Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Francesca Duraturo | -- | 1402 | 2022-12-27 15:08:07 | | | |
| 2 | Conner Chen | Meta information modification | 1402 | 2022-12-28 07:03:11 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nolano, A.; Medugno, A.; Trombetti, S.; Liccardo, R.; Rosa, M.D.; Izzo, P.; Duraturo, F. Mismatch Repair Genes in Lynch Syndrome. Encyclopedia. Available online: https://encyclopedia.pub/entry/39456 (accessed on 25 July 2026).
Nolano A, Medugno A, Trombetti S, Liccardo R, Rosa MD, Izzo P, et al. Mismatch Repair Genes in Lynch Syndrome. Encyclopedia. Available at: https://encyclopedia.pub/entry/39456. Accessed July 25, 2026.
Nolano, Antonio, Alessia Medugno, Silvia Trombetti, Raffaella Liccardo, Marina De Rosa, Paola Izzo, Francesca Duraturo. "Mismatch Repair Genes in Lynch Syndrome" Encyclopedia, https://encyclopedia.pub/entry/39456 (accessed July 25, 2026).
Nolano, A., Medugno, A., Trombetti, S., Liccardo, R., Rosa, M.D., Izzo, P., & Duraturo, F. (2022, December 27). Mismatch Repair Genes in Lynch Syndrome. In Encyclopedia. https://encyclopedia.pub/entry/39456
Nolano, Antonio, et al. "Mismatch Repair Genes in Lynch Syndrome." Encyclopedia. Web. 27 December, 2022.
Copy Citation
Hereditary non-polyposis colorectal cancer is also known as Lynch syndrome. Lynch syndrome is associated with pathogenetic variants in one of the mismatch repair (MMR) genes.
Lynch syndrome
MMR genes
VUS MMR genes
MSI-status
lynch like syndrome
1. Introduction
Lynch syndrome (LS) is the most common form of hereditary colorectal cancer, with an incidence of between 2% and 3% of all colorectal cancers (CRCs) [1], followed by familial adenomatous polyposis (FAP), which accounts for less than 1% of total CRCs [2] and other inherited syndromes, such as hamartomatous polyposis [3], Table 1. LS is also known as hereditary non-polyposis colorectal cancer (HNPCC); however, colorectal cancer develops due to a malignant transformation of adenomatous polyps, but they are not numerous and widespread as instead observed in FAP, which is characterized by 100–1000 polyps [4].
Table 1. Hereditary syndromes with known genetic predisposition to CRC.
| Syndrome | Genes | Hereditary | Incidence | Lifetime crc Risk |
|---|---|---|---|---|
| Lynch Syndrome LS |
MLH1, MSH2, MSH6, PMS2, EPCAM | AD | 3–5% | 15–90% |
| Familial Adenomatous Polyposis FAP |
APC | AD | 1% | Classic forms 100%; Attenuated forms until 70% |
| Mutyh-Associated Polyposis MAP |
MUTYH | AR | 1% | 43–99% |
| Peutz-jeghers Syndrome PJS |
STK11 | AD | <1% | 39% |
| Juvenile Polyposis JPS |
SMAD4, BMPR1A | AD | <1% | 39–69% |
Although the incidence of early-onset colorectal cancer, which occurs in individuals <50 years of age, has been increasing worldwide and particularly in high-income countries [5], LS patients generally develop colorectal cancer at an early age (on average about 45 years), with a predominance of 70% in the proximal/right colon [6].
Affected patients also present with synchronous tumors (multiple malignant tumors) and metachronous tumors (the appearance of a second tumor in one or more colorectal segments in patients who have already undergone resection surgery for cancer).
The precursor lesion of CRC in individuals with LS is an adenoma, which occasionally may be flat rather than raised or polypoid. Compared to patients with attenuated polyposis syndromes, patients with LS develop fewer colorectal adenomas by 50 years of age (usually less than three neoplasms) [6]. Colorectal adenomas in patients with LS exhibit accelerated carcinogenesis, leading to transition to carcinoma within 2 to 3 years, in contrast to the 8 to 10 years this process may take in the general population [7].
In addition to CRC, patients with LS have a significantly increased risk for a wide variety of cancers in other body sites, such as the endometrium, ovary, stomach, small intestine, hepatobiliary tract, pancreas, urinary tract, prostate, brain, and skin [7][8].
CRC associated with LS has clinical features distinct from those of sporadic CRC, often showing a combination of the presence of prominent tumor-infiltrating lymphocytes with marked lymphocytic inflammation that resembles the “Crohn’s-like reaction,” poor differentiation, and presence of mucinous and/or ring-like cells [9][10].
Although fewer studies have been published on non-colorectal LS-associated cancers, LS-associated endometrial cancers may be seen more frequently than their sporadic counterparts in the lower uterine segment; the majority are of the endometrioid type and often show poor differentiation, with tumor-infiltrating lymphocytes [11].
2. MMR Genes
LS is inherited in an autosomal dominant fashion and develops due to a germline mutation in one allele of one of the DNA MMR genes.
In the human mismatch repair (MMR) system, MSH2, MSH3, and MSH6 proteins associate in two heterodimeric complexes as MSH2-MSH6 (MutSα) and MSH2-MSH3 (MutSβ), which is homologous to the bacterial MutS protein [12], Table 2.
Table 2. MMR proteins and their functions.
| Bacterial MMR System | Yeast MMR System | Human MMR System | Functions |
|---|---|---|---|
| MutS | MutSα (MSH2/MSH6) MutSβ (MSH2/MSH3) |
MutSα (MSH2/MSH6) MutSβ (MSH2/MSH3) |
Mismatch recognition |
| MUTL | MutLα (MLH1/PMS1) MutLβ (MLH1/MLH2) MutLγ (MLH1/MLH3) |
MutLα (MLH1/PMS2) MutLβ (MLH1/PMS1) MutLγ (MLH1/MLH3) |
Match making |
| MutH | PCNA | PCNA | Strand incision |
| RFC | RFC | ||
| MutLα (MLH1/PMS1) MutLγ (MLH1/MLH3) |
MutLα (MLH1/PMS2) MutLγ (MLH1/MLH3) |
||
| RecJ | EXO1 | EXO1 | Strand excision (exonuclease) |
| ExoI | |||
| ExoVII | |||
| ExoX | |||
| UvrD | - | - | Strand excision (helicase) |
| DNA polymerase III |
DNA polymerase δ | DNA polymerase δ | Repair synthesis |
On the one hand, the first complex is able to recognize and bind DNA at the site of a mismatch due to substitution, insertion, or deletion of a single base. On the other hand, the second complex is responsible for the identification of insertions or deletions of a few nucleotides (2–4 bases). MSH2 protein is essential for the functional constitution of both complexes.
The heteroduplex formed by MLH1 and PMS2 (MutLα) or by MLH1 and MLH3 (MutLγ) interacts with the MutSα or MutSβ complex and stimulates the excision and resynthesis of DNA [13]. As already pointed out for the role of MSH2 protein within the MutSα–MutSβ complex, MLH1 protein is essential for the functional constitution of the MutLα and MutLγ complexes.
As a result, the MutLα–MutLγ complex coordinates the reciprocal action between the “mismatch” recognition complex and the other proteins necessary for the excision and resynthesis of the wrong strand. These additional proteins include DNA polymerases δ and ε (Polδ and Polε), the proliferating cell nuclear antigen factor (PCNA), an exonuclease (EXO1), and a replication factor C (RFC), Table 2.
The ATPase activity of the MutSα complex is important for the interaction with the unpaired DNA and the initiation of repair activity. MutSα binding stimulates the hydrolysis of ATP, leading to a conformational change that consequently triggers the recruitment of the MutLα complex. The tetrameric complex moving along the DNA looks for the mismatch present on the newly synthesized strand, which in turn activates the PCNA factor and RFC. MutLα possesses intrinsic ATP-mediated endonuclease activity, which is activated by PCNA. This activation causes an incision in the newly synthesized strand containing the error. This is followed by the recruitment of EXO1, which removes the newly synthesized strand containing the pairing error, in order that the strand can be synthesized again by DNA polymerase δ, while ligase 1 joins the previously created ends [14][15], Figure 1.
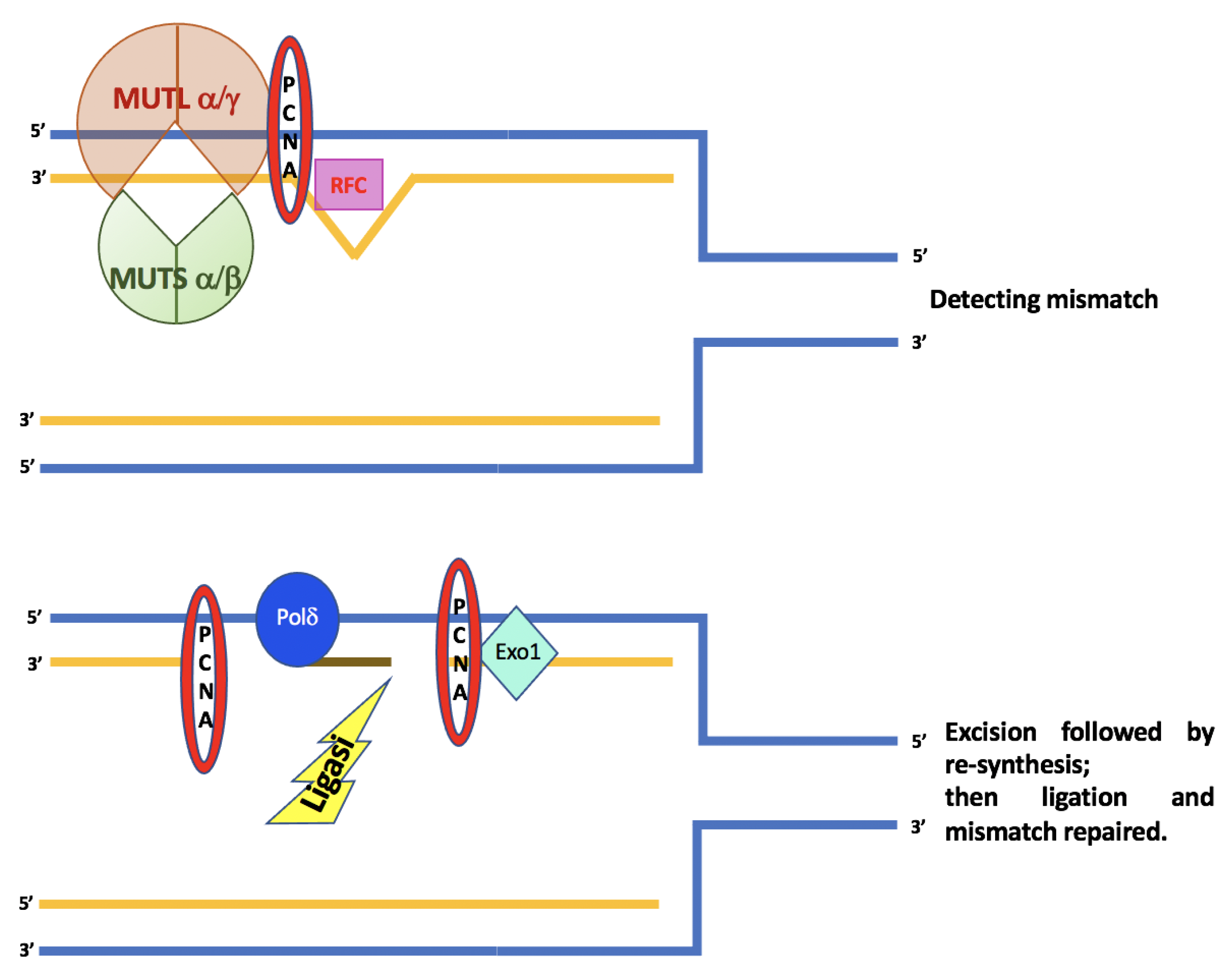
Figure 1. A schematic diagram for mechanisms and functions of human DNA mismatch repair.
3. Other Functions of MMR Genes
In addition to fulfilling their role in repairing DNA damage, MMR proteins perform other highly relevant functions in carcinogenesis [13]. As shown in Figure 2, these roles include:
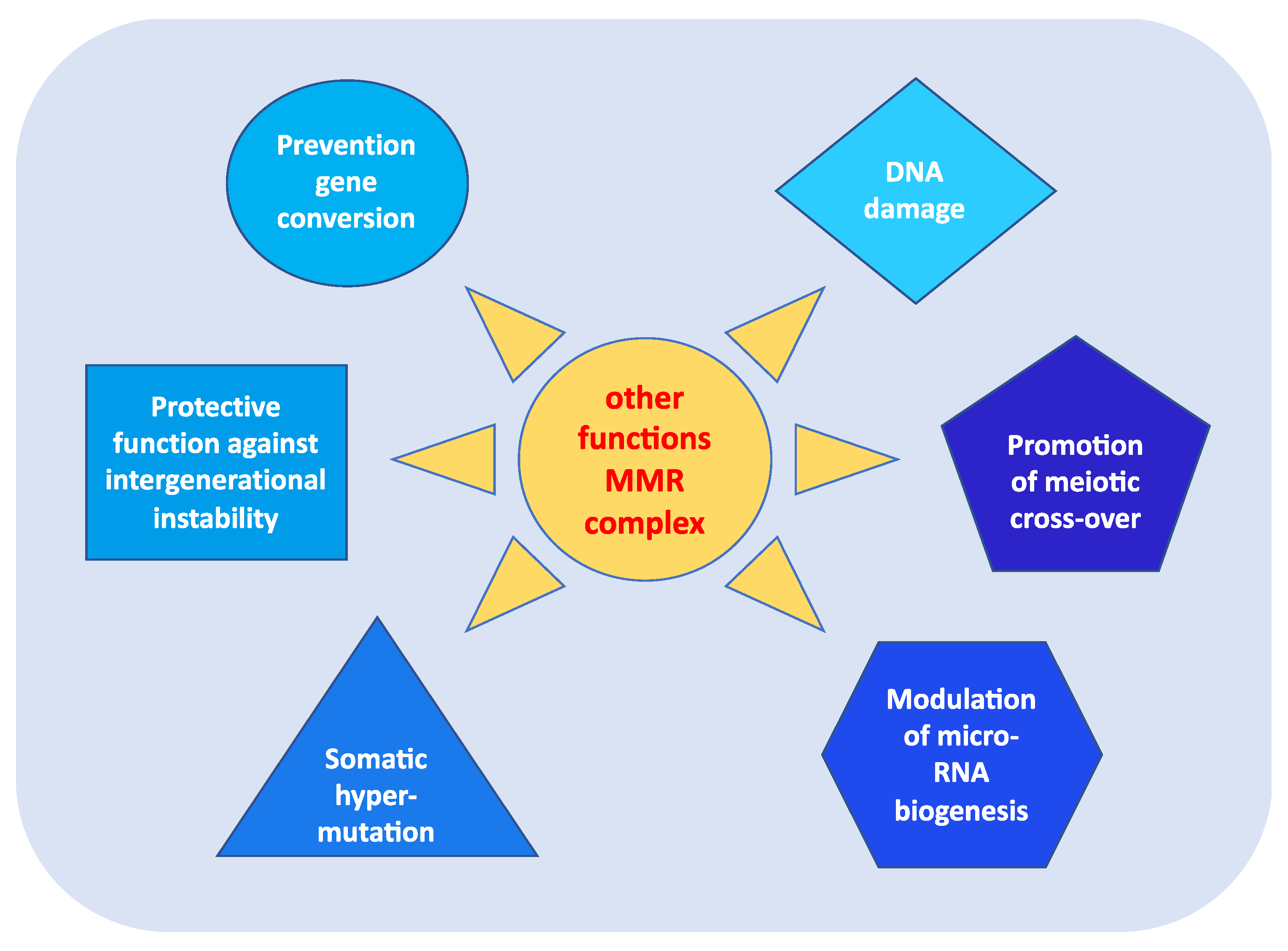
Figure 2. Other functions of MMR complex.
- prevention of reparative recombination (gene conversion) between non-identical sequences [16];
- promotion of meiotic cross-over, which involves the MLH1, PMS2, and MLH3 proteins in particular [16][17];
- protection against intergenerational instability resulting from the phenomenon of trinucleotide repeat expansion, which is the basis of the pathogenesis of various neurodegenerative diseases [18];
- the immunoglobulin (Ig) differentiation process based on “somatic hypermutation”, regulated by the MutSα–MutLα complex in combination with two other proteins, AID (activation-induced cytidine deaminase) and Polμ (error-prone DNA polymerase) [19];
- modulation of microRNA (miRNA) biogenesis through the interaction of MMR proteins with the microprocessor complex; in particular, MutLα specifically binds to pri-miRNAs and the Drosha–DGCR8 complex to stimulate the processing of pri-miRNAs into pre-miRNAs in a manner dependent on the ATPase activity of MutLα [20];
- reporting of DNA damage caused by exogenous carcinogens (heterocyclic amines, oxidizing agents, and UV radiation) obtained through a synergistic action between the homologous proteins of p53 (p53, p63, and p73) and the MutSα–MutLα complex. Moreover, in response to exogenous damage, MLH1 interacts with the MRE11 protein, a component of the BRCA1-associated surveillance complex (BASC), and regulates the cell cycle and the apoptotic pathway; indeed, there is a correlation between the MMR system and the G2/M phase of the cell cycle [21].
In particular, during carcinogenesis, the apoptotic mechanism is deregulated and thus the cells tend to escape the programmed death process and to bypass any cellular damage. Therefore, cells cannot fulfill their normal growth function if a mutation is present in some genes related to tumor development, and the MMR system may be representative of only this scenario. Indeed, several studies show how the MMR system plays an important role in the apoptotic machinery and in the activation of cell cycle check points [21][22][23]. Among the MMR genes, MLH1 and MSH2 are, above all, the most studied in relation to the anomalies found in apoptotic processes. In particular, the MSH2 gene plays a key role in genomic stability. In addition to its DNA damage repair function, it acts as a “sensor” for DNA replication errors caused by DNA base analogs and binds to various damage-induced DNA adducts to trigger cell cycle arrest or apoptosis [13][21][22][23].
The combination of all of these functions makes MMR proteins extremely important in maintaining the integrity of the genetic material, in the regulation of the cell cycle, and in the development of an effective immune system. Consequently, when the functionality of the MMR is lost or defective, there is a decrease in apoptosis and an increase in cell survival and mutagenesis induced by the damage, which leads to a selective growth of the defective cells with a parallel increase in tumor susceptibility.
References
- Abu-Ghazaleh, N.; Kaushik, V.; Gorelik, A.; Jenkins, M.; Macrae, F. Worldwide prevalence of Lynch syndrome in patients with colorectal cancer: Systematic review and meta-analysis. Anesthesia Analg. 2022, 24, 971–985.
- Marina, D.R.; Ugo, P.; Daniela, R.; Valeria, C.; Francesca, D.; Paola, I.; Paola, D. Genetics, diagnosis and management of colorectal cancer (Review). Oncol. Rep. 2015, 34, 1087–1096.
- Carlomagno, N.; Duraturo, F.; Candida, M.; De Rosa, M.; Varone, V.; Ciancia, G.; Calogero, A.; Santangelo, M.L. Multiple splenic hamartomas and familial adenomatous polyposis: A case report and review of the literature. J. Med Case Rep. 2015, 9, 1–6.
- Dodaro, C.; Grifasi, C.; Florio, J.; Santangelo, M.L.; Duraturo, F.; De Rosa, M.; Izzo, P.; Renda, A. The role of mutation analysis of the APC gene in the management of FAP patients. A controversial issue. Ann. Ital. di Chir. 2016, 87.
- Akimoto, N.; Ugai, T.; Zhong, R.; Hamada, T.; Fujiyoshi, K.; Giannakis, M.; Wu, K.; Cao, Y.; Ng, K.; Ogino, S. Rising incidence of early-onset colorectal cancer - a call to action. Nat. Rev. Clin. Oncol. 2021, 18, 230–243.
- Boyle, T.; Keegel, T.; Bull, F.; Heyworth, J.; Fritschi, L. Physical Activity and Risks of Proximal and Distal Colon Cancers: A Systematic Review and Meta-analysis. Gynecol. Oncol. 2012, 104, 1548–1561.
- Duraturo, F.; Liccardo, R.; De Rosa, M.; Izzo, P. Genetics, diagnosis and treatment of Lynch syndrome: Old lessons and current challenges (Review). Oncol. Lett. 2019, 17, 3048–3054.
- Li, X.; Liu, G.; Wu, W. Recent advances in Lynch syndrome. Exp. Hematol. Oncol. 2021, 10, 1–8.
- Aune, D.; Chan, D.S.M.; Lau, R.; Vieira, R.; Greenwood, D.C.; Kampman, E.; Norat, T. Dietary fibre, whole grains, and risk of colorectal cancer: Systematic review and dose-response meta-analysis of prospective studies. BMJ 2011, 343, d6617.
- Lichtenstein, P.; Holmm, N.V.; Verkasalom, P.K.; Iliadoum, A.; Kapriom, J.; Koskenvuom, M.; Pukkalam, E.; Skytthem, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer—Analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000, 343, 78–85.
- De la Chapelle, A. Genetic predisposition to colorectal cancer. Nat. Rev. Cancer 2004, 4, 769–780.
- Jeon, Y.; Kim, D.; Martín-López, J.V.; Lee, R.; Oh, J.; Hanne, J.; Fishel, R.; Lee, J.-B. Dynamic control of strand excision during human DNA mismatch repair. Proc. Natl. Acad. Sci. USA 2016, 113, 3281–3286.
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346.
- Hsieh, P.; Yamane, K. DNA mismatch repair: Molecular mechanism, cancer, and ageing. Mech. Ageing Dev. 2008, 129, 391–407.
- Ijsselsteijn, R.; Jansen, J.G.; de Wind, N. DNA mismatch repair-dependent DNA damage responses and cancer. DNA Repair 2020, 93, 102923.
- Nicholsonm, A.; Hendrixm, M.; Jinks-Robertsonm, S.; Crouse, G.F. Regulation of mitotic homeologous recombination in yeast: Functions of mismatch repair and nucleotide excision repair genes. Genetics 2000, 154, 133–146.
- Ji, G.; Long, Y.; Zhou, Y.; Huang, C.; Gu, A.; Wang, X. Common variants in mismatch repair genes associated with increased risk of sperm DNA damage and male infertility. BMC Med. 2012, 10, 49.
- Tomé, S.; Holt, I.; Edelmann, W.; Morris, G.E.; Munnich, A.; Pearson, C.; Gourdon, G. MSH2 ATPase Domain Mutation Affects CTG•CAG Repeat Instability in Transgenic Mice. PLoS Genet. 2009, 5, e1000482.
- Zanotti, K.J.; Gearhart, P.J. Antibody diversification caused by disrupted mismatch repair and promiscuous DNA polymerases. DNA Repair. (Amst.) 2016, 38, 110–116.
- Mao, G.; Pan, X.; Gu, L. Evidence that a mutation in the MLH1 3’-untranslated region confers a mutator phenotype and mismatch repair deficiency in patients with relapsed leukemia. JBC 2008, 283, 3211–3216.
- O’Brien, V.; Brown, R. Signalling cell cycle arrest and cell death through the MMR System. Carcinog 2005, 27, 682–692.
- Jiricny, J.; Nyström-Lahti, M. Mismatch repair defects in cancer. Curr. Opin. Genet. Dev. 2000, 10, 157–161.
- Li, Z.; Pearlman, A.H.; Hsieh, P. DNA mismatch repair and the DNA damage response. DNA Repair 2015, 38, 94–101.
More
Information
Subjects:
Genetics & Heredity; Pathology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
886
Revisions:
2 times
(View History)
Update Date:
28 Dec 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No