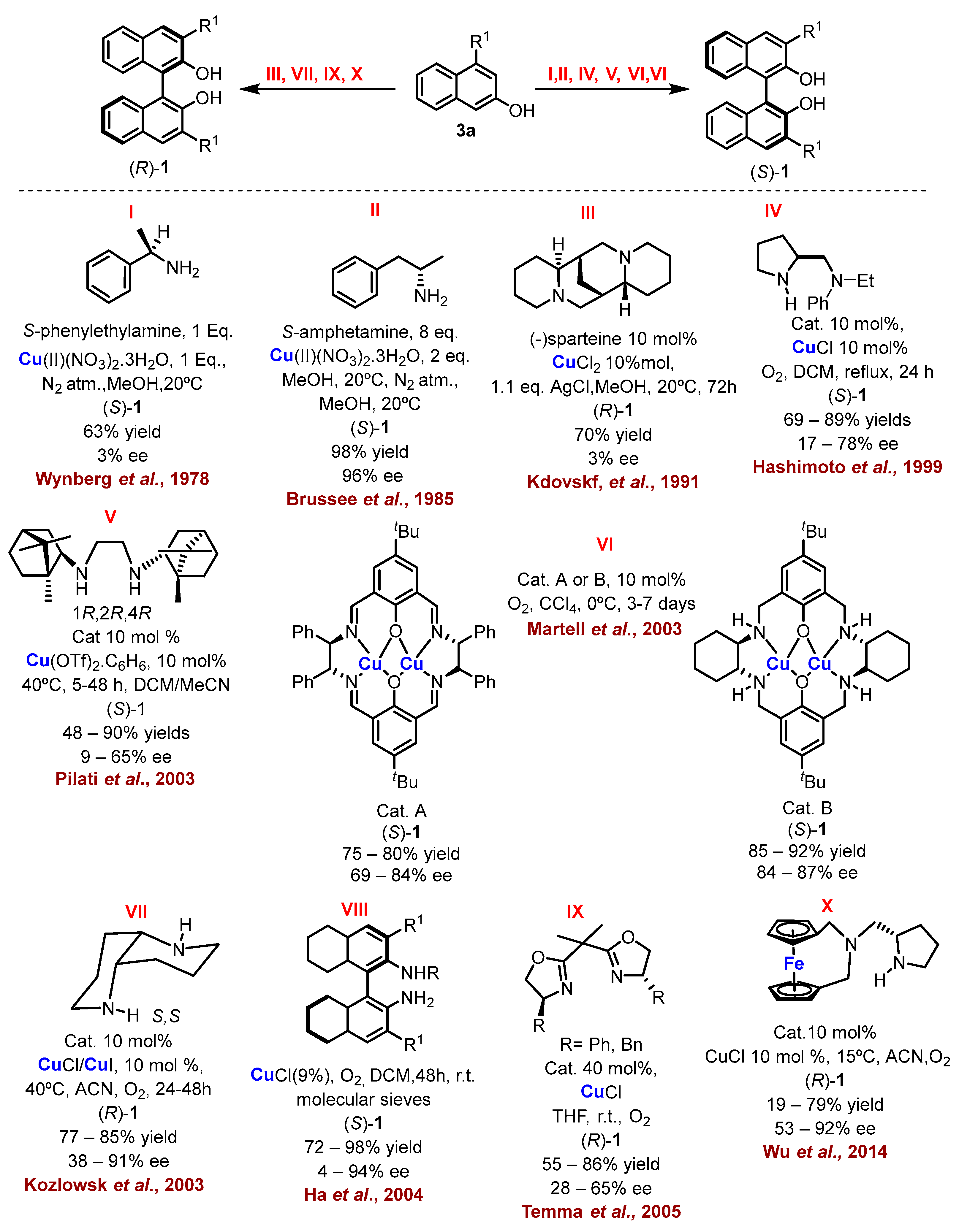
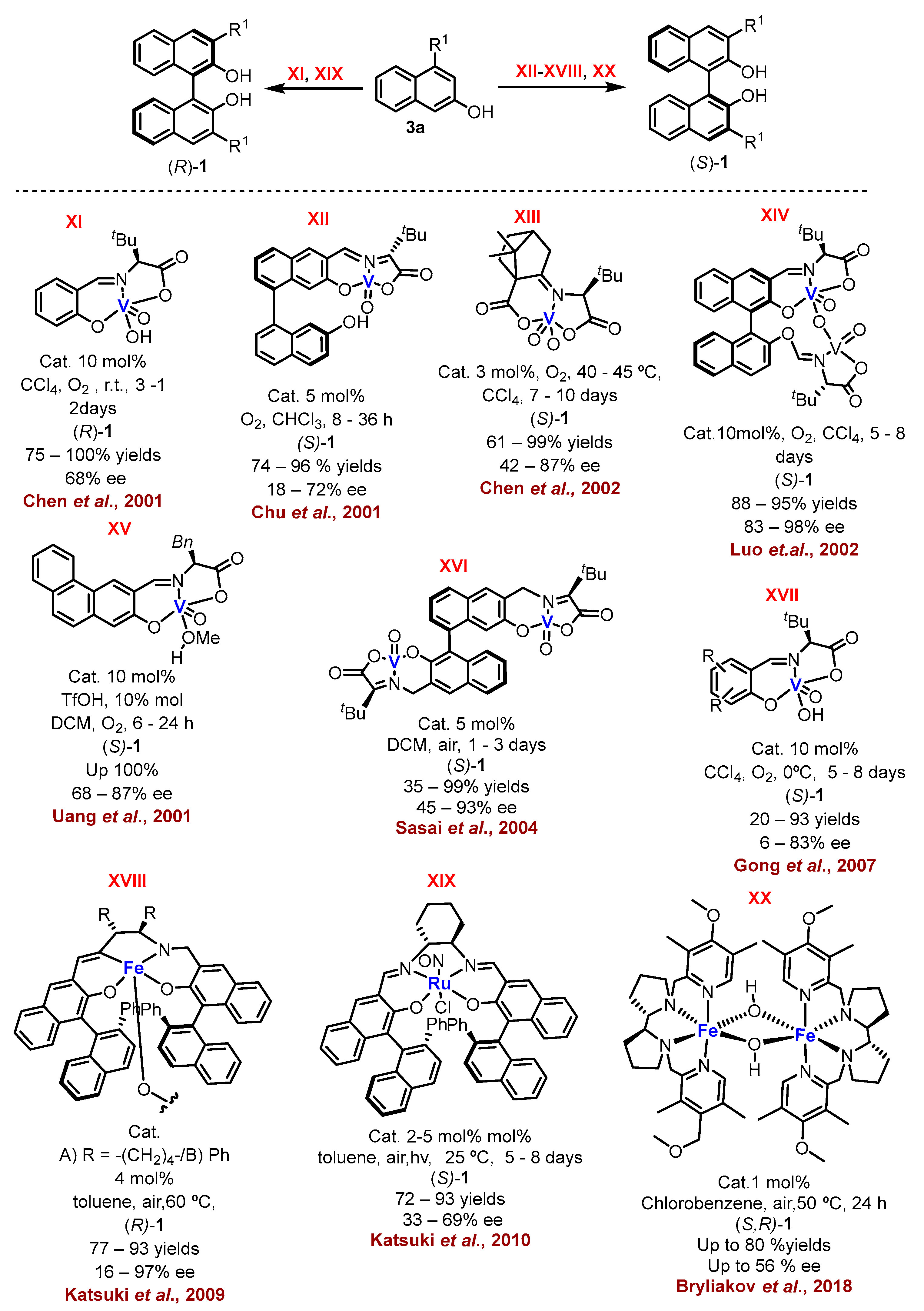
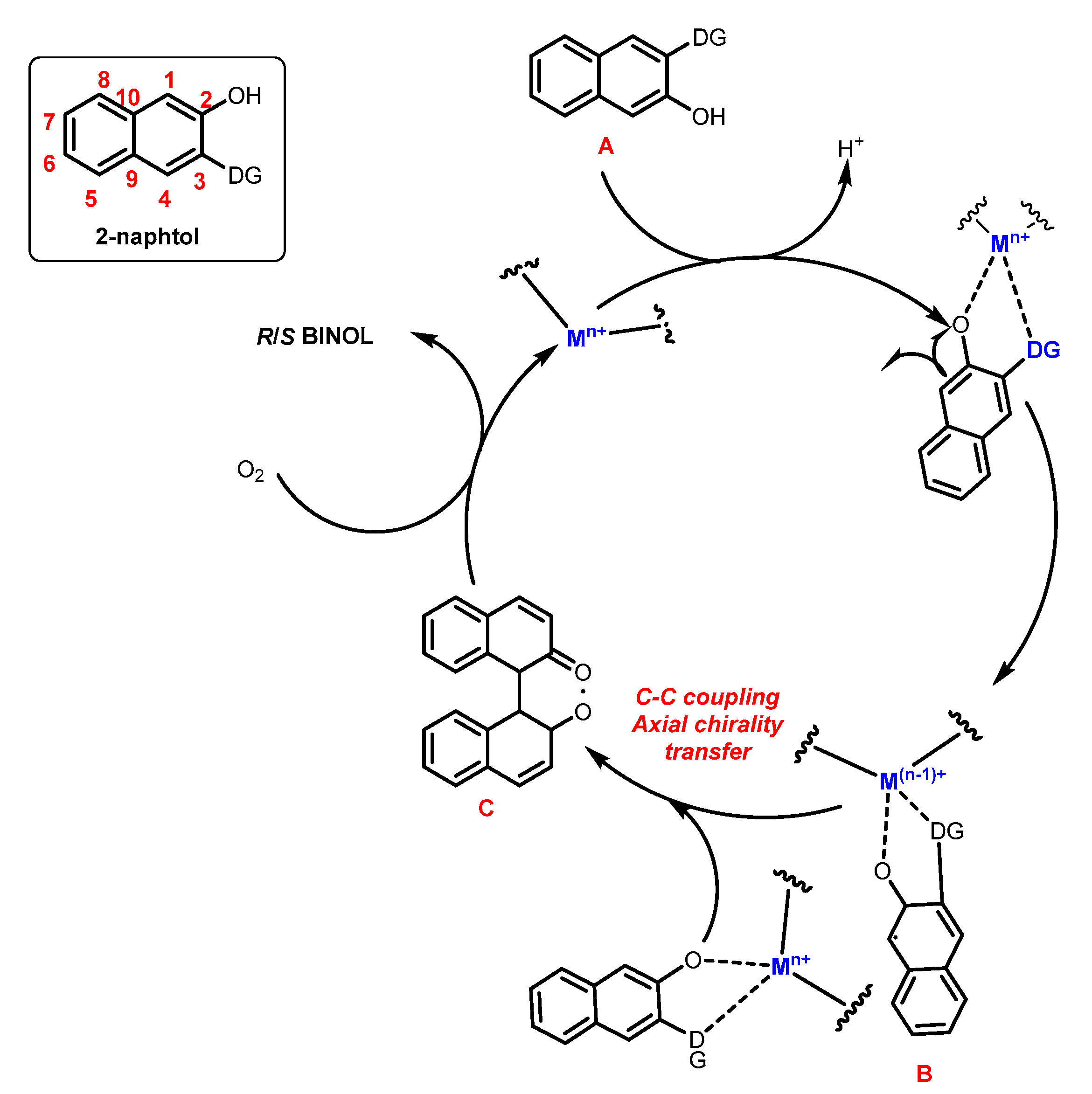
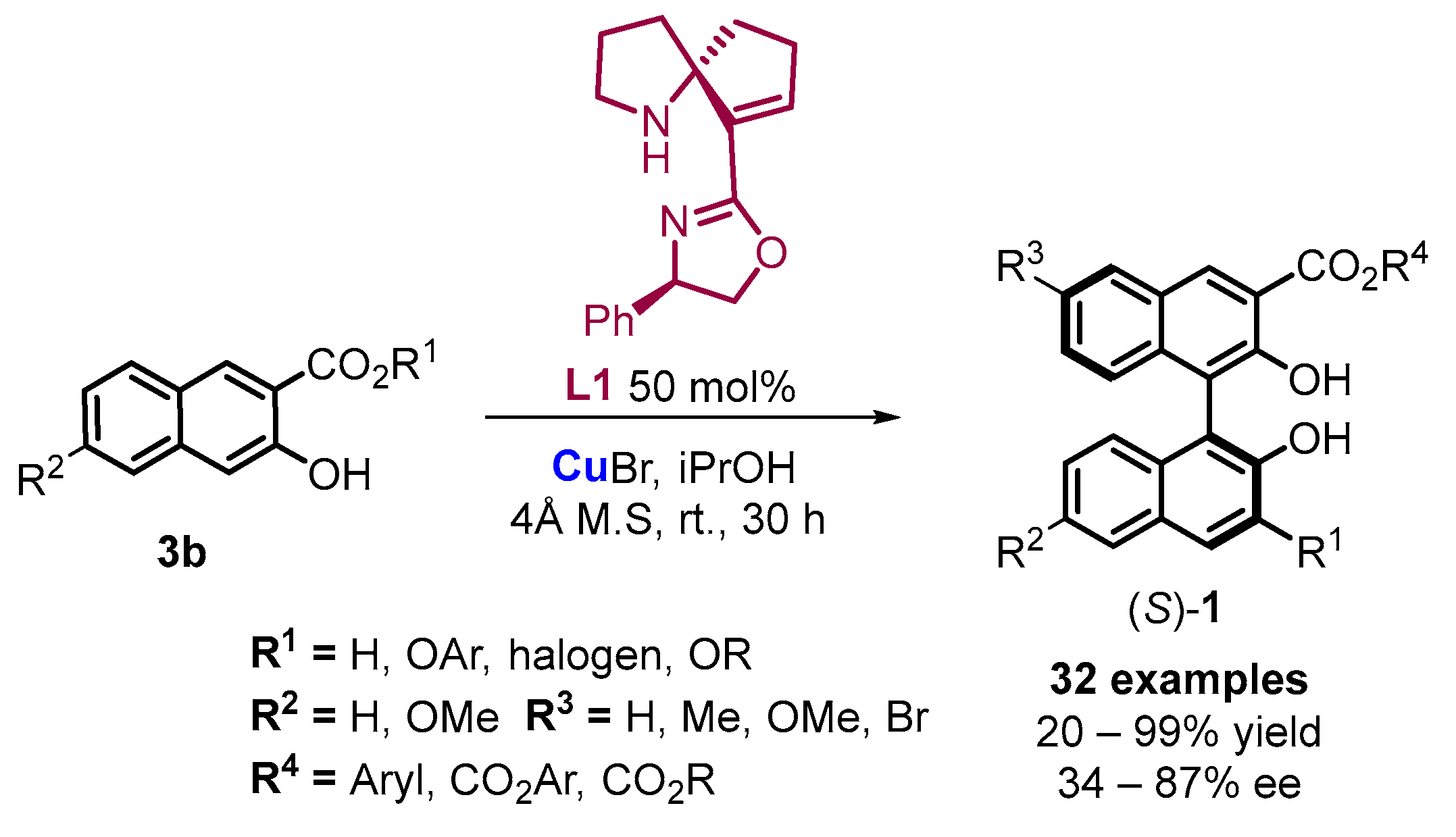
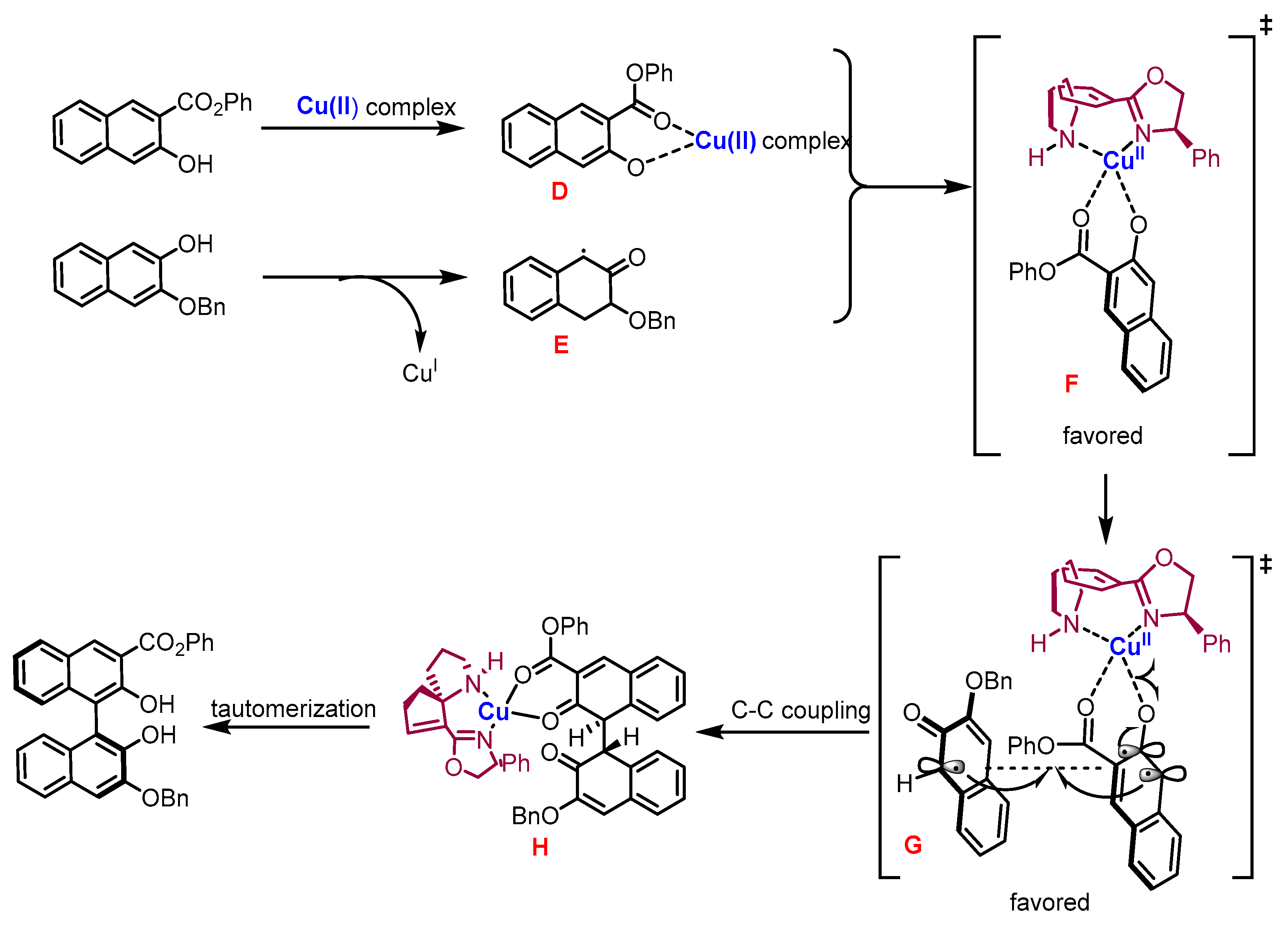
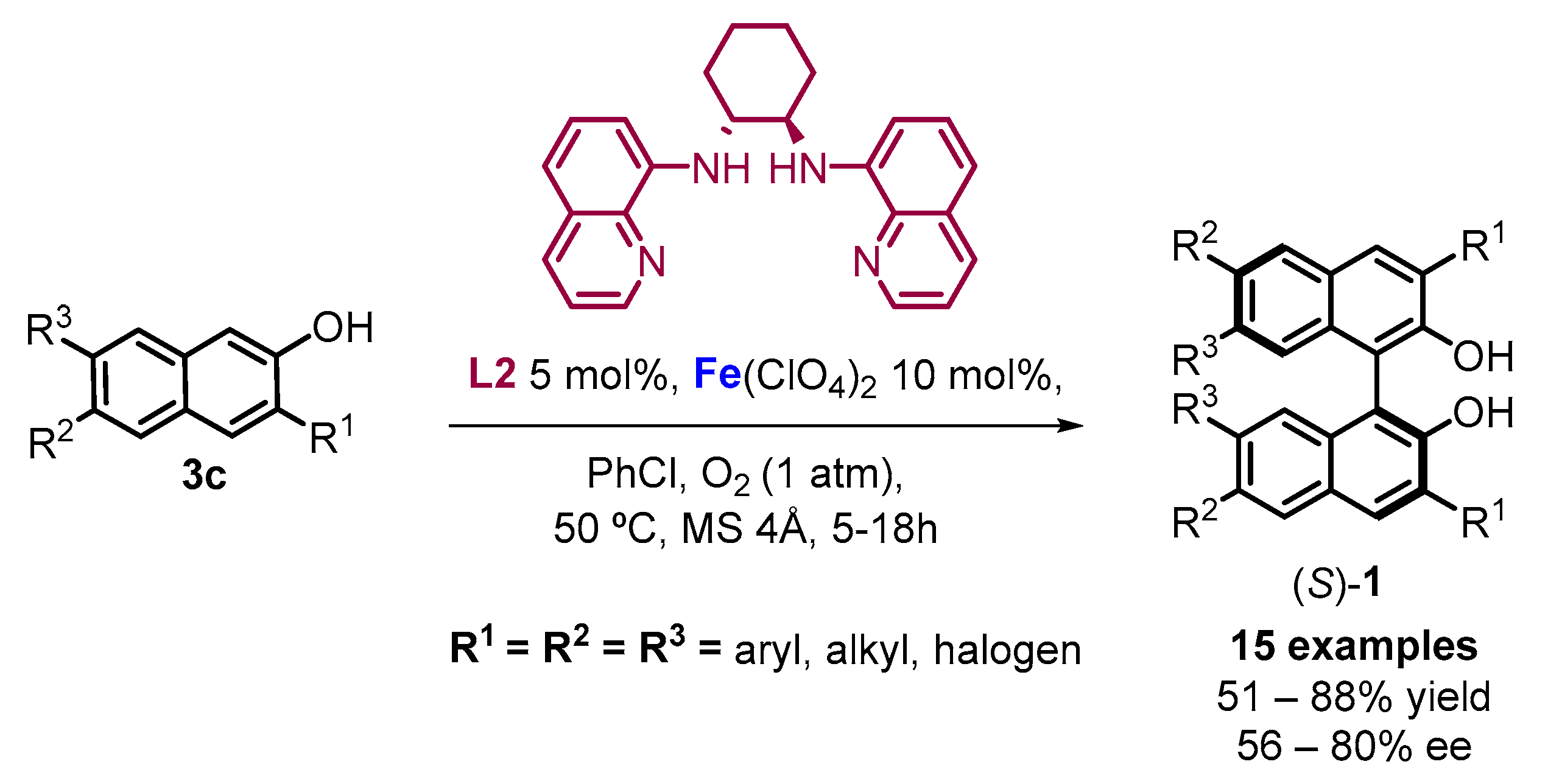
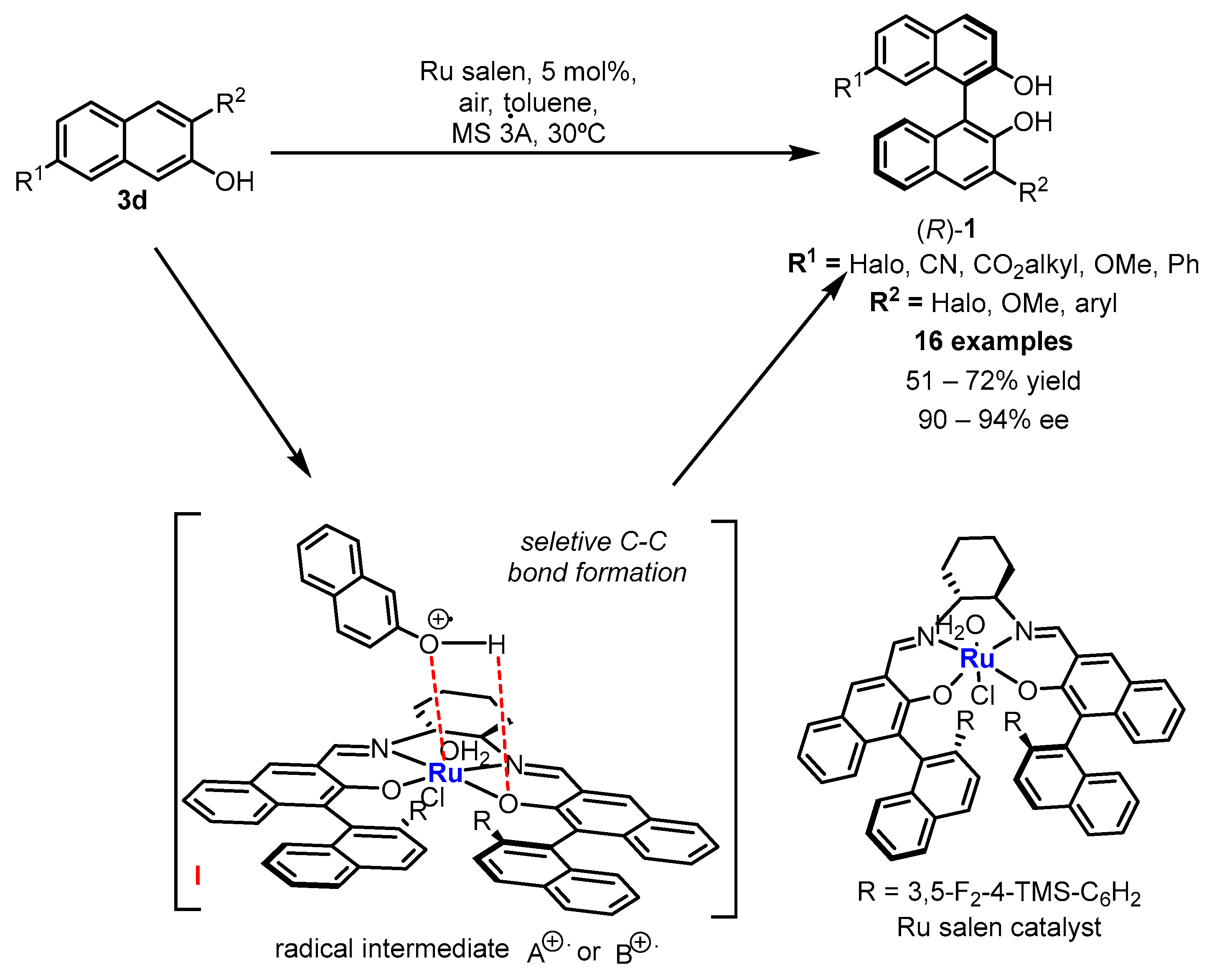

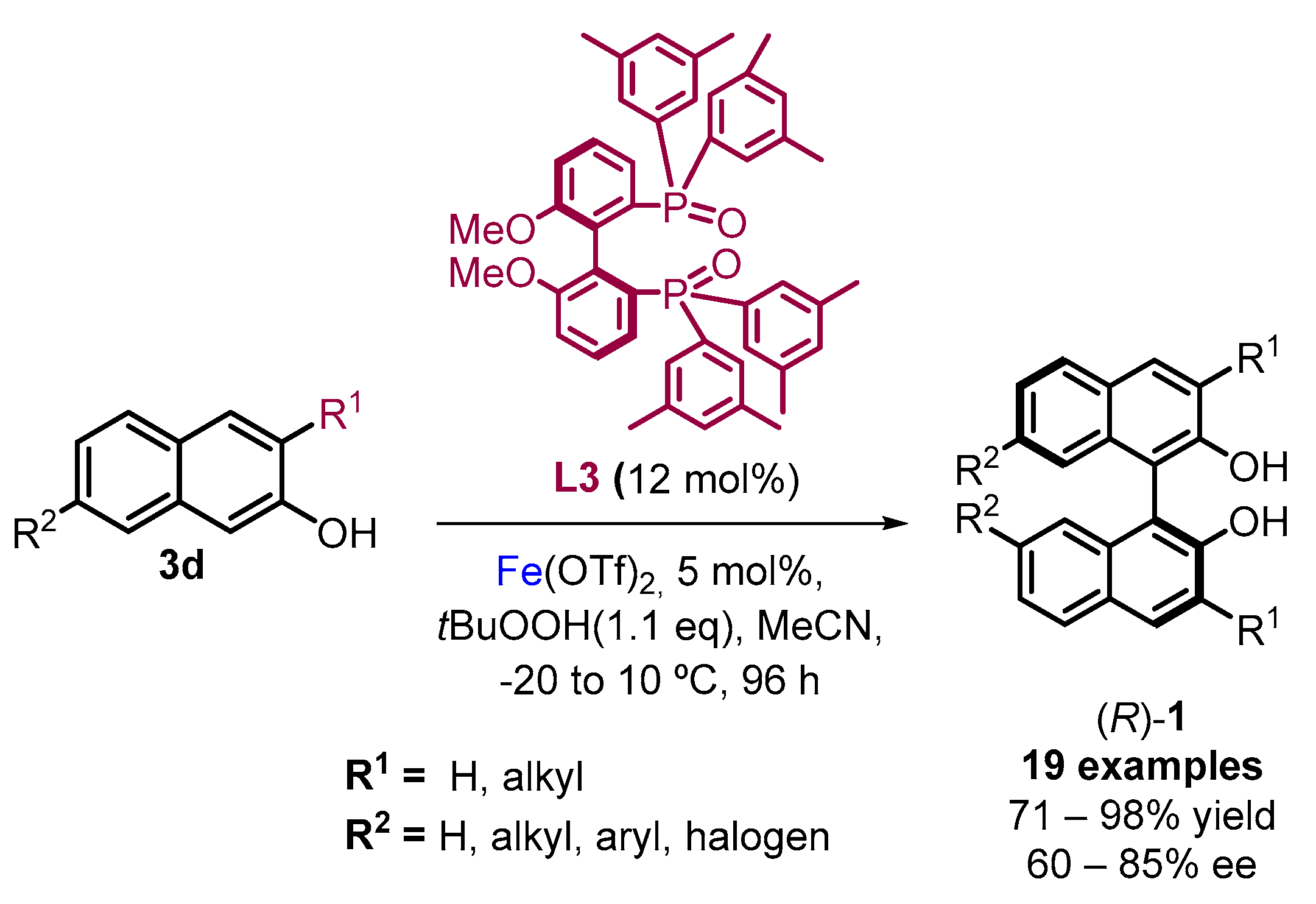

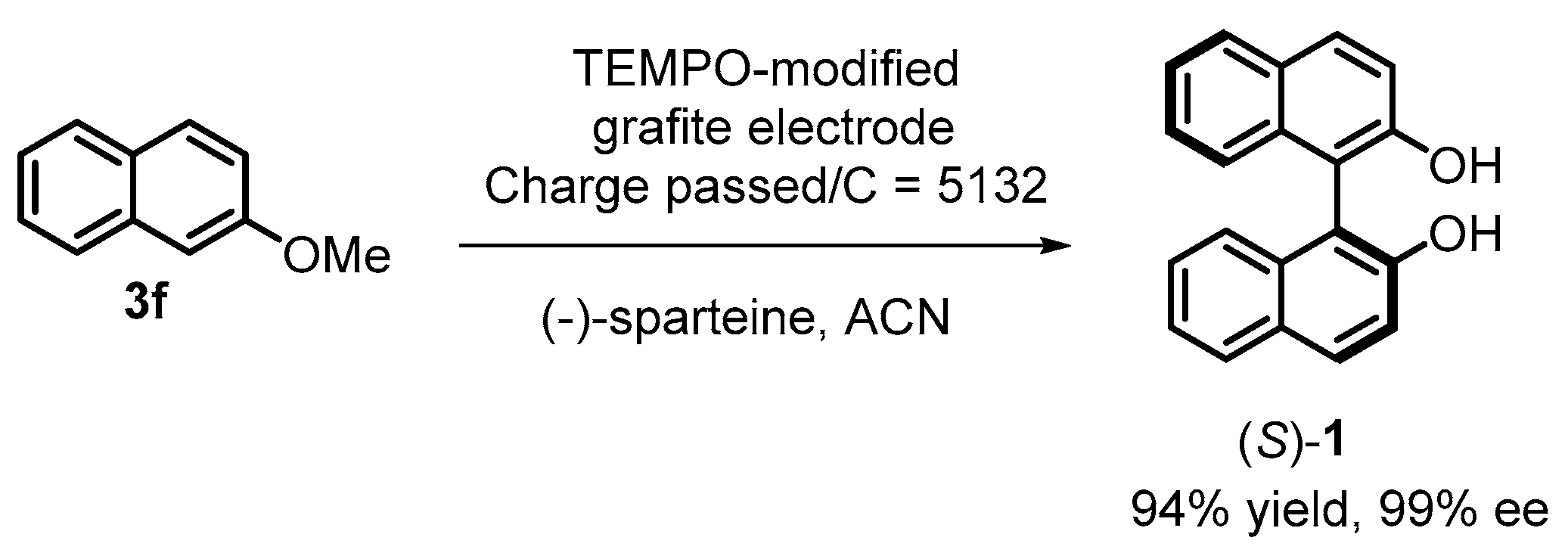
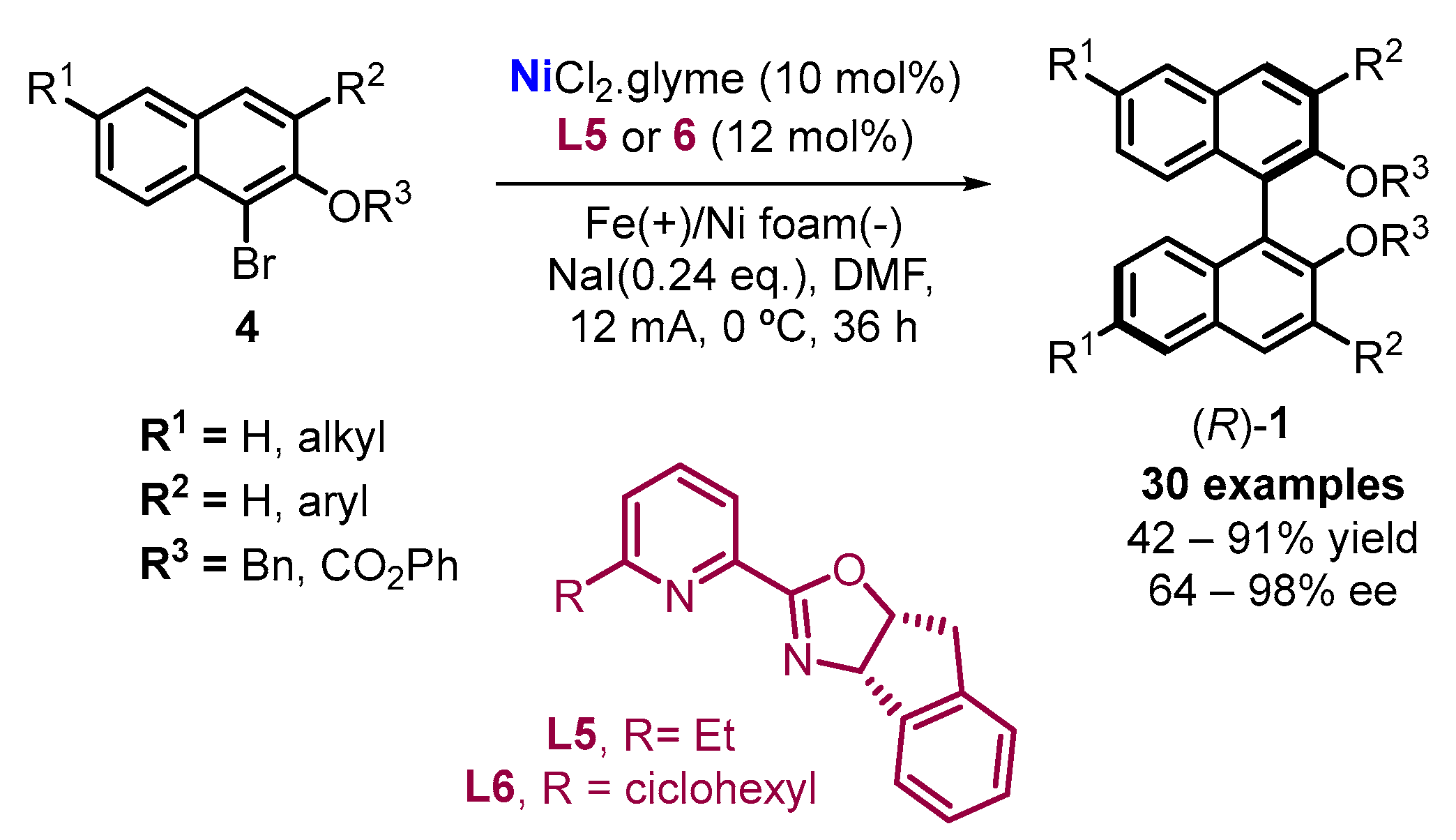
2. Electrochemical Synthesis
References
- Feringa, B.; Wynberg, H. Biomimetic Asymmetric Oxidative Coupling of Phenols. Bioorg. Chem. 1978, 7, 397–408.
- Brussee, J.; Groenendijk, J.L.G.; te Koppele, J.M.; Jansen, A.C.A. On the Mechanism of the Formation of s(−)-(1, 1’-Binaphthalene)-2,2’-Diol via Copper(II)Amine Complexes. Tetrahedron 1985, 41, 3313–3319.
- Nakajima, M.; Miyoshi, I.; Kanayama, K.; Hashimoto, S.; Noji, M.; Koga, K. Enantioselective Synthesis of Binaphthol Derivatives by Oxidative Coupling of Naphthol Derivatives Catalyzed by Chiral Diamine Copper Complexes. J. Org. Chem. 1999, 64, 2264–2271.
- Caselli, A.; Giovenzana, G.B.; Palmisano, G.; Sisti, M.; Pilati, T. Synthesis of C2-Symmetrical Diamine Based on (1R)-(+)-Camphor and Application to Oxidative Aryl Coupling of Naphthols. Tetrahedron Asymmetry 2003, 14, 1451–1454.
- Gao, J.; Reibenspies, J.H.; Martell, A.E. Structurally Defined Catalysts for Enantioselective Oxidative Coupling Reactions. Angew. Chem. Int. Ed. 2003, 42, 6008–6012.
- Li, X.; Hewgley, J.B.; Mulrooney, C.A.; Yang, J.; Kozlowski, M.C. Enantioselective Oxidative Biaryl Coupling Reactions Catalyzed by 1,5-Diazadecalin Metal Complexes: Efficient Formation of Chiral Functionalized BINOL Derivatives. J. Org. Chem. 2003, 68, 5500–5511.
- Kim, K.H.; Lee, D.-W.; Lee, Y.-S.; Ko, D.-H.; Ha, D.-C. Enantioselective Oxidative Coupling of Methyl 3-Hydroxy-2-Naphthoate Using Mono-N-Alkylated Octahydrobinaphthyl-2,2′-Diamine Ligand. Tetrahedron 2004, 60, 9037–9042.
- Temma, T.; Habaue, S. Highly Selective Oxidative Cross-Coupling of 2-Naphthol Derivatives with Chiral Copper(I)–Bisoxazoline Catalysts. Tetrahedron Lett. 2005, 46, 5655–5657.
- Zhang, Q.; Cui, X.; Chen, L.; Liu, H.; Wu, Y. Syntheses of Chiral Ferrocenophanes and Their Application to Asymmetric Catalysis. Eur. J. Org. Chem. 2014, 2014, 7823–7829.
- Egami, H.; Katsuki, T. Iron-Catalyzed Asymmetric Aerobic Oxidation: Oxidative Coupling of 2-Naphthols. J. Am. Chem. Soc. 2009, 131, 6082–6083.
- Tkachenko, N.V.; Lyakin, O.Y.; Samsonenko, D.G.; Talsi, E.P.; Bryliakov, K.P. Highly Efficient Asymmetric Aerobic Oxidative Coupling of 2-Naphthols in the Presence of Bioinspired Iron Aminopyridine Complexes. Catal. Commun. 2018, 104, 112–117.
- Hon, S.-W.; Li, C.-H.; Kuo, J.-H.; Barhate, N.B.; Liu, Y.-H.; Wang, Y.; Chen, C.-T. Catalytic Asymmetric Coupling of 2-Naphthols by Chiral Tridentate Oxovanadium(IV) Complexes. Org. Lett. 2001, 3, 869–872.
- Chu, C.-Y.; Hwang, D.-R.; Wang, S.-K.; Uang, B.-J. Chiral Oxovanadium Complex Catalyzed Enantioselective Oxidative Coupling of 2-Naphthols. Chem. Commun. 2001, 11, 980–981.
- Barhate, N.B.; Chen, C.-T. Catalytic Asymmetric Oxidative Couplings of 2-Naphthols by Tridentate N-Ketopinidene-Based Vanadyl Dicarboxylates. Org. Lett. 2002, 4, 2529–2532.
- Luo, Z.; Liu, Q.; Gong, L.; Cui, X.; Mi, A.; Jiang, Y. The Rational Design of Novel Chiral Oxovanadium(IV) Complexes for Highly Enantioselective Oxidative Coupling of 2-Naphthols. Chem. Commun. 2002, 8, 914–915.
- Somei, H.; Asano, Y.; Yoshida, T.; Takizawa, S.; Yamataka, H.; Sasai, H. Dual Activation in a Homolytic Coupling Reaction Promoted by an Enantioselective Dinuclear Vanadium(IV) Catalyst. Tetrahedron Lett. 2004, 45, 1841–1844.
- Guo, Q.-X.; Wu, Z.-J.; Luo, Z.-B.; Liu, Q.-Z.; Ye, J.-L.; Luo, S.-W.; Cun, L.-F.; Gong, L.-Z. Highly Enantioselective Oxidative Couplings of 2-Naphthols Catalyzed by Chiral Bimetallic Oxovanadium Complexes with Either Oxygen or Air as Oxidant. J. Am. Chem. Soc. 2007, 129, 13927–13938.
- Tanaka, H.; Nishikawa, H.; Uchida, T.; Katsuki, T. Photopromoted Ru-Catalyzed Asymmetric Aerobic Sulfide Oxidation and Epoxidation Using Water as a Proton Transfer Mediator. J. Am. Chem. Soc. 2010, 132, 12034–12041.
- Brunel, J.M. BINOL: A Versatile Chiral Reagent. Chem. Rev. 2005, 105, 857–898.
- Wang, H. Recent Advances in Asymmetric Oxidative Coupling of 2-Naphthol and Its Derivatives. Chirality 2010, 22, 827–837.
- Tkachenko, N.V.; Bryliakov, K.P. Transition Metal Catalyzed Aerobic Asymmetric Coupling of 2-Naphthols. Mini. Rev. Org. Chem. 2019, 16, 392–398.
- Liao, G.; Zhou, T.; Yao, Q.-J.; Shi, B.-F. Recent Advances in the Synthesis of Axially Chiral Biaryls via Transition Metal-Catalysed Asymmetric C–H Functionalization. Chem. Commun. 2019, 55, 8514–8523.
- Matsushita, M.; Kamata, K.; Yamaguchi, K.; Mizuno, N. Heterogeneously Catalyzed Aerobic Oxidative Biaryl Coupling of 2-Naphthols and Substituted Phenols in Water. J. Am. Chem. Soc. 2005, 127, 6632–6640.
- Egami, H.; Matsumoto, K.; Oguma, T.; Kunisu, T.; Katsuki, T. Enantioenriched Synthesis of C1-Symmetric BINOLs: Iron-Catalyzed Cross-Coupling of 2-Naphthols and Some Mechanistic Insight. J. Am. Chem. Soc. 2010, 132, 13633–13635.
- Narute, S.; Parnes, R.; Toste, F.D.; Pappo, D. Enantioselective Oxidative Homocoupling and Cross-Coupling of 2-Naphthols Catalyzed by Chiral Iron Phosphate Complexes. J. Am. Chem. Soc. 2016, 138, 16553–16560.
- Libman, A.; Shalit, H.; Vainer, Y.; Narute, S.; Kozuch, S.; Pappo, D. Synthetic and Predictive Approach to Unsymmetrical Biphenols by Iron-Catalyzed Chelated Radical–Anion Oxidative Coupling. J. Am. Chem. Soc. 2015, 137, 11453–11460.
- Shibasaki, M.; Matsunaga, S. Design and Application of Linked-BINOL Chiral Ligands in Bifunctional Asymmetric Catalysis. Chem. Soc. Rev. 2006, 35, 269.
- Tian, J.; Wang, A.; Yang, J.; Zhao, X.; Tu, Y.; Zhang, S.; Chen, Z. Copper-Complex-Catalyzed Asymmetric Aerobic Oxidative Cross-Coupling of 2-Naphthols: Enantioselective Synthesis of 3,3′-Substituted C1 -Symmetric BINOLs. Angew. Chem. 2019, 131, 11139–11143.
- Zang, C.; Liu, Y.; Xu, Z.-J.; Tse, C.-W.; Guan, X.; Wei, J.; Huang, J.-S.; Che, C.-M. Highly Enantioselective Iron-Catalyzed Cis -Dihydroxylation of Alkenes with Hydrogen Peroxide Oxidant via an Fe III -OOH Reactive Intermediate. Angew. Chem. 2016, 128, 10409–10413.
- Wu, L.Y.; Usman, M.; Liu, W.B. Enantioselective Iron/Bisquinolyldiamine Ligand-catalyzed Oxidative Coupling Reaction of 2-naphthols. Molecules 2020, 25, 852.
- Hayashi, H.; Ueno, T.; Kim, C.; Uchida, T. Ruthenium-Catalyzed Cross-Selective Asymmetric Oxidative Coupling of Arenols. Org. Lett. 2020, 22, 1469–1474.
- Chinnaraja, E.; Arunachalam, R.; Pillai, R.S.; Peuronen, A.; Rissanen, K.; Subramanian, P.S. One-Pot Synthesis of -Helicate-like Macrocycle and 2+4-Μ4-Oxo Tetranuclear Open Frame Complexes: Chiroptical Properties and Asymmetric Oxidative Coupling of 2-Naphthols. Appl. Organomet. Chem. 2020, 34, e5666.
- Horibe, T.; Nakagawa, K.; Hazeyama, T.; Takeda, K.; Ishihara, K. An Enantioselective Oxidative Coupling Reaction of 2-Naphthol Derivatives Catalyzed by Chiral Diphosphine Oxide-Iron(II) Complexes. Chem. Commun. 2019, 55, 13677–13680.
- Wang, P.; Cen, S.; Gao, J.; Shen, A.; Zhang, Z. Novel Axially Chiral Ligand-Enabled Copper-Catalyzed Asymmetric Oxidative Coupling of 2-Naphthols for the Synthesis of 6,6′-Disubstituted BINOLs. Org. Lett. 2022, 24, 2321–2326.
- Kumar, A.; Sasai, H.; Takizawa, S. Atroposelective Synthesis of C-C Axially Chiral Compounds via Mono- and Dinuclear Vanadium Catalysis. Acc. Chem. Res. 2022, 55, 2949–2965.
- Qiu, H.; Shuai, B.; Wang, Y.Z.; Liu, D.; Chen, Y.G.; Gao, P.-S.; Ma, H.X.; Ma, H.X.; Chen, S.; Mei, T.S. Enantioselective Ni-Catalyzed Electrochemical Synthesis of Biaryl Atropisomers. J. Am. Chem. Soc. 2020, 142, 9872–9878.
- Osa, T.; Kashiwagi, Y.; Yanagisawa, Y.; Bobbitt, J.M. Enantioselective, Electrocatalytic Oxidative Coupling of Naphthol, Naphthyl Ether and Phenanthrol on a TEMPO-Modified Graphite Felt Electrode in the Presence of (-)-Sparteine (TEMPO = 2,2,6,6-Tetramethylpiperidin-1-Yloxyl). J. Chem. Soc. Chem. Commun. 1994, 1, 2535–2537.

